
Targeting Cartilage Degradation in Osteoarthritis

Abstract
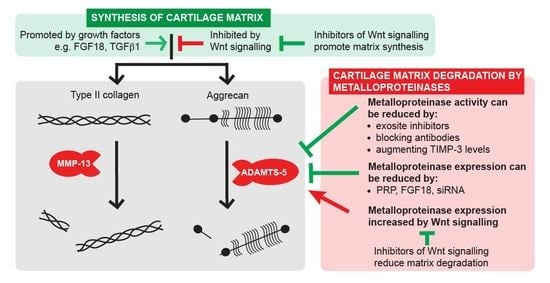
1. Introduction
2. Novel Strategies for Inhibiting Cartilage Breakdown
2.1. Inhibiting ADAMTS Activity
2.2. Augmenting Levels of Endogenous TIMPs
3. Promoting Cartilage Repair with Anabolic Growth Factors
3.1. Platelet-Rich Plasma Therapy
3.2. FGF18 Promotes Cartilage Anabolism
3.3. Wnt Pathway Inhibition
3.4. TGF-β1 Supplementation
3.5. Considerations around Receptor Expression and Downstream Signaling
4. Targeting Therapies to Cartilage
4.1. Strategies for Delivery to the Cartilage Matrix or to Chondrocytes
4.1.1. Targeting Type II Collagen
4.1.2. Targeting Aggrecan
4.1.3. Targeting Chondrocytes
4.2. Increasing Sophistication to Tailor Avidity and Enable DMOAD Latency
5. Broader Challenges in DMOAD Development
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- James, S.L.; Abate, D.; Abate, K.H.; Abay, S.M.; Abbafati, C.; Abbasi, N.; Abbastabar, H.; Abd-Allah, F.; Abdela, J.; Abdelalim, A.; et al. Global, regional, and national incidence, prevalence, and years lived with disability for 354 Diseases and Injuries for 195 countries and territories, 1990–2017: A systematic analysis for the Global Burden of Disease Study 2017. Lancet 2018, 392, 1789–1858. [Google Scholar] [CrossRef]
- Hunter, D.J.; Bierma-Zeinstra, S. Osteoarthritis. Lancet 2019, 393, 1745–1759. [Google Scholar] [CrossRef]
- Prieto-Alhambra, D.; Judge, A.; Javaid, M.K.; Cooper, C.; Diez-Perez, A.; Arden, N.K. Incidence and risk factors for clinically diagnosed knee, hip and hand osteoarthritis: Influences of age, gender and osteoarthritis affecting other joints. Ann. Rheum. Dis. 2014, 73, 1659–1664. [Google Scholar] [CrossRef] [PubMed]
- Donell, S. Subchondral bone remodelling in osteoarthritis. EFORT Open Rev. 2019, 4, 221–229. [Google Scholar] [CrossRef]
- Belluzzi, E.; Stocco, E.; Pozzuoli, A.; Granzotto, M.; Porzionato, A.; Vettor, R.; De Caro, R.; Ruggieri, P.; Ramonda, R.; Rossato, M.; et al. Contribution of Infrapatellar Fat Pad and Synovial Membrane to Knee Osteoarthritis Pain. Biomed. Res. Int. 2019, 2019, 6390182. [Google Scholar] [CrossRef] [PubMed]
- Englund, M.; Guermazi, A.; Lohmander, S.L. The Role of the Meniscus in Knee Osteoarthritis: A Cause or Consequence? Radiol. Clin. N. Am. 2009, 47, 703–712. [Google Scholar] [CrossRef] [PubMed]
- Latourte, A.; Kloppenburg, M.; Richette, P. Emerging pharmaceutical therapies for osteoarthritis. Nat. Rev. Rheumatol. 2020, 16, 673–688. [Google Scholar] [CrossRef] [PubMed]
- Heinegård, D.; Saxne, T. The role of the cartilage matrix in osteoarthritis. Nat. Rev. Rheumatol. 2011, 7, 50–56. [Google Scholar] [CrossRef] [PubMed]
- Burleigh, A.; Chanalaris, A.; Gardiner, M.D.; Driscoll, C.; Boruc, O.; Saklatvala, J.; Vincent, T.L. Joint immobilization prevents murine osteoarthritis and reveals the highly mechanosensitive nature of protease expression in vivo. Arthritis Rheumatol. 2012, 64, 2278–2288. [Google Scholar] [CrossRef]
- Troeberg, L.; Nagase, H. Proteases involved in cartilage matrix degradation in osteoarthritis. Biochim. Biophys. Acta 2012, 1824, 133–145. [Google Scholar] [CrossRef] [PubMed]
- Coryell, P.R.; Diekman, B.O.; Loeser, R.F. Mechanisms and therapeutic implications of cellular senescence in osteoarthritis. Nat. Rev. Rheumatol. 2020, 17, 47–57. [Google Scholar] [CrossRef]
- Ansari, M.Y.; Ahmad, N.; Voleti, S.; Wase, S.J.; Novak, K.; Haqqi, T.M. Mitochondrial dysfunction triggers a catabolic response in chondrocytes via ROS-mediated activation of the JNK/AP1 pathway. J. Cell Sci. 2020, 133, jcs247353. [Google Scholar] [CrossRef]
- Zahan, O.M.; Serban, O.; Gherman, C.; Fodor, D. The evaluation of oxidative stress in osteoarthritis. Med. Pharm. Rep. 2020, 93, 12–22. [Google Scholar] [CrossRef]
- Ansari, M.Y.; Ahmad, N.; Haqqi, T.M. Oxidative stress and inflammation in osteoarthritis pathogenesis: Role of polyphenols. Biomed. Pharmacother. 2020, 129, 110452. [Google Scholar] [CrossRef] [PubMed]
- Bolduc, J.A.; Collins, J.A.; Loeser, R.F. Reactive oxygen species, aging and articular cartilage homeostasis. Free Radic. Biol. Med. 2019, 132, 73–82. [Google Scholar] [CrossRef]
- Billinghurst, R.C.C.; Dahlberg, L.; Ionescu, M.; Reiner, A.; Bourne, R.; Rorabeck, C.; Mitchell, P.; Hambor, J.; Diekmann, O.; Tschesche, H.; et al. Enhanced cleavage of type II collagen by collagenases in osteoarthritic articular cartilage. J. Clin. Investig. 1997, 99, 1534–1545. [Google Scholar] [CrossRef] [PubMed]
- Little, C.B.; Barai, A.; Burkhardt, D.; Smith, S.M.; Fosang, A.J.; Werb, Z.; Shah, M.; Thompson, E.W. Matrix metalloproteinase 13-deficient mice are resistant to osteoarthritic cartilage erosion but not chondrocyte hypertrophy or osteophyte development. Arthritis Rheum. 2009, 60, 3723–3733. [Google Scholar] [CrossRef] [PubMed]
- Neuhold, L.A.; Killar, L.; Zhao, W.; Sung, M.L.A.; Warner, L.; Kulik, J.; Turner, J.; Wu, W.; Billinghurst, C.; Meijers, T.; et al. Postnatal expression in hyaline cartilage of constitutively active human collagenase-3 (MMP-13) induces osteoarthritis in mice. J. Clin. Investig. 2001, 107, 35–44. [Google Scholar] [CrossRef]
- Stanton, H.; Rogerson, F.M.; East, C.J.; Golub, S.B.; Lawlor, K.E.; Meeker, C.T.; Little, C.B.; Last, K.; Farmer, P.J.; Campbell, I.K.; et al. ADAMTS5 is the major aggrecanase in mouse cartilage in vivo and in vitro. Nature 2005, 434, 648–652. [Google Scholar] [CrossRef]
- Glasson, S.S.; Askew, R.; Sheppard, B.; Carito, B.; Blanchet, T.; Ma, H.L.; Flannery, C.R.; Peluso, D.; Kanki, K.; Yang, Z.; et al. Deletion of active ADAMTS5 prevents cartilage degradation in a murine model of osteoarthritis. Nature 2005, 434, 644–648. [Google Scholar] [CrossRef] [PubMed]
- Bau, B.; Gebhard, P.M.; Haag, J.; Knorr, T.; Bartnik, E.; Aigner, T. Relative messenger RNA expression profiling of collagenases and aggrecanases in human articular chondrocytes in vivo and in vitro. Arthritis Rheum. 2002, 46, 2648–2657. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, K.; Troeberg, L.; Scilabra, S.D.; Pelosi, M.; Murphy, C.L.; Strickland, D.K.; Nagase, H. LRP-1-mediated endocytosis regulates extracellular activity of ADAMTS-5 in articular cartilage. FASEB J. 2013, 27, 511–521. [Google Scholar] [CrossRef]
- Walling, H.W.; Raggatt, L.J.; Irvine, D.W.; Barmina, O.Y.; Toledano, J.E.; Goldring, M.B.; Hruska, K.A.; Adkisson, H.D.; Burdge, R.E.; Gatt, C.J.; et al. Impairment of the collagenase-3 endocytotic receptor system in cells from patients with osteoarthritis. Osteoarthr. Cartil. 2003, 11, 854–863. [Google Scholar] [CrossRef][Green Version]
- Morris, K.J.; Cs-Szabo, G.; Cole, A.A. Characterization of TIMP-3 in human articular talar cartilage. Connect. Tissue Res. 2010, 51, 478–490. [Google Scholar] [CrossRef] [PubMed]
- Blaney Davidson, E.N.; van der Kraan, P.M.; van den Berg, W.B. TGF-β and osteoarthritis. Osteoarthr. Cartil. 2007, 15, 597–604. [Google Scholar] [CrossRef] [PubMed]
- Malfait, A.M.; Tortorella, M.D. The “elusive DMOAD”: Aggrecanase inhibition from laboratory to clinic. Clin. Exp. Rheumatol. 2019, 120, 130–134. [Google Scholar]
- Dancevic, C.M.; McCulloch, D.R. Current and emerging therapeutic strategies for preventing inflammation and aggrecanase-mediated cartilage destruction in arthritis. Arthritis Res. Ther. 2014, 16, 429. [Google Scholar] [CrossRef] [PubMed]
- Santamaria, S. ADAMTS-5: A difficult teenager turning 20. Int. J. Exp. Pathol. 2020, 101, 4–20. [Google Scholar] [CrossRef] [PubMed]
- Mehana, E.S.E.; Khafaga, A.F.; El-Blehi, S.S. The role of matrix metalloproteinases in osteoarthritis pathogenesis: An updated review. Life Sci. 2019, 234, 116786. [Google Scholar] [CrossRef]
- Xie, X.W.; Wan, R.Z.; Liu, Z.P. Recent Research Advances in Selective Matrix Metalloproteinase-13 Inhibitors as Anti-Osteoarthritis Agents. ChemMedChem 2017, 12, 1157–1168. [Google Scholar] [CrossRef] [PubMed]
- Pratta, M.A.; Yao, W.; Decicco, C.; Tortorella, M.D.; Liu, R.Q.; Copeland, R.A.; Magolda, R.; Newton, R.C.; Trzaskos, J.M.; Arner, E.C. Aggrecan Protects Cartilage Collagen from Proteolytic Cleavage. J. Biol. Chem. 2003, 278, 45539–45545. [Google Scholar] [CrossRef]
- Karsdal, M.A.; Madsen, S.H.; Christiansen, C.; Henriksen, K.; Fosang, A.J.; Sondergaard, B.C. Cartilage degradation is fully reversible in the presence of aggrecanase but not matrix metalloproteinase activity. Arthritis Res. Ther. 2008, 10, R63. [Google Scholar] [CrossRef]
- Ismail, H.M.; Yamamoto, K.; Vincent, T.L.; Nagase, H.; Troeberg, L.; Saklatvala, J. Interleukin-1 acts via the JNK-2 signaling pathway to induce aggrecan degradation by human chondrocytes. Arthritis Rheumatol. 2015, 67, 1826–1836. [Google Scholar] [CrossRef] [PubMed]
- Coussens, L.M.; Fingleton, B.; Matrisian, L.M. Matrix metalloproteinase inhibitors and cancer: Trials and tribulations. Science 2002, 295, 2387–2392. [Google Scholar] [CrossRef] [PubMed]
- Fields, G.B. The Rebirth of Matrix Metalloproteinase Inhibitors: Moving Beyond the Dogma. Cells 2019, 8, 984. [Google Scholar] [CrossRef]
- Brenneis, C.; Serruys, B.; Van Belle, T.; Poelmans, S.; Kleinschmidt-Doerr, K.; Guehring, H.; Michaelis, M.; Lindemann, S. Structural and symptomatic benefit of a half-live extended, systemically applied anti-ADAMTS-5 inhibitor (M6495). Osteoarthr. Cartil. 2018, 26, S299–S300. [Google Scholar] [CrossRef]
- Santamaria, S.; Cuffaro, D.; Nuti, E.; Ciccone, L.; Tuccinardi, T.; Liva, F.; D’Andrea, F.; de Groot, R.; Rossello, A.; Ahnström, J. Exosite inhibition of ADAMTS-5 by a glycoconjugated arylsulfonamide. Sci. Rep. 2021, 11, 949. [Google Scholar] [CrossRef]
- Larkin, J.; Lohr, T.; Elefante, L.; Shearin, J.; Matico, R.; Su, J.-L.; Xue, Y.; Liu, F.; Rossman, E.I.; Renninger, J.; et al. The highs and lows of translational drug development: Antibody-mediated inhibition of ADAMTS-5 for osteoarthritis disease modification. Osteoarthr. Cartil. 2014, 22, S483–S484. [Google Scholar] [CrossRef]
- Hattori, N.; Mochizuki, S.; Kishi, K.; Nakajima, T.; Takaishi, H.; D’Armiento, J.; Okada, Y. MMP-13 plays a role in keratinocyte migration, angiogenesis, and contraction in mouse skin wound healing. Am. J. Pathol. 2009, 175, 533–546. [Google Scholar] [CrossRef] [PubMed]
- Smith, L.R.; Kok, H.J.; Zhang, B.; Chung, D.; Spradlin, R.A.; Rakoczy, K.D.; Lei, H.; Boesze-Battaglia, K.; Barton, E.R. Matrix metalloproteinase 13 from satellite cells is required for efficient muscle growth and regeneration. Cell. Physiol. Biochem. 2020, 54, 333–353. [Google Scholar] [CrossRef]
- Kosaki, N.; Takaishi, H.; Kamekura, S.; Kimura, T.; Okada, Y.; Minqi, L.; Amizuka, N.; Chung, U.-I.; Nakamura, K.; Kawaguchi, H.; et al. Impaired bone fracture healing in matrix metalloproteinase-13 deficient mice. Biochem. Biophys. Res. Commun. 2007, 354, 846–851. [Google Scholar] [CrossRef] [PubMed]
- Behonick, D.J.; Xing, Z.; Lieu, S.; Buckley, J.M.; Lotz, J.C.; Marcucio, R.S.; Werb, Z.; Miclau, T.; Colnot, C. Role of matrix metalloproteinase 13 in both endochondral and intramembranous ossification during skeletal regeneration. PLoS ONE 2007, 2, e1150. [Google Scholar] [CrossRef] [PubMed]
- Chu, X.; You, H.; Yuan, X.; Zhao, W.; Li, W.; Guo, X. Protective effect of lentivirus-mediated sirna targeting adamts-5 on cartilage degradation in a rat model of osteoarthritis. Int. J. Mol. Med. 2013, 31, 1222–1228. [Google Scholar] [CrossRef] [PubMed]
- Hoshi, H.; Akagi, R.; Yamaguchi, S.; Muramatsu, Y.; Akatsu, Y.; Yamamoto, Y.; Sasaki, T.; Takahashi, K.; Sasho, T. Effect of inhibiting MMP13 and ADAMTS5 by intra-articular injection of small interfering RNA in a surgically induced osteoarthritis model of mice. Cell Tissue Res. 2017, 368, 379–387. [Google Scholar] [CrossRef]
- Yamamoto, K.; Okano, H.; Miyagawa, W.; Visse, R.; Shitomi, Y.; Santamaria, S.; Dudhia, J.; Troeberg, L.; Strickland, D.K.; Hirohata, S.; et al. MMP-13 is constitutively produced in human chondrocytes and co-endocytosed with ADAMTS-5 and TIMP-3 by the endocytic receptor LRP1. Matrix Biol. 2016, 56, 57–73. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, K.; Scavenius, C.; Santamaria, S.; Botkjaer, K.A.; Dudhia, J.; Troeberg, L.; Itoh, Y.; Murphy, G.; Enghild, J.J.; Nagase, H. Inhibition of LRP1 shedding reverses cartilage degradation in osteoarthritis. Osteoarthr. Cartil. 2018, 26 (Suppl. S1), S22. [Google Scholar] [CrossRef]
- Kashiwagi, M.; Tortorella, M.; Nagase, H.; Brew, K. TIMP-3 Is a Potent Inhibitor of Aggrecanase 1 (ADAM-TS4) and Aggrecanase 2 (ADAM-TS5). J. Biol. Chem. 2001, 276, 12501–12504. [Google Scholar] [CrossRef]
- Brew, K.; Nagase, H. The tissue inhibitors of metalloproteinases (TIMPs): An ancient family with structural and functional diversity. Biochim. Biophys. Acta Mol. Cell Res. 2010, 1803, 55–71. [Google Scholar] [CrossRef] [PubMed]
- Davidson, R.K.; Waters, J.G.; Kevorkian, L.; Darrah, C.; Cooper, A.; Donell, S.T.; Clark, I.M. Expression profiling of metalloproteinases and their inhibitors in synovium and cartilage. Arthritis Res. Ther. 2006, 8, R124. [Google Scholar] [CrossRef]
- Scilabra, S.D.; Troeberg, L.; Yamamoto, K.; Emonard, H.; Thøgersen, I.; Enghild, J.J.; Strickland, D.K.; Nagase, H. Differential regulation of extracellular tissue inhibitor of metalloproteinases-3 levels by cell membrane-bound and shed low density lipoprotein receptor-related protein 1. J. Biol. Chem. 2013, 288, 332–342. [Google Scholar] [CrossRef]
- Troeberg, L.; Lazenbatt, C.; Anower-E-Khuda, M.F.; Freeman, C.; Federov, O.; Habuchi, H.; Habuchi, O.; Kimata, K.; Nagase, H. Sulfated glycosaminoglycans control the extracellular trafficking and the activity of the metalloprotease inhibitor TIMP-3. Chem. Biol. 2014, 21, 1300–1309. [Google Scholar] [CrossRef] [PubMed]
- Carreca, A.P.; Pravatà, V.M.; Markham, M.; Bonelli, S.; Murphy, G.; Nagase, H.; Troeberg, L.; Scilabra, S.D. TIMP-3 facilitates binding of target metalloproteinases to the endocytic receptor LRP-1 and promotes scavenging of MMP-1. Sci. Rep. 2020, 10, 12067. [Google Scholar] [CrossRef] [PubMed]
- Doherty, C.M.; Visse, R.; Dinakarpandian, D.; Strickland, D.K.; Nagase, H.; Troeberg, L. Engineered tissue inhibitor of metalloproteinases-3 variants resistant to endocytosis have prolonged chondroprotective activity. J. Biol. Chem. 2016, 291, 22160–22172. [Google Scholar] [CrossRef] [PubMed]
- Troeberg, L.; Fushimi, K.; Khokha, R.; Emonard, H.; Ghosh, P.; Nagase, H. Calcium pentosan polysulfate is a multifaceted exosite inhibitor of aggrecanases. FASEB J. 2008, 22, 3515–3524. [Google Scholar] [CrossRef] [PubMed]
- Chanalaris, A.; Doherty, C.M.; Marsden, B.D.; Bambridge, G.; Wren, S.P.; Nagase, H.; Troeberg, L. Suramin inhibits osteoarthritic cartilage degradation by increasing extracellular levels of chondroprotective tissue inhibitor of metalloproteinases 3. Mol. Pharmacol. 2017, 92, 459–468. [Google Scholar] [CrossRef]
- Guns, L.-A.; Monteagudo, S.; Kvasnytsia, M.; Kerckhofs, G.; Vandooren, J.; Opdenakker, G.; Lories, R.J.; Cailotto, F. Suramin increases cartilage proteoglycan accumulation in vitro and protects against joint damage triggered by papain injection in mouse knees in vivo. RMD Open 2017, 3, e000604. [Google Scholar] [CrossRef]
- Patel, J.M.; Saleh, K.S.; Burdick, J.A.; Mauck, R.L. Bioactive factors for cartilage repair and regeneration: Improving delivery, retention, and activity. Acta Biomater. 2019, 93, 222–238. [Google Scholar] [CrossRef]
- Everts, P.; Onishi, K.; Jayaram, P.; Lana, J.F.; Mautner, K. Platelet-rich plasma: New performance understandings and therapeutic considerations in 2020. Int. J. Mol. Sci. 2020, 21, 7794. [Google Scholar] [CrossRef] [PubMed]
- Akeda, K.; An, H.S.; Okuma, M.; Attawia, M.; Miyamoto, K.; Thonar, E.J.M.A.; Lenz, M.E.; Sah, R.L.; Masuda, K. Platelet-rich plasma stimulates porcine articular chondrocyte proliferation and matrix biosynthesis. Osteoarthr. Cartil. 2006, 14, 1272–1280. [Google Scholar] [CrossRef]
- Van Buul, G.M.; Koevoet, W.L.M.; Kops, N.; Bos, P.K.; Verhaar, J.A.N.; Weinans, H.; Bernsen, M.R.; Van Osch, G.J.V.M. Platelet-rich plasma releasate inhibits inflammatory processes in osteoarthritic chondrocytes. Am. J. Sports Med. 2011, 39, 2362–2370. [Google Scholar] [CrossRef] [PubMed]
- Chien, C.S.; Ho, H.O.; Liang, Y.C.; Ko, P.H.; Sheu, M.T.; Chen, C.H. Incorporation of exudates of human platelet-rich fibrin gel in biodegradable fibrin scaffolds for tissue engineering of cartilage. J. Biomed. Mater. Res. Part B Appl. Biomater. 2012, 100B, 948–955. [Google Scholar] [CrossRef]
- Pettersson, S.; Wetterö, J.; Tengvall, P.; Kratz, G. Human articular chondrocytes on macroporous gelatin microcarriers form structurally stable constructs with blood-derived biological glues in vitro. J. Tissue Eng. Regen. Med. 2009, 3, 450–460. [Google Scholar] [CrossRef]
- Gaissmaier, C.; Fritz, J.; Krackhardt, T.; Flesch, I.; Aicher, W.K.; Ashammakhi, N. Effect of human platelet supernatant on proliferation and matrix synthesis of human articular chondrocytes in monolayer and three-dimensional alginate cultures. Biomaterials 2005, 26, 1953–1960. [Google Scholar] [CrossRef]
- Sundman, E.A.; Cole, B.J.; Fortier, L.A. Growth factor and catabolic cytokine concentrations are influenced by the cellular composition of platelet-rich plasma. Am. J. Sports Med. 2011, 39, 2135–2140. [Google Scholar] [CrossRef] [PubMed]
- Patel, S.; Dhillon, M.S.; Aggarwal, S.; Marwaha, N.; Jain, A. Treatment with platelet-rich plasma is more effective than placebo for knee osteoarthritis: A prospective, double-blind, randomized trial. Am. J. Sports Med. 2013, 41, 356–364. [Google Scholar] [CrossRef] [PubMed]
- Cerza, F.; Carnì, S.; Carcangiu, A.; Di Vavo, I.; Schiavilla, V.; Pecora, A.; De Biasi, G.; Ciuffreda, M. Comparison between hyaluronic acid and platelet-rich plasma, intra-articular infiltration in the treatment of gonarthrosis. Am. J. Sports Med. 2012, 40, 2822–2827. [Google Scholar] [CrossRef] [PubMed]
- Sánchez, M.; Guadilla, J.; Fiz, N.; Andia, I. Ultrasound-guided platelet-rich plasma injections for the treatment of osteoarthritis of the hip. Rheumatology 2012, 51, 144–150. [Google Scholar] [CrossRef] [PubMed]
- Filardo, G.; Kon, E.; Di Martino, A.; Di Matteo, B.; Merli, M.L.; Cenacchi, A.; Fornasari, P.M.; Marcacci, M. Platelet-rich plasma vs hyaluronic acid to treat knee degenerative pathology: Study design and preliminary results of a randomized controlled trial. BMC Musculoskelet. Disord. 2012, 13, 229. [Google Scholar] [CrossRef] [PubMed]
- Dhurat, R.; Sukesh, M. Principles and methods of preparation of platelet-rich plasma: A review and author′s perspective. J. Cutan. Aesthet. Surg. 2014, 7, 189–197. [Google Scholar] [CrossRef] [PubMed]
- Ellsworth, J.L.; Berry, J.; Bukowski, T.; Claus, J.; Feldhaus, A.; Holderman, S.; Holdren, M.S.; Lum, K.D.; Moore, E.E.; Raymond, F.; et al. Fibroblast growth factor-18 is a trophic factor for mature chondrocytes and their progenitors. Osteoarthr. Cartil. 2002, 10, 308–320. [Google Scholar] [CrossRef]
- Mori, Y.; Saito, T.; Chang, S.H.; Kobayashi, H.; Ladel, C.H.; Guehring, H.; Chung, U.-I.; Kawaguchi, H. Identification of fibroblast growth factor-18 as a molecule to protect adult articular cartilage by gene expression profiling. J. Biol. Chem. 2014, 289, 10192–101200. [Google Scholar] [CrossRef] [PubMed]
- Moore, E.E.; Bendele, A.M.; Thompson, D.L.; Littau, A.; Waggie, K.S.; Reardon, B.; Ellsworth, J.L. Fibroblast growth factor-18 stimulates chondrogenesis and cartilage repair in a rat model of injury-induced osteoarthritis. Osteoarthr. Cartil. 2005, 13, 623–631. [Google Scholar] [CrossRef] [PubMed]
- Gigout, A.; Guehring, H.; Froemel, D.; Meurer, A.; Ladel, C.; Reker, D.; Bay-Jensen, A.C.; Karsdal, M.A.; Lindemann, S. Sprifermin (rhFGF18) enables proliferation of chondrocytes producing a hyaline cartilage matrix. Osteoarthr. Cartil. 2017, 25, 1858–1867. [Google Scholar] [CrossRef] [PubMed]
- Dahlberg, L.E.; Aydemir, A.; Muuraheinen, N.; Gühring, H.; Edebo, H.F.; Krarup-Jensen, N.; Ladel, C.H.; Jurvelin, J.S. A first-in-human, double-blind, randomised, placebo-controlled, dose ascending study of intra-articular rhFGF18 (sprifermin) in patients with advanced knee osteoarthritis. Clin. Exp. Rheumatol. 2016, 34, 445–450. [Google Scholar] [PubMed]
- Lohmander, L.S.; Hellot, S.; Dreher, D.; Krantz, E.F.W.; Kruger, D.S.; Guermazi, A.; Eckstein, F. Intraarticular sprifermin (recombinant human fibroblast growth factor 18) in knee osteoarthritis: A randomized, double-blind, placebo-controlled trial. Arthritis Rheumatol. 2014, 66, 1820–1831. [Google Scholar] [CrossRef]
- Hochberg, M.C.; Guermazi, A.; Guehring, H.; Aydemir, A.; Wax, S.; Fleuranceau-Morel, P.; Reinstrup Bihlet, A.; Byrjalsen, I.; Ragnar Andersen, J.; Eckstein, F. Effect of Intra-Articular Sprifermin vs Placebo on Femorotibial Joint Cartilage Thickness in Patients with Osteoarthritis: The FORWARD Randomized Clinical Trial. JAMA J. Am. Med. Assoc. 2019, 322, 1360–1370. [Google Scholar] [CrossRef] [PubMed]
- Usami, Y.; Gunawardena, A.T.; Iwamoto, M.; Enomoto-Iwamoto, M. Wnt signaling in cartilage development and diseases: Lessons from animal studies. Lab. Investig. 2016, 96, 186–196. [Google Scholar] [CrossRef] [PubMed]
- Lories, R.J.; Corr, M.; Lane, N.E. To Wnt or not to Wnt: The bone and joint health dilemma. Nat. Rev. Rheumatol. 2013, 9, 328–339. [Google Scholar] [CrossRef]
- Dell’Accio, F.; Cailotto, F. Pharmacological blockade of the WNT-beta-catenin signaling: A possible first-in-kind DMOAD. Osteoarthr. Cartil. 2018, 26, 4–6. [Google Scholar] [CrossRef] [PubMed]
- Hill, T.P.; Später, D.; Taketo, M.M.; Birchmeier, W.; Hartmann, C. Canonical Wnt/β-catenin signaling prevents osteoblasts from differentiating into chondrocytes. Dev. Cell 2005, 8, 727–738. [Google Scholar] [CrossRef]
- Kim, Y.S.; Lee, H.J.; Yeo, J.E.; Kim, Y.I.; Choi, Y.J.; Koh, Y.G. Isolation and Characterization of Human Mesenchymal Stem Cells Derived from Synovial Fluid in Patients with Osteochondral Lesion of the Talus. Am. J. Sports Med. 2015, 43, 399–406. [Google Scholar] [CrossRef] [PubMed]
- Barroga, C.; Hu, Y.; Deshmukh, V.; Hood, J. Discovery of an intra-articular injection small molecule inhibitor of the Wnt pathway (SM04690) as a potential disease modifying treatment for knee osteoarthritis. Arthritis Rheumatol. 2015, 67 (Suppl. S10). Available online: https://acrabstracts.org/abstract/discovery-of-an-intra-articular-injection-small-molecule-inhibitor-of-the-wnt-pathway-sm04690-as-a-potential-disease-modifying-treatment-for-knee-osteoarthritis/ (accessed on 23 January 2021).
- Deshmukh, V.; Hu, H.; Barroga, C.; Bossard, C.; KC, S.; Dellamary, L.; Stewart, J.; Chiu, K.; Ibanez, M.; Pedraza, M.; et al. A small-molecule inhibitor of the Wnt pathway (SM04690) as a potential disease modifying agent for the treatment of osteoarthritis of the knee. Osteoarthr. Cartil. 2018, 26, 18–27. [Google Scholar] [CrossRef] [PubMed]
- Deshmukh, V.; O’Green, A.L.; Bossard, C.; Seo, T.; Lamangan, L.; Ibanez, M.; Ghias, A.; Lai, C.; Do, L.; Cho, S.; et al. Modulation of the Wnt pathway through inhibition of CLK2 and DYRK1A by lorecivivint as a novel, potentially disease-modifying approach for knee osteoarthritis treatment. Osteoarthr. Cartil. 2019, 27, 1347–1360. [Google Scholar] [CrossRef] [PubMed]
- Yazici, Y.; McAlindon, T.E.; Fleischmann, R.; Gibofsky, A.; Lane, N.E.; Kivitz, A.J.; Skrepnik, N.; Armas, E.; Swearingen, C.J.; DiFrancesco, A.; et al. A novel Wnt pathway inhibitor, SM04690, for the treatment of moderate to severe osteoarthritis of the knee: Results of a 24-week, randomized, controlled, phase 1 study. Osteoarthr. Cartil. 2017, 25, 1598–1606. [Google Scholar] [CrossRef]
- Yazici, Y.; McAlindon, T.E.; Gibofsky, A.; Lane, N.E.; Clauw, D.; Jones, M.; Bergfeld, J.; Swearingen, C.J.; DiFrancesco, A.; Simsek, I.; et al. Lorecivivint, a Novel Intraarticular CDC-like Kinase 2 and Dual-Specificity Tyrosine Phosphorylation-Regulated Kinase 1A Inhibitor and Wnt Pathway Modulator for the Treatment of Knee Osteoarthritis: A Phase II Randomized Trial. Arthritis Rheumatol. 2020, 72, 1694–1706. [Google Scholar] [CrossRef] [PubMed]
- Held, A.; Glas, A.; Dietrich, L.; Bollmann, M.; Brandstädter, K.; Grossmann, T.N.; Lohmann, C.H.; Pap, T.; Bertrand, J. Targeting β-catenin dependent Wnt signaling via peptidomimetic inhibitors in murine chondrocytes and OA cartilage. Osteoarthr. Cartil. 2018, 26, 818–823. [Google Scholar] [CrossRef] [PubMed]
- Nalesso, G.; Sherwood, J.; Bertrand, J.; Pap, T.; Ramachandran, M.; de Bari, C.; Pitzalis, C.; Dell’Accio, F. WNT-3A modulates articular chondrocyte phenotype by activating both canonical and noncanonical pathways. J. Cell Biol. 2011, 193, 511–564. [Google Scholar] [CrossRef] [PubMed]
- Kizawa, H.; Kou, I.; Iida, A.; Sudo, A.; Miyamoto, Y.; Fukuda, A.; Mabuchi, A.; Kotani, A.; Kawakami, A.; Yamamoto, S.; et al. An aspartic acid repeat polymorphism in asporin inhibits chondrogenesis and increases susceptibility to osteoarthritis. Nat. Genet. 2005, 37, 138–144. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Q.; Shi, D.; Yi, L.; Ikegawa, S.; Wang, Y.; Nakamura, T.; Qiao, D.; Liu, C.; Dai, J. Replication of the association of the aspartic acid repeat polymorphism in the asporin gene with knee-osteoarthritis susceptibility in Han Chinese. J. Hum. Genet. 2006, 51, 1068–1072. [Google Scholar] [CrossRef]
- Valdes, A.M.; Spector, T.D.; Tamm, A.A.; Kisand, K.; Doherty, S.A.; Dennison, E.M.; Mangino, M.; Tamm, A.A.; Kerna, I.; Hart, D.J.; et al. Genetic variation in the SMAD3 gene is associated with hip and knee osteoarthritis. Arthritis Rheum. 2010, 62, 2347–2352. [Google Scholar] [CrossRef]
- Serra, R.; Johnson, M.; Filvaroff, E.H.; LaBorde, J.; Sheehan, D.M.; Derynck, R.; Moses, H.L. Expression of a truncated, kinase-defective TGF-β type II receptor in mouse skeletal tissue promotes terminal chondrocyte differentiation and osteoarthritis. J. Cell Biol. 1997, 139, 541–545. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.; Li, J.; Wang, B.; Jin, H.; Wang, M.; Zhang, Y.; Yang, Y.; Im, H.J.; O’Keefe, R.; Chen, D. Deletion of the transforming growth factor β Receptor type II gene in articular chondrocytes leads to a progressive osteoarthritis-like phenotype in mice. Arthritis Rheum. 2013, 65, 3107–3119. [Google Scholar] [CrossRef]
- Yang, X.; Chen, L.; Xu, X.; Li, C.; Huang, C.; Deng, C.X. TGF-β/Smad3 signals repress chondrocyte hypertrophic differentiation and are required for maintaining articular cartilage. J. Cell Biol. 2001, 153, 35–46. [Google Scholar] [CrossRef]
- Ha, C.W.; Noh, M.J.; Choi, K.B.; Lee, K.H. Initial phase i safety of retrovirally transduced human chondrocytes expressing transforming growth factor-beta-1 in degenerative arthritis patients. Cytotherapy 2012, 14, 247–256. [Google Scholar] [CrossRef]
- Ha, C.W.; Cho, J.J.; Elmallah, R.K.; Cherian, J.J.; Kim, T.W.; Lee, M.C.; Mont, M.A. A Multicenter, Single-Blind, Phase IIa Clinical Trial to Evaluate the Efficacy and Safety of a Cell-Mediated Gene Therapy in Degenerative Knee Arthritis Patients. Hum. Gene Ther. Clin. Dev. 2015, 26, 125–130. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.C.; Ha, C.-W.; Elmallah, R.K.; Cherian, J.J.; Cho, J.J.; Kim, T.W.; Bin, S.-I.; Mont, M.A. A placebo-controlled randomised trial to assess the effect of TGF-β1-expressing chondrocytes in patients with arthritis of the knee. Bone Jt. J. 2015, 97-B, 924–932. [Google Scholar] [CrossRef] [PubMed]
- Osiecka-Iwan, A.; Hyc, A.; Moskalewski, S. Immunosuppression and rejection of cartilage formed by allogeneic chondrocytes in rats. Cell Transplant. 1999, 8, 627–636. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.; Parvizi, J.; Bramlet, D.; Romness, D.W.; Guermazi, A.; Noh, M.; Sodhi, N.; Khlopas, A.; Mont, M.A. Results of a Phase II Study to Determine the Efficacy and Safety of Genetically Engineered Allogeneic Human Chondrocytes Expressing TGF-β1. J. Knee Surg. 2020, 33, 167–172. [Google Scholar] [CrossRef] [PubMed]
- Guermazi, A.; Kalsi, G.; Niu, J.; Crema, M.D.; Copeland, R.O.; Orlando, A.; Noh, M.J.; Roemer, F.W. Structural effects of intra-articular TGF-β1 in moderate to advanced knee osteoarthritis: MRI-based assessment in a randomized controlled trial. BMC Musculoskelet. Disord. 2017, 18, 461. [Google Scholar] [CrossRef] [PubMed]
- Blaney Davidson, E.N.; Vitters, E.L.; van den Berg, W.B.; van der Kraan, P.M. TGF β-induced cartilage repair is maintained but fibrosis is blocked in the presence of Smad7. Arthritis Res. Ther. 2006, 8, R65. [Google Scholar] [CrossRef] [PubMed]
- Im, H.J.; Li, X.; Muddasani, P.; Kim, G.H.; Davis, F.; Rangan, J.; Forsyth, C.B.; Ellman, M.; Thonar, E.J.M.A. Basic fibroblast growth factor accelerates matrix degradation via a neuro-endocrine pathway in human adult articular chondrocytes. J. Cell. Physiol. 2008, 215, 452–463. [Google Scholar] [CrossRef] [PubMed]
- Yan, D.; Chen, D.; Im, H.J. Fibroblast growth factor-2 promotes catabolism via FGFR1-Ras-Raf-MEK1/2-ERK1/2 axis that coordinates with the PKCδ pathway in human articular chondrocytes. J. Cell. Biochem. 2012, 113, 2856–2865. [Google Scholar] [CrossRef]
- Wang, X.; Manner, P.A.; Horner, A.; Shum, L.; Tuan, R.S.; Nuckolls, G.H. Regulation of MMP-13 expression by RUNX2 and FGF2 in osteoarthritic cartilage. Osteoarthr. Cartil. 2004, 12, 963–973. [Google Scholar] [CrossRef]
- Chia, S.L.; Sawaji, Y.; Burleigh, A.; McLean, C.; Inglis, J.; Saklatvala, J.; Vincent, T. Fibroblast growth factor 2 is an intrinsic chondroprotective agent that suppresses ADAMTS-5 and delays cartilage degradation in murine osteoarthritis. Arthritis Rheum. 2009, 60, 2019–2027. [Google Scholar] [CrossRef]
- Sawaji, Y.; Hynes, J.; Vincent, T.; Saklatvala, J. Fibroblast growth factor 2 inhibits induction of aggrecanase activity in human articular cartilage. Arthritis Rheum. 2008, 58, 3498–3509. [Google Scholar] [CrossRef]
- Yan, D.; Chen, D.; Cool, S.M.; van Wijnen, A.J.; Mikecz, K.; Murphy, G.; Im, H.J. Fibroblast growth factor receptor 1 is principally responsible for fibroblast growth factor 2-induced catabolic activities in human articular chondrocytes. Arthritis Res. Ther. 2011, 13, R130. [Google Scholar] [CrossRef] [PubMed]
- Chuang, C.Y.; Lord, M.S.; Melrose, J.; Rees, M.D.; Knox, S.M.; Freeman, C.; Iozzo, R.V.; Whitelock, J.M. Heparan sulfate-dependent signaling of fibroblast growth factor 18 by chondrocyte-derived perlecan. Biochemistry 2010, 49, 5524–5532. [Google Scholar] [CrossRef]
- Chanalaris, A.; Clarke, H.; Guimond, S.E.; Vincent, T.L.; Turnbull, J.E.; Troeberg, L. Heparan Sulfate Proteoglycan Synthesis Is Dysregulated in Human Osteoarthritic Cartilage. Am. J. Pathol. 2019, 189, 632–647. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Tao, H.; Jin, C.; Liu, Y.; Lu, X.; Hu, X.; Wang, X. Transforming growth factor-β1 induces type II collagen and aggrecan expression via activation of extracellular signal-regulated kinase 1/2 and Smad2/3 signaling pathways. Mol. Med. Rep. 2015, 12, 5573–5579. [Google Scholar] [CrossRef] [PubMed]
- Blaney Davidson, E.N.; Remst, D.F.G.; Vitters, E.L.; van Beuningen, H.M.; Blom, A.B.; Goumans, M.-J.; van den Berg, W.B.; van der Kraan, P.M. Increase in ALK1/ALK5 Ratio as a Cause for Elevated MMP-13 Expression in Osteoarthritis in Humans and Mice. J. Immunol. 2009, 182, 7937–7945. [Google Scholar] [CrossRef] [PubMed]
- Van Der Kraan, P.M.; Goumans, M.J.; Blaney Davidson, E.; Ten Dijke, P. Age-dependent alteration of TGF-β signalling in osteoarthritis. Cell Tissue Res. 2012, 347, 257–265. [Google Scholar] [CrossRef] [PubMed]
- Schultz, C. Targeting the extracellular matrix for delivery of bioactive molecules to sites of arthritis. Br. J. Pharmacol. 2019, 176, 26–37. [Google Scholar] [CrossRef]
- Rothenfluh, D.A.; Bermudez, H.; O’Neil, C.P.; Hubbell, J.A. Biofunctional polymer nanoparticles for intra-articular targeting and retention in cartilage. Nat. Mater. 2008, 7, 248–254. [Google Scholar] [CrossRef]
- Formica, F.A.; Barreto, G.; Zenobi-Wong, M. Cartilage-targeting dexamethasone prodrugs increase the efficacy of dexamethasone. J. Control. Release 2019, 295, 118–129. [Google Scholar] [CrossRef] [PubMed]
- Hu, H.Y.; Lim, N.H.; Ding-Pfennigdorff, D.; Saas, J.; Wendt, K.U.; Ritzeler, O.; Nagase, H.; Plettenburg, O.; Schultz, C.; Nazare, M. DOTAM Derivatives as Active Cartilage-Targeting Drug Carriers for the Treatment of Osteoarthritis. Bioconj. Chem. 2015, 26, 383–388. [Google Scholar] [CrossRef]
- Singh, A.; Corvelli, M.; Unterman, S.A.; Wepasnick, K.A.; McDonnell, P.; Elisseeff, J.H. Enhanced lubrication on tissue and biomaterial surfaces through peptide-mediated binding of hyaluronic acid. Nat. Mater. 2014, 13, 988–995. [Google Scholar] [CrossRef] [PubMed]
- Faust, H.J.; Sommerfeld, S.D.; Rathod, S.; Rittenbach, A.; Ray Banerjee, S.; Tsui, B.M.W.; Pomper, M.; Amzel, M.L.; Singh, A.; Elisseeff, J.H. A hyaluronic acid binding peptide-polymer system for treating osteoarthritis. Biomaterials 2018, 183, 93–101. [Google Scholar] [CrossRef]
- Brown, S.B.; Wang, L.; Jungels, R.R.; Sharma, B. Effects of cartilage-targeting moieties on nanoparticle biodistribution in healthy and osteoarthritic joints. Acta Biomater. 2020, 101, 469–483. [Google Scholar] [CrossRef]
- Shah, R.N.; Shah, N.A.; Lim, M.M.D.R.; Hsieh, C.; Nuber, G.; Stupp, S.I. Supramolecular design of self-assembling nanofibers for cartilage regeneration. Proc. Natl. Acad. Sci. USA 2010, 107, 3293–3298. [Google Scholar] [CrossRef] [PubMed]
- Pi, Y.; Zhang, X.; Shi, J.; Zhu, J.; Chen, W.; Zhang, C.; Gao, W.; Zhou, C.; Ao, Y. Targeted delivery of non-viral vectors to cartilage in vivo using a chondrocyte-homing peptide identified by phage display. Biomaterials 2011, 32, 6324–6332. [Google Scholar] [CrossRef] [PubMed]
- Pi, Y.; Zhang, X.; Shao, Z.; Zhao, F.; Hu, X.; Ao, Y. Intra-articular delivery of anti-Hif-2α siRNA by chondrocyte-homing nanoparticles to prevent cartilage degeneration in arthritic mice. Gene Ther. 2015, 22, 439–448. [Google Scholar] [CrossRef] [PubMed]
- Cheung, C.S.F.; Lui, J.C.; Baron, J. Identification of chondrocyte-binding peptides by phage display. J. Orthop. Res. 2013, 31, 1053–1058. [Google Scholar] [CrossRef] [PubMed]
- Loffredo, F.S.; Pancoast, J.R.; Cai, L.; Vannelli, T.; Dong, J.Z.; Lee, R.T.; Patwari, P. Targeted delivery to cartilage is critical for in vivo efficacy of insulin-like growth factor 1 in a rat model of osteoarthritis. Arthritis Rheumatol. 2014, 66, 1247–1255. [Google Scholar] [CrossRef] [PubMed]
- Silverman, J.; Lu, Q.; Bakker, A.; To, W.; Duguay, A.; Alba, B.M.; Smith, R.; Rivas, A.; Li, P.; Le, H.; et al. Multivalent avimer proteins evolved by exon shuffling of a family of human receptor domains. Nat. Biotechnol. 2005, 23, 1556–1561. [Google Scholar] [CrossRef] [PubMed]
- Hulme, J.T.; D’Souza, W.N.; McBride, H.J.; Yoon, B.R.P.; Willee, A.M.; Duguay, A.; Thomas, M.; Fan, B.; Dayao, M.R.; Rottman, J.B.; et al. Novel protein therapeutic joint retention strategy based on collagen-binding Avimers. J. Orthop. Res. 2018, 36, 1238–1247. [Google Scholar] [CrossRef]
- Hughes, C.; Faurholm, B.; Dell’Accio, F.; Manzo, A.; Seed, M.; Eltawil, N.; Marrelli, A.; Gould, D.; Subang, C.; Al-Kashi, A.; et al. Human single-chain variable fragment that specifically targets arthritic cartilage. Arthritis Rheum. 2010, 62, 1007–1016. [Google Scholar] [CrossRef]
- Hughes, C.; Sette, A.; Seed, M.; D’Acquisto, F.; Manzo, A.; Vincent, T.L.; Lim, N.H.; Nissim, A. Targeting of viral interleukin-10 with an antibody fragment specific to damaged arthritic cartilage improves its therapeutic potency. Arthritis Res. Ther. 2014, 16, R151. [Google Scholar] [CrossRef]
- Lim, N.H.; Vincent, T.L.; Nissim, A. In vivo optical imaging of early osteoarthritis using an antibody specific to damaged arthritic cartilage. Arthritis Res. Ther. 2015, 17, 376. [Google Scholar] [CrossRef]
- Bajpayee, A.G.; Wong, C.R.; Bawendi, M.G.; Frank, E.H.; Grodzinsky, A.J. Avidin as a model for charge driven transport into cartilage and drug delivery for treating early stage post-traumatic osteoarthritis. Biomaterials 2014, 35, 538–549. [Google Scholar] [CrossRef]
- Vedadghavami, A.; Wagner, E.K.; Mehta, S.; He, T.; Zhang, C.; Bajpayee, A.G. Cartilage penetrating cationic peptide carriers for applications in drug delivery to avascular negatively charged tissues. Acta Biomater. 2019, 93, 258–269. [Google Scholar] [CrossRef]
- Sterner, B.; Harms, M.; Wöll, S.; Weigandt, M.; Windbergs, M.; Lehr, C.M. The effect of polymer size and charge of molecules on permeation through synovial membrane and accumulation in hyaline articular cartilage. Eur. J. Pharm. Biopharm. 2016, 101. [Google Scholar] [CrossRef] [PubMed]
- Bajpayee, A.G.; Grodzinsky, A.J. Cartilage-targeting drug delivery: Can electrostatic interactions help? Nat. Rev. Rheumatol. 2017, 13, 183–193. [Google Scholar] [CrossRef]
- Bajpayee, A.G.; Quadir, M.A.; Hammond, P.T.; Grodzinsky, A.J. Charge based intra-cartilage delivery of single dose dexamethasone using Avidin nano-carriers suppresses cytokine-induced catabolism long term. Osteoarthr. Cartil. 2016, 24, 71–81. [Google Scholar] [CrossRef]
- Bajpayee, A.G.; Scheu, M.; Grodzinsky, A.J.; Porter, R.M. A rabbit model demonstrates the influence of cartilage thickness on intra-articular drug delivery and retention within cartilage. J. Orthop. Res. 2015, 33, 660–667. [Google Scholar] [CrossRef] [PubMed]
- Topping, L.M.; Thomas, B.L.; Rhys, H.I.; Tremoleda, J.L.; Foster, M.; Seed, M.; Voisin, M.B.; Vinci, C.; Law, H.L.; Perretti, M.; et al. Targeting Extracellular Vesicles to the Arthritic Joint Using a Damaged Cartilage-Specific Antibody. Front. Immunol. 2020, 11, 10. [Google Scholar] [CrossRef] [PubMed]
- Adams, G.; Vessillier, S.; Dreja, H.; Chernajovsky, Y. Targeting cytokines to inflammation sites. Nat. Biotechnol. 2003, 21, 1314–1320. [Google Scholar] [CrossRef] [PubMed]
- Mullen, L.; Adams, G.; Foster, J.; Vessillier, S.; Köster, M.; Hauser, H.; Layward, L.; Gould, D.; Chernajovsky, Y. A comparative study of matrix metalloproteinase and aggrecanase mediated release of latent cytokines at arthritic joints. Ann. Rheum. Dis. 2014, 79, 1728–1736. [Google Scholar] [CrossRef]
- Alberts, B.M.; Sacre, S.M.; Bush, P.G.; Mullen, L.M. Engineering of TIMP-3 as a LAP-fusion protein for targeting to sites of inflammation. J. Cell. Mol. Med. 2019, 23, 1617–1621. [Google Scholar] [CrossRef] [PubMed]
- Hu, H.Y.; Vats, D.; Vizovisek, M.; Kramer, L.; Germanier, C.; Wendt, K.U.; Rudin, M.; Turk, B.; Plettenburg, O.; Schultz, C. In vivo imaging of mouse tumors by a lipidated cathepsin S substrate. Angew. Chem. Int. Ed. 2014, 53, 7669–7673. [Google Scholar] [CrossRef]
- Hu, H.Y.; Lim, N.H.; Juretschke, H.P.; Ding-Pfennigdorff, D.; Florian, P.; Kohlmann, M.; Kandira, A.; Peter Von Kries, J.; Saas, J.; Rudolphi, K.A.; et al. In vivo visualization of osteoarthritic hypertrophic lesions. Chem. Sci. 2015, 6, 6256–6261. [Google Scholar] [CrossRef] [PubMed]
- Kavanaugh, T.E.; Werfel, T.A.; Cho, H.; Hasty, K.A.; Duvall, C.L. Particle-based technologies for osteoarthritis detection and therapy. Drug Deliv. Transl. Res. 2016, 6, 132–147. [Google Scholar] [CrossRef]
- Brown, S.; Kumar, S.; Sharma, B. Intra-articular targeting of nanomaterials for the treatment of osteoarthritis. Acta Biomater. 2019, 93, 239–257. [Google Scholar] [CrossRef]
- Geiger, B.C.; Wang, S.; Padera, R.F.; Grodzinsky, A.J.; Hammond, P.T. Cartilage-penetrating nanocarriers improve delivery and efficacy of growth factor treatment of osteoarthritis. Sci. Transl. Med. 2018, 10, eaat8800. [Google Scholar] [CrossRef]
- Hu, Q.; Chen, Q.; Yan, X.; Ding, B.; Chen, D.; Cheng, L. Chondrocyte affinity peptide modified PAMAM conjugate as a nanoplatform for targeting and retention in cartilage. Nanomedicine 2018, 13, 749–767. [Google Scholar] [CrossRef] [PubMed]
- Didomenico, C.D.; Lintz, M.; Bonassar, L.J. Molecular transport in articular cartilage—What have we learned from the past 50 years? Nat. Rev. Rheumatol. 2018, 14, 393–403. [Google Scholar] [CrossRef]
- Byun, S.; Sinskey, Y.L.; Lu, Y.C.S.; Ort, T.; Kavalkovich, K.; Sivakumar, P.; Hunziker, E.B.; Frank, E.H.; Grodzinsky, A.J. Transport of anti-il-6 antigen binding fragments into cartilage and the effects of injury. Arch. Biochem. Biophys. 2013, 532, 15–22. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Owen, S.; Francis, H.; Roberts, M. Disappearance kinetics of solutes from synovial fluid after intra-articular injection. Br. J. Clin. Pharmacol. 1994, 38, 349–355. [Google Scholar] [CrossRef]
- Vincent, T.L. Of mice and men: Converging on a common molecular understanding of osteoarthritis. Lancet Rheumatol. 2020, 2, e633–e645. [Google Scholar] [CrossRef]
- Mobasheri, A.; Bay-Jensen, A.C.; van Spil, W.E.; Larkin, J.; Levesque, M.C. Osteoarthritis Year in Review 2016: Biomarkers (biochemical markers). Osteoarthr. Cartil. 2017, 25, 199–208. [Google Scholar] [CrossRef] [PubMed]
- Lim, N.H.; Wen, C.; Vincent, T.L. Molecular and structural imaging in surgically induced murine osteoarthritis. Osteoarthr. Cartil. 2020, 28, 874–884. [Google Scholar] [CrossRef]
- Oo, W.M.; Hunter, D.J. Disease modification in osteoarthritis: Are we there yet? Clin. Exp. Rheumatol. 2019, 37, S135–S140. [Google Scholar]
- Bacon, K.; Lavalley, M.P.; Jafarzadeh, S.R.; Felson, D. Does cartilage loss cause pain in osteoarthritis and if so, how much? Ann. Rheum. Dis. 2020, 79, 1105–1110. [Google Scholar] [CrossRef] [PubMed]
- Rosseland, L.A.; Helgesen, K.G.; Breivik, H.; Stubhaug, A. Moderate-to-severe pain after knee arthroscopy is relieved by intraarticular saline: A randomized controlled trial. Anesth. Analg. 2004, 98, 1546–1551. [Google Scholar] [CrossRef] [PubMed]
- Abhishek, A.; Doherty, M. Mechanisms of the placebo response in pain in osteoarthritis. Osteoarthr. Cartil. 2013, 21, 1229–1235. [Google Scholar] [CrossRef] [PubMed]
- Copsey, B.; Thompson, J.Y.; Vadher, K.; Ali, U.; Dutton, S.J.; Fitzpatrick, R.; Lamb, S.E.; Cook, J.A. Problems persist in reporting of methods and results for the WOMAC measure in hip and knee osteoarthritis trials. Qual. Life Res. 2019, 28, 335–343. [Google Scholar] [CrossRef] [PubMed]
- Woolacott, N.F.; Corbett, M.S.; Rice, S.J.C. The use and reporting of WOMAC in the assessment of the benefit of physical therapies for the pain of osteoarthritis of the knee: Findings from a systematic review of clinical trials. Rheumatology 2012, 51, 1440–1446. [Google Scholar] [CrossRef]
- Miller, R.E.; Tran, P.B.; Ishihara, S.; Larkin, J.; Malfait, A.M. Therapeutic effects of an anti-ADAMTS-5 antibody on joint damage and mechanical allodynia in a murine model of osteoarthritis. Osteoarthr. Cartil. 2016, 24, 299–306. [Google Scholar] [CrossRef]
- Miller, R.E.; Ishihara, S.; Tran, P.B.; Golub, S.B.; Last, K.; Miller, R.J.; Fosang, A.J.; Malfait, A.M. An aggrecan fragment drives osteoarthritis pain through Toll-like receptor 2. JCI Insight 2018, 3, e95704. [Google Scholar] [CrossRef]

{kind=link}
| Peptide | Identified by | Binds to | Delivers | In Vivo Efficacy | Delivery |
|---|---|---|---|---|---|
| WYRGRL [114] | Screening of phage display peptide libraries against collagenase D-treated bovine cartilage pieces | Type II collagen | Nanoparticles [114,119]; dexamethasone [115]; pepstatin A via DOTAM scaffold [116]; HA-binding peptide [117,118] | 72-fold higher cartilage targeting compared with scrambled peptide [114]; 14-fold more retention of DOTAM-pepstatin A in murine knee joints, with ex vivo reduction in cathepsin D activity [116] | Intra-articular [114,116,118] |
| HSNGLPL [120] | Screening of phage display peptide libraries against TGFβ1 | TGFβ1 | TGFβ1 [120] | Increases cartilage regeneration in rabbit full thickness defect model [120] | Intra-articular during surgery to create cartilage defect [120] |
| DWRVIIPPRPSA [121] | Screening of phage display peptide libraries against rabbit cartilage pieces | Chondrocytes | DNA vector [121]; siRNA targeting Hif2a [122] | Higher uptake by chondrocytes than scrambled peptide [121]. Reduced cartilage damage in murine OA model than scrambled peptide [122] | Intra-articular [122] |
| RLDPTSYLRTFW and HDSQLEALIKFM [123] | Screening of phage display peptide libraries against cultured chondrocytes | Chondrocytes, at least in part via binding to aggrecan | |||
| KRKKKGKGLGKKRDPSLRKYK [124] | Sequence taken from heparin-binding domain of HB-EGF [124] | Heparin in vitro, binding to HS in vivo not shown [124] | Fusion protein consisting of IGF-1 fused with a heparin-binding domain [124] | Increased in retention of IGF-1 and proteoglycan synthesis in cartilage in vivo. Reduced cartilage damage in rat knee OA model [124] | Intra-articular [124] |
| Strategy | Identified by | Binds to | Delivers | In Vivo Efficacy | Delivery |
|---|---|---|---|---|---|
| Cationic carriers (avidin, peptides, etc.) [130,131,132,134] | Various | Negatively-charged cartilage matrix | Dexamethasone [134] | In vitro: Improved retention of cargo in cartilage explants [134] | Intra-articular [135] |
| scFv | Screening of scFv phage display library against ROS-modified type II collagen [127] | ROS-modified type II collagen [127] | MMP-cleavable form of viral IL-10 [128]; soluble TNF receptor II [127]; anti-inflammatory extracellular vesicles [136] | Reduced inflammation in RA models [127,128]; in vivo imaging of murine OA [129] | Intra-peritoneal [127,128]; intravenous [136] |
| Avimer [126] | Screening of avimer phage display library against rat and human type II collagen | Type II collagen | IL-1Ra [126] | Blocked IL-1 activity in rat knee joints when administered at same time as IL-1, and also when administered 1 week before | Intra-articular [126] |
| Metalloproteinase-activatable prodrugs | Use of latency-associated peptide of TGFβ1 [137] | Cleaved by activating MMPs and ADAMTSs | IFNβ [137,138]; TIMP-3 [139] | Reduced joint swelling [137], in vivo targeting, and therapeutic efficacy in CIA model [138] | Intramuscular [136], intraperitoneal [138] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
McClurg, O.; Tinson, R.; Troeberg, L. Targeting Cartilage Degradation in Osteoarthritis. Pharmaceuticals 2021, 14, 126. https://doi.org/10.3390/ph14020126
McClurg O, Tinson R, Troeberg L. Targeting Cartilage Degradation in Osteoarthritis. Pharmaceuticals. 2021; 14(2):126. https://doi.org/10.3390/ph14020126
Chicago/Turabian StyleMcClurg, Oliver, Ryan Tinson, and Linda Troeberg. 2021. "Targeting Cartilage Degradation in Osteoarthritis" Pharmaceuticals 14, no. 2: 126. https://doi.org/10.3390/ph14020126
APA StyleMcClurg, O., Tinson, R., & Troeberg, L. (2021). Targeting Cartilage Degradation in Osteoarthritis. Pharmaceuticals, 14(2), 126. https://doi.org/10.3390/ph14020126