
Thymoquinone Inhibits Growth of Acute Myeloid Leukemia Cells through Reversal SHP-1 and SOCS-3 Hypermethylation: In Vitro and In Silico Evaluation

 , , and
, , and 
Abstract
:1. Introduction
2. Results
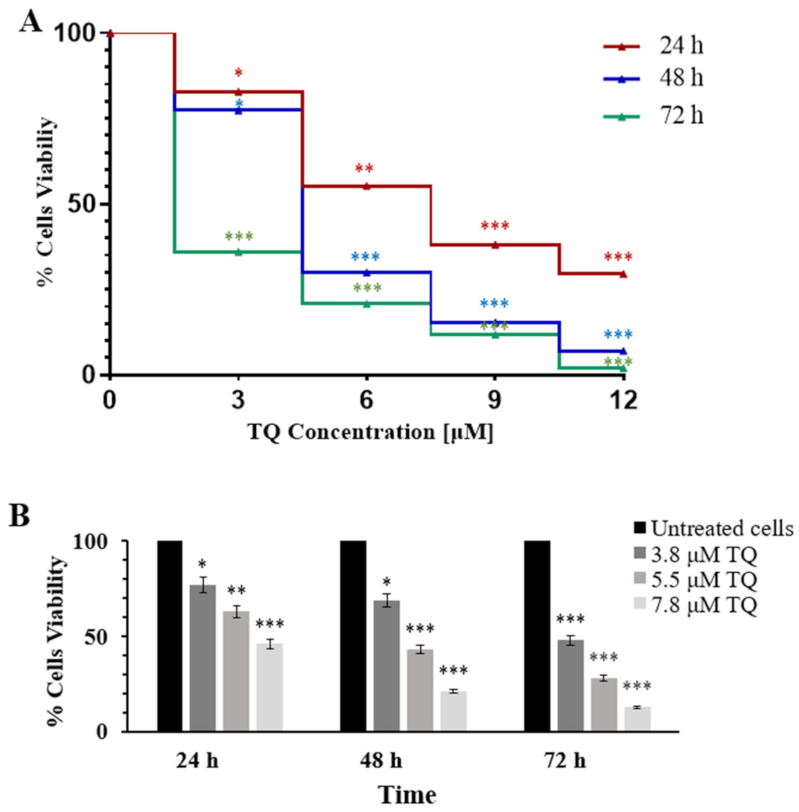
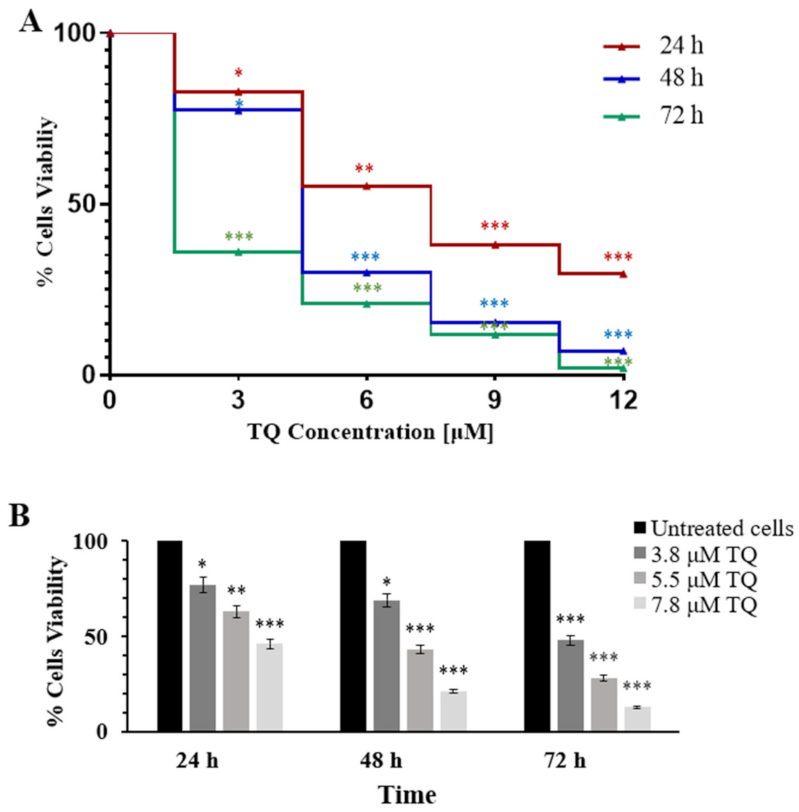
2.1. Thymoquinone Inhibits Cell Proliferation in MV4-11 Cells
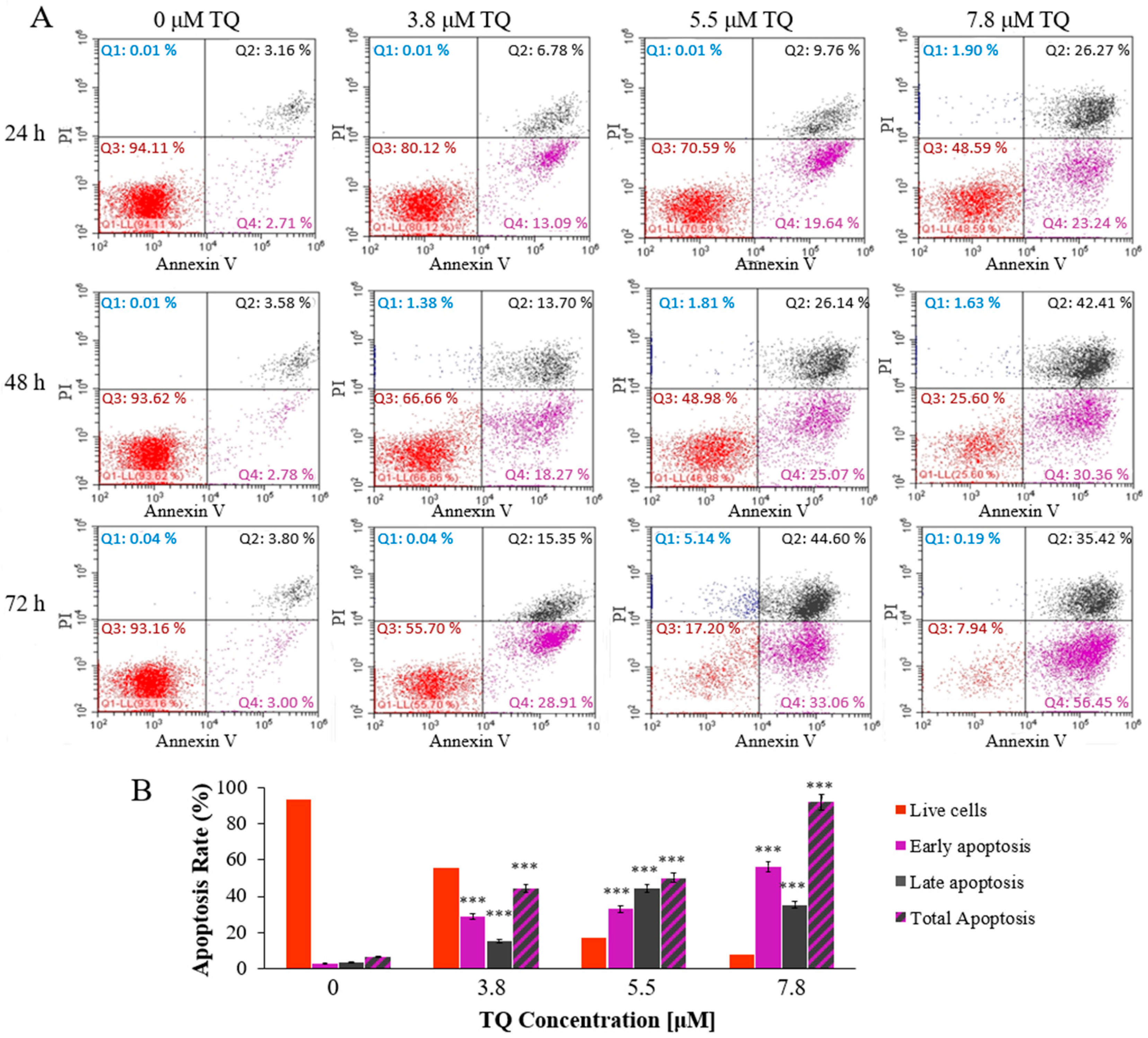
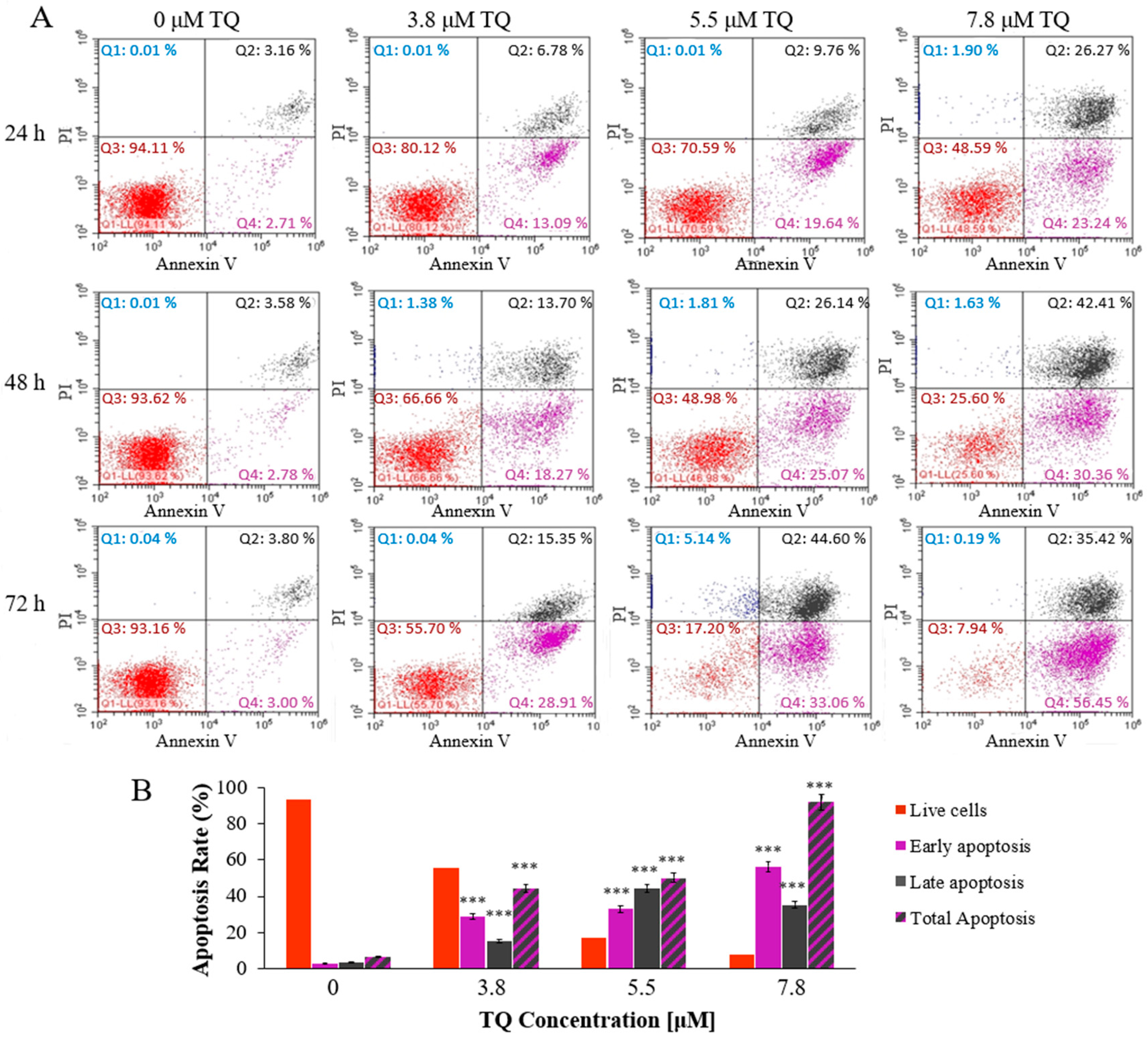
2.2. Thymoquinone Induces Dose and Time-Dependent Apoptosis in MV4-11 Cells
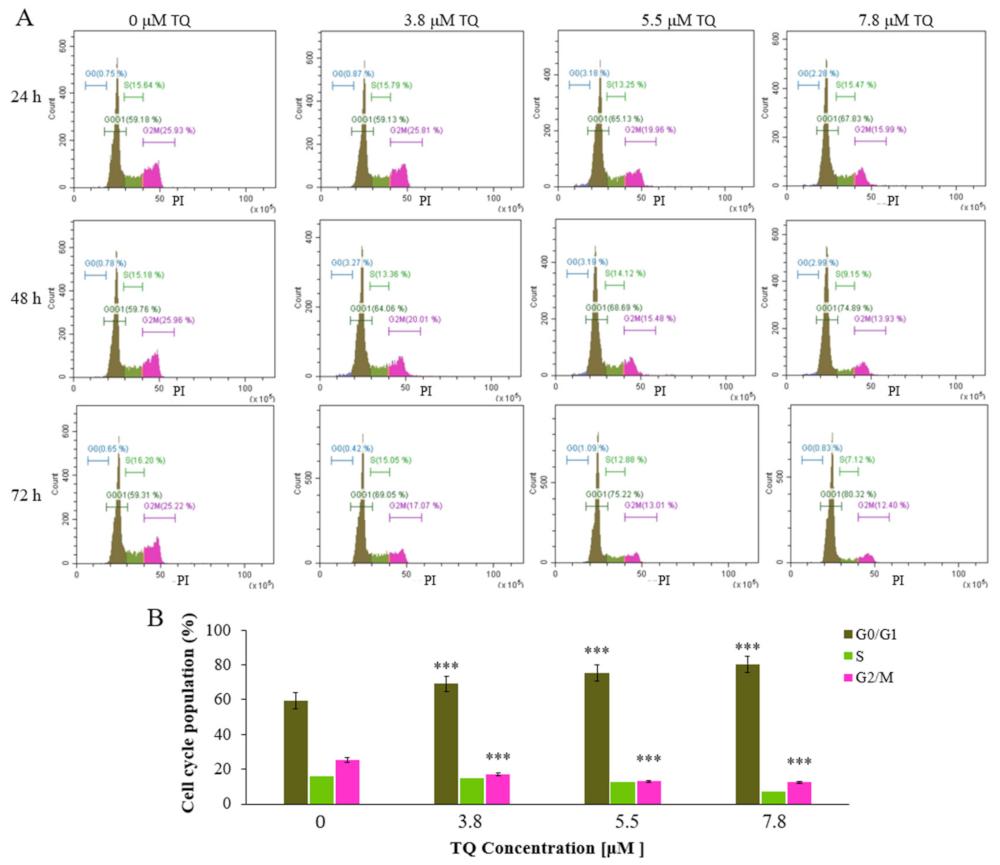
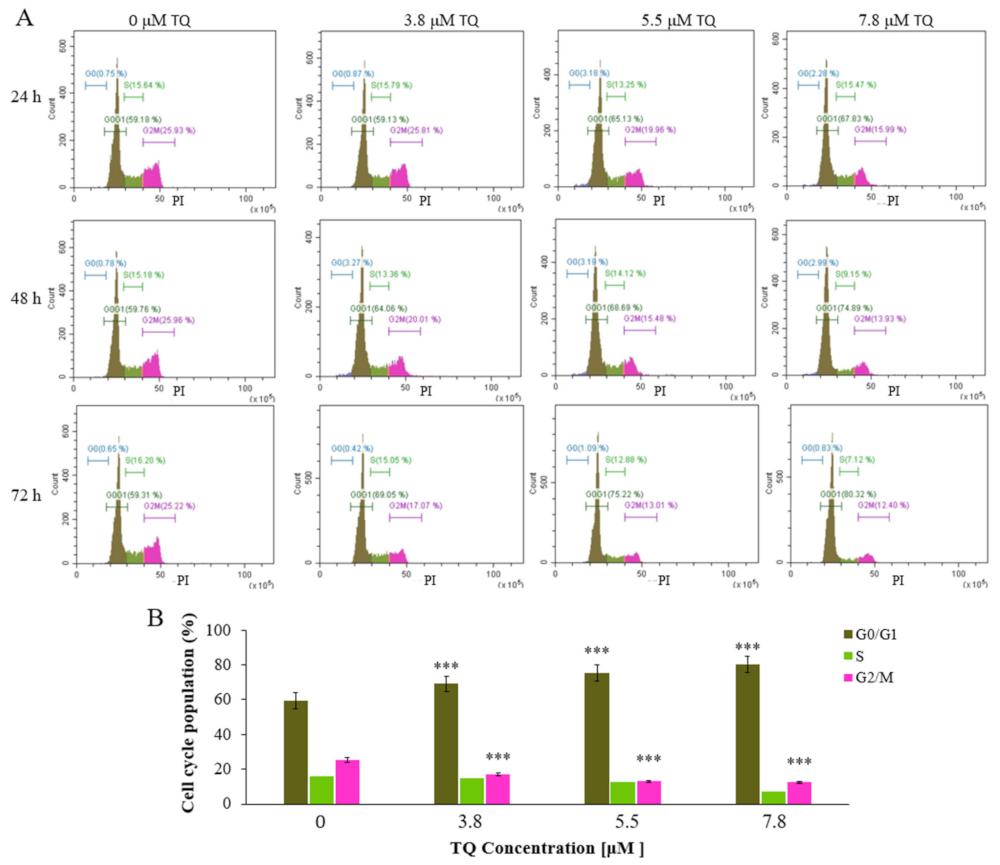
2.3. Thymoquinone Induces Cell Cycle Arrest at G0/G1 Phase in MV4-11 Cells
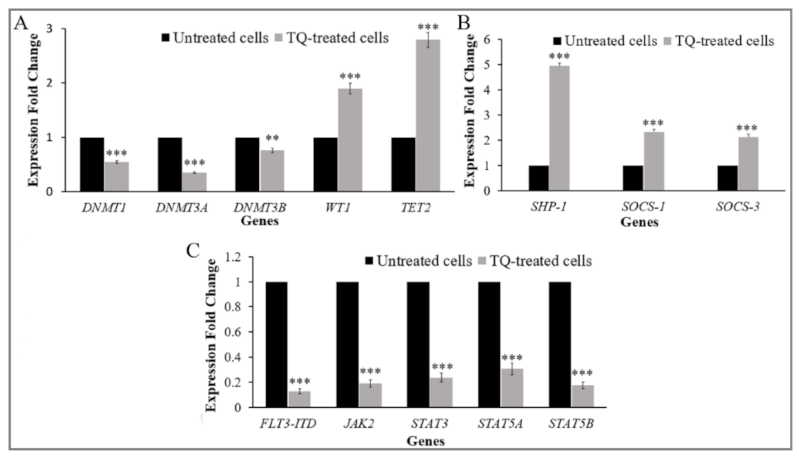
2.4. Thymoquinone Creates a Balance in the Expression of the Regulators of DNA Methylation Genes in MV4-11 Cells
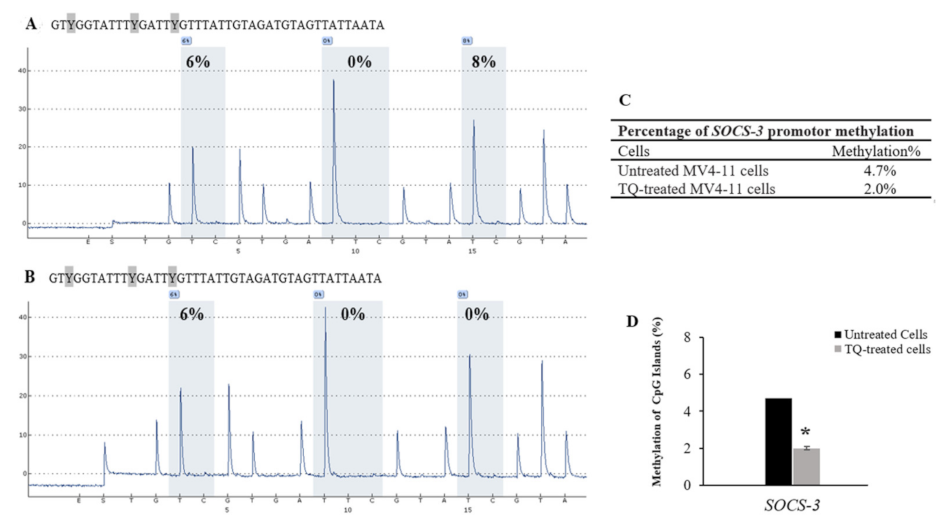
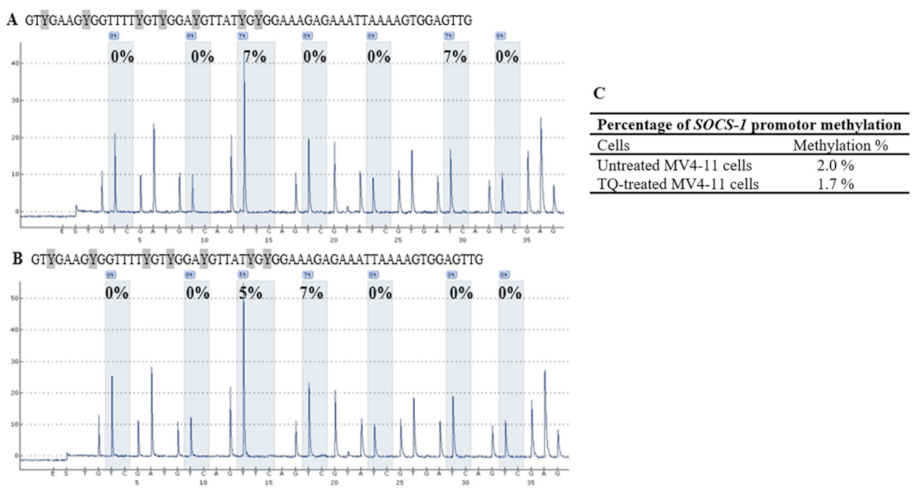
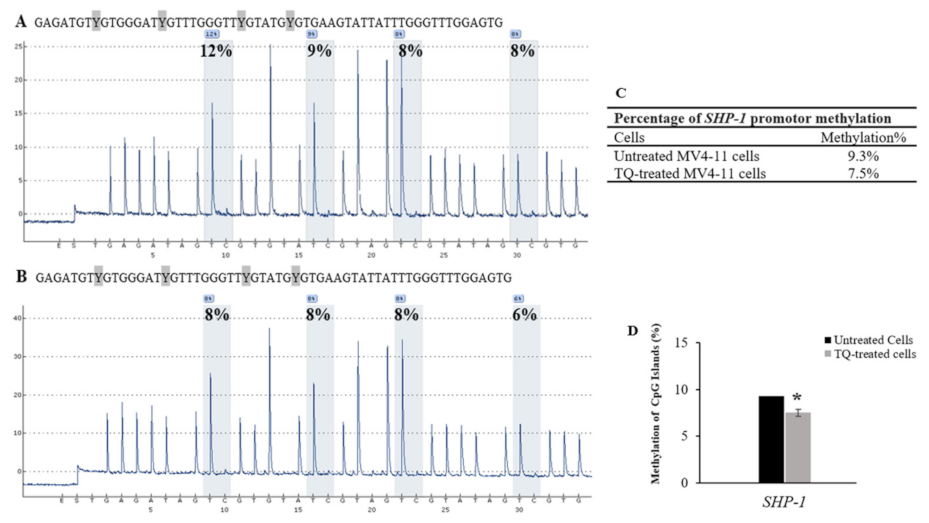
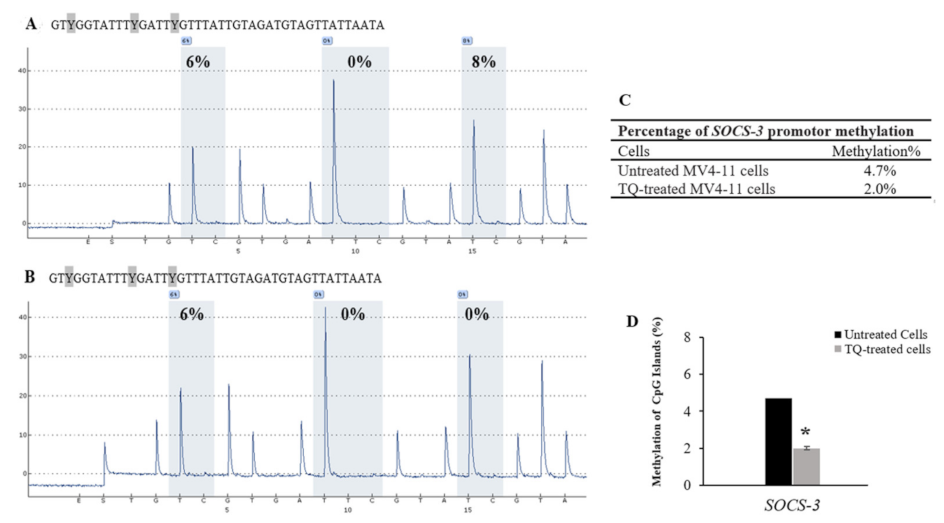
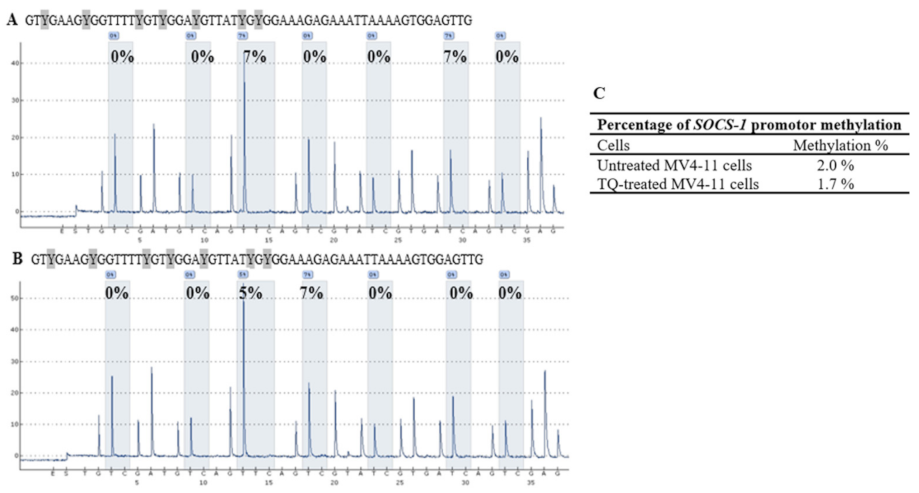
2.5. Thymoquinone Induces SHP-1 and SOCS-3 Promoter Hypomethylation in MV4-11 Cells
2.6. Thymoquinone Enhances Re-Expression of SHP-1, SOCS-1, and SOCS-3 in MV4-11 Cells
2.7. Thymoquinone Downregulates FLT3-ITD and JAK/STAT Signaling
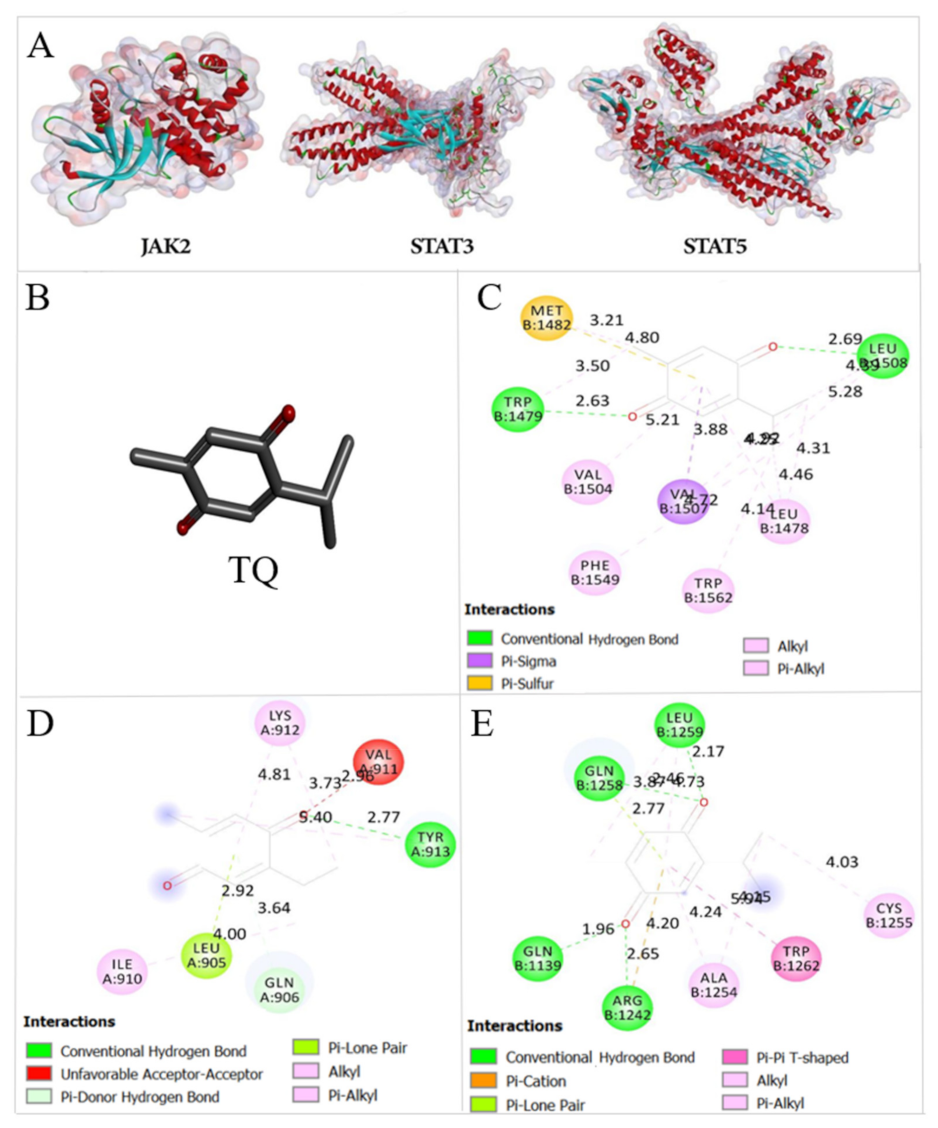
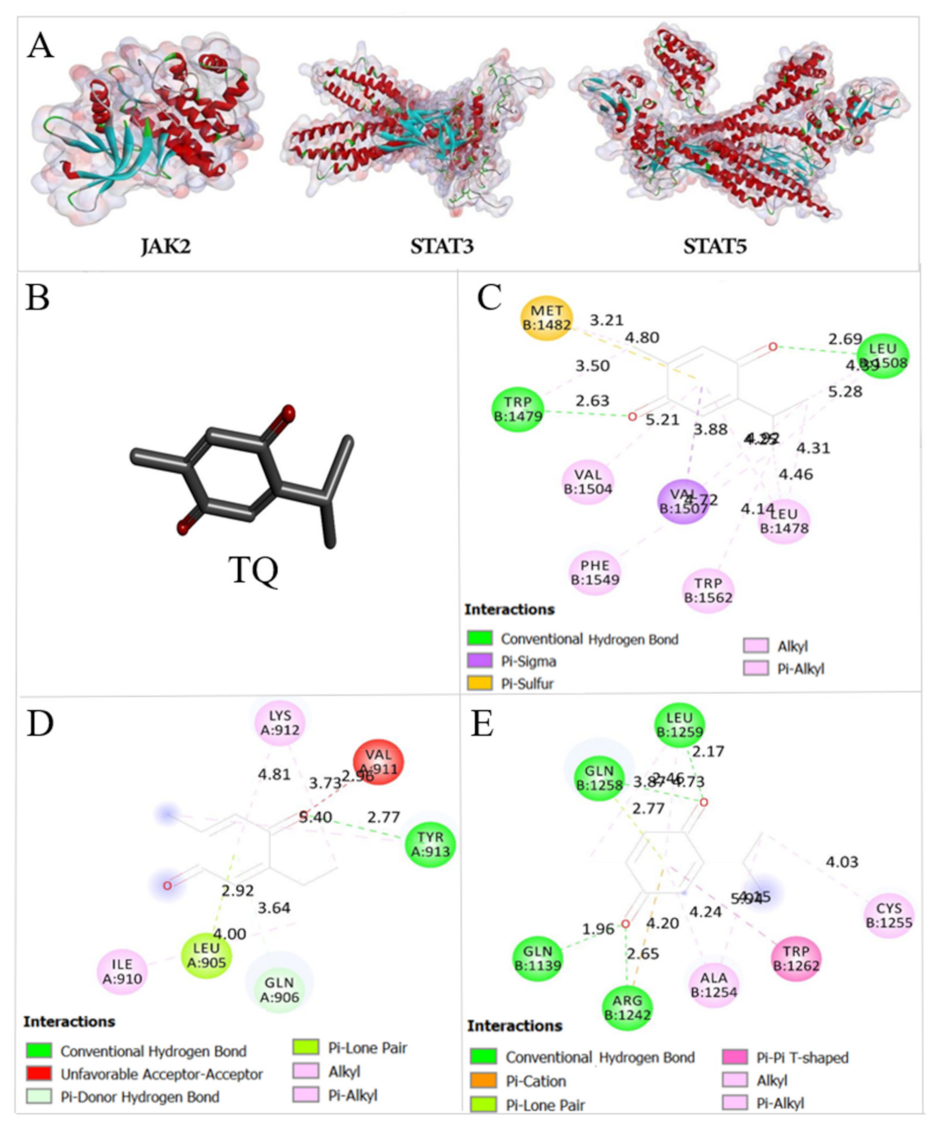
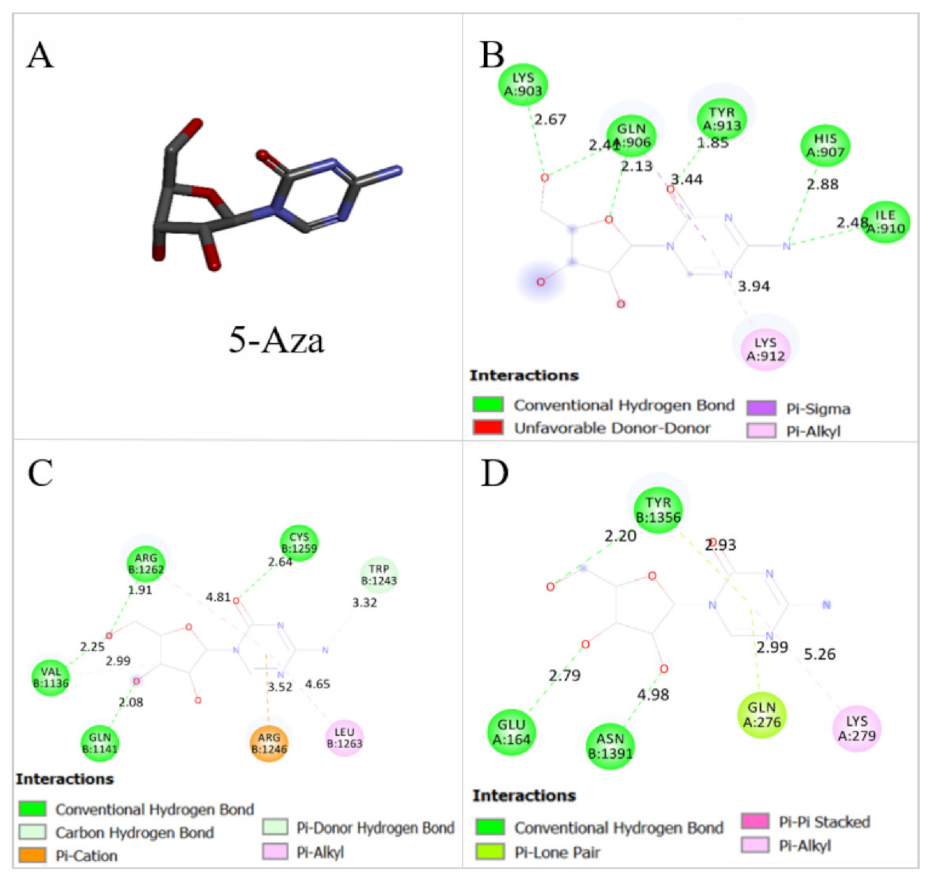
2.8. Thymoquinone Binds the Active Pocket of JAK2, STAT3, and STAT5
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. TQ and 5-Aza Treatment
4.3. Cytotoxicity Assay
4.4. Cell Viability Assay
4.5. Apoptosis Assay
4.6. Cell Cycle Analysis Using Flow Cytometry
4.7. RNA Extraction
4.8. Quantitative Reverse Transcriptase PCR (RT-qPCR)
4.9. DNA Extraction and Bisulfite Treatment
4.10. Primer Design for Pyrosequencing Assay
4.11. Docking of Thymoquinone
4.12. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Yang, X.; Wong, M.P.M.; Ng, R.K. Aberrant DNA methylation in acute myeloid leukemia and its clinical implications. Int. J. Mol. Sci. 2019, 20, 4576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, S.; Garrett-Bakelman, F.E.; Chung, S.S.; Sanders, M.A.; Hricik, T.; Rapaport, F.; Patel, J.; Dillon, R.; Vijay, P.; Brown, A.L. Distinct evolution and dynamics of epigenetic and genetic heterogeneity in acute myeloid leukemia. Nat. Med. 2016, 22, 792–799. [Google Scholar] [CrossRef] [Green Version]
- Hoang, N.M.; Rui, L. DNA methyltransferases in hematological malignancies. J. Genet. Genom. 2020, 47, 361–372. [Google Scholar] [CrossRef]
- Morinishi, L.; Kochanowski, K.; Levine, R.L.; Wu, L.F.; Altschuler, S.J. Loss of TET2 affects proliferation and drug sensitivity through altered dynamics of cell-state transitions. Cell Syst. 2020, 11, 86–94.e85. [Google Scholar] [CrossRef] [PubMed]
- Sinha, S.; Thomas, D.; Yu, L.; Gentles, A.J.; Jung, N.; Corces-Zimmerman, M.R.; Chan, S.M.; Reinisch, A.; Feinberg, A.P.; Dill, D.L. Mutant WT1 is associated with DNA hypermethylation of PRC2 targets in AML and responds to EZH2 inhibition. Blood 2015, 125, 316–326. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Weng, W.-J.; Zhou, D.-H.; Fang, J.-P.; Mishra, S.; Chai, L.; Xu, L.-H. Wilms tumor 1 mutations are independent poor prognostic factors in pediatric acute myeloid leukemia. Front. Oncol. 2021, 11, 632094. [Google Scholar] [CrossRef]
- Memari, F.; Joneidi, Z.; Taheri, B.; Aval, S.F.; Roointan, A.; Zarghami, N. Epigenetics and Epi-miRNAs: Potential markers/therapeutics in leukemia. Biomed. Pharmacother. 2018, 106, 1668–1677. [Google Scholar] [CrossRef]
- Asmaa, M.J.S.; Al-Jamal, H.A.; Hussein, A.R.; Yahaya, B.H.; Hassan, R.; Hussain, F.A.; Shamsuddin, S.; Johan, M.F. Transcriptomic profiles of MV4-11 and Kasumi 1 acute myeloid leukemia cell lines modulated by epigenetic modifiers Trichostatin A and 5-Azacytidine. Int. J. Hematol. Oncol. Stem Cell Res. 2020, 14, 72. [Google Scholar] [CrossRef]
- Liu, K.; Wu, Z.; Chu, J.; Yang, L.; Wang, N. Promoter methylation and expression of SOCS3 affect the clinical outcome of pediatric acute lymphoblastic leukemia by JAK/STAT pathway. Biomed. Pharmacother. 2019, 115, 108913. [Google Scholar] [CrossRef]
- Middeldorf, I.; Galm, O.; Osieka, R.; Jost, E.; Herman, J.G.; Wilop, S. Sequence of administration and methylation of SOCS3 may govern response to gemtuzumab ozogamicin in combination with conventional chemotherapy in patients with refractory or relapsed acute myelogenous leukemia (AML). Am. J. Hematol. 2010, 85, 477–481. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Liu, X.; Guo, X.; Liu, X.; Luo, J. DNA methyltransferase 1 mediated aberrant methylation and silencing of SHP-1 gene in chronic myelogenous leukemia cells. Leuk. Res. 2017, 58, 9–13. [Google Scholar] [CrossRef] [PubMed]
- Al-Jamal, H.A.N.; Jusoh, S.A.M.; Hassan, R.; Johan, M.F. Enhancing SHP-1 expression with 5-azacytidine may inhibit STAT3 activation and confer sensitivity in lestaurtinib (CEP-701)-resistant FLT3-ITD positive acute myeloid leukemia. BMC Cancer 2015, 15, 869. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.-J.; Chen, H.; Zheng, X.-Q.; Li, H.-Y.; Wu, J.-B.; Tang, L.-Y.; Gao, S.-M. Methylated alteration of SHP1 complements mutation of JAK2 tyrosine kinase in patients with myeloproliferative neoplasm. Asian Pac. J. Cancer Prev. 2015, 16, 2219–2225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brakensiek, K.; Länger, F.; Schlegelberger, B.; Kreipe, H.; Lehmann, U. Hypermethylation of the suppressor of cytokine signalling-1 (SOCS-1) in myelodysplastic syndrome. Br. J. Haematol. 2005, 130, 209–217. [Google Scholar] [CrossRef]
- Chim, C.-S.; Fung, T.-K.; Cheung, W.-C.; Liang, R.; Kwong, Y.-L. SOCS1 and SHP1 hypermethylation in multiple myeloma: Implications for epigenetic activation of the Jak/STAT pathway. Blood 2004, 103, 4630–4635. [Google Scholar] [CrossRef]
- Chim, C.; Wong, A.; Kwong, Y. Epigenetic dysregulation of the Jak/STAT pathway by frequent aberrant methylation of SHP1 but not SOCS1 in acute leukaemias. Ann. Hematol. 2004, 83, 527–532. [Google Scholar] [CrossRef]
- Johan, M.; Bowen, D.; Frew, M.; Goodeve, A.; Reilly, J. Aberrant methylation of the negative regulators RASSFIA, SHP-1 and SOCS-1 in myelodysplastic syndromes and acute myeloid leukaemia. Br. J. Haematol. 2005, 129, 60–65. [Google Scholar] [CrossRef]
- Chen, C.Y.; Tsay, W.; Tang, J.L.; Shen, H.L.; Lin, S.W.; Huang, S.Y.; Yao, M.; Chen, Y.C.; Shen, M.C.; Wang, C.H. SOCS1 methylation in patients with newly diagnosed acute myeloid leukemia. Genes Chromosomes Cancer 2003, 37, 300–305. [Google Scholar] [CrossRef]
- Agrawal, K.; Das, V.; Vyas, P.; Hajdúch, M. Nucleosidic DNA demethylating epigenetic drugs–a comprehensive review from discovery to clinic. Pharmacol. Ther. 2018, 188, 45–79. [Google Scholar] [CrossRef]
- Yan, F.; Shen, N.; Pang, J.; Molina, J.R.; Yang, P.; Liu, S. The DNA methyltransferase DNMT1 and tyrosine-protein kinase KIT cooperatively promote resistance to 5-Aza-2′-deoxycytidine (decitabine) and midostaurin (PKC412) in lung cancer cells. J. Biol. Chem. 2015, 290, 18480–18494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vela-Ojeda, J.; Cardenas, P.V.; Esparza, M.A.G.-R.; Cervantes, L.A.M.; Chavez, J.G.; Caballero, A.H.; Majluf-Cruz, A.; Vega-López, A.; Reyes-Maldonado, E. FLT3-ITD and CD135 over-expression are frequent findings of poor survival in adult patients with acute leukemias. Arch. Med. Res. 2021, 52, 217–223. [Google Scholar] [CrossRef]
- Pang, J.; Shen, N.; Yan, F.; Zhao, N.; Dou, L.; Wu, L.-C.; Seiler, C.L.; Yu, L.; Yang, K.; Bachanova, V. Thymoquinone exerts potent growth-suppressive activity on leukemia through DNA hypermethylation reversal in leukemia cells. Oncotarget 2017, 8, 34453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perl, A.E.; Altman, J.K.; Cortes, J.; Smith, C.; Litzow, M.; Baer, M.R.; Claxton, D.; Erba, H.P.; Gill, S.; Goldberg, S. Selective inhibition of FLT3 by gilteritinib in relapsed or refractory acute myeloid leukaemia: A multicentre, first-in-human, open-label, phase 1–2 study. Lancet Oncol. 2017, 18, 1061–1075. [Google Scholar] [CrossRef]
- Knapper, S.; Burnett, A.K.; Littlewood, T.; Kell, W.J.; Agrawal, S.; Chopra, R.; Clark, R.; Levis, M.J.; Small, D. A phase 2 trial of the FLT3 inhibitor lestaurtinib (CEP701) as first-line treatment for older patients with acute myeloid leukemia not considered fit for intensive chemotherapy. Blood 2006, 108, 3262–3270. [Google Scholar] [CrossRef]
- Tima, S.; Okonogi, S.; Ampasavate, C.; Berkland, C.; Anuchapreeda, S. FLT3-specific curcumin micelles enhance activity of curcumin on FLT3-ITD overexpressing MV4-11 leukemic cells. Drug Dev. Ind. Pharm. 2019, 45, 498–505. [Google Scholar] [CrossRef]
- Singh, S.; Vaishnav, N.; Saha, D.; Ahsan, Z.; Mathur, R.; Jha, A.K. Demethylation of tumor suppressor genes in leukemia by using natural compounds. Int. J. Bio. Sci. Bio. Technol. 2020, 12, 2233–7849. [Google Scholar]
- Khan, M.A.; Tania, M.; Fu, J. Epigenetic role of thymoquinone: Impact on cellular mechanism and cancer therapeutics. Drug Discov. Today 2019, 24, 2315–2322. [Google Scholar] [CrossRef]
- Li, J.; Khan, M.; Wei, C.; Cheng, J.; Chen, H.; Yang, L.; Ijaz, I.; Fu, J. Thymoquinone inhibits the migration and invasive characteristics of cervical cancer cells SiHa and CaSki in vitro by targeting epithelial to mesenchymal transition associated transcription factors Twist1 and Zeb1. Molecules 2017, 22, 2105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hassan, F.-U.; Rehman, M.S.-U.; Khan, M.S.; Ali, M.A.; Javed, A.; Nawaz, A.; Yang, C. Curcumin as an alternative epigenetic modulator: Mechanism of action and potential effects. Front. Genet. 2019, 10, 514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Houssein, M.; Fatfat, M.; Habli, Z.; Ghazal, N.; Moodad, S.; Khalife, H.; Khalil, M.; Gali-Muhtasib, H. Thymoquinone synergizes with arsenic and interferon alpha to target human T-cell leukemia/lymphoma. Life Sci. 2020, 251, 117639. [Google Scholar] [CrossRef]
- Pal, R.R.; Rajpal, V.; Singh, P.; Saraf, S.A. Recent findings on thymoquinone and its applications as a nanocarrier for the treatment of cancer and rheumatoid arthritis. Pharmaceutics 2021, 13, 775. [Google Scholar] [CrossRef]
- Almajali, B.; Al-Jamal, H.A.N.; Wan Taib, W.R.; Ismail, I.; Johan, M.F.; Doolaanea, A.A.; Ibrahim, W.N.; Tajudin, S.A. Thymoquinone suppresses cell proliferation and enhances apoptosis of HL60 leukemia cells through re-expression of JAK/STAT negative regulators. Asian Pac. J. Cancer Prev. 2021, 22, 879–885. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; He, C.; Wang, M.; Ma, X.; Mo, F.; Yang, S.; Han, J.; Wei, X. Targeting epigenetic regulators for cancer therapy: Mechanisms and advances in clinical trials. Signal Transduct. Target. Ther. 2019, 4, 62. [Google Scholar] [CrossRef] [Green Version]
- Qadi, S.A.; Hassan, M.A.; Sheikh, R.A.; Baothman, O.A.; Zamzami, M.A.; Choudhry, H.; Al-Malki, A.L.; Albukhari, A.; Alhosin, M. Thymoquinone-induced reactivation of tumor suppressor genes in cancer cells involves epigenetic mechanisms. Epigenetics Insights 2019, 12, 2516865719839011. [Google Scholar] [CrossRef]
- Amartey, J.; Gapper, S.; Hussein, N.; Morris, K.; Withycombe, C.E. Nigella sativa extract and thymoquinone regulate inflammatory cytokine and TET-2 expression in endothelial cells. Artery Res. 2019, 25, 157–163. [Google Scholar] [CrossRef] [Green Version]
- Yu, J.; Peng, Y.; Wu, L.-C.; Xie, Z.; Deng, Y.; Hughes, T.; He, S.; Mo, X.; Chiu, M.; Wang, Q.-E. Curcumin down-regulates DNA methyltransferase 1 and plays an anti-leukemic role in acute myeloid leukemia. PLoS ONE 2013, 8, e55934. [Google Scholar] [CrossRef] [PubMed]
- Jiang, M.; Zhang, W.-W.; Liu, P.; Yu, W.; Liu, T.; Yu, J. Dysregulation of SOCS-mediated negative feedback of cytokine signaling in carcinogenesis and its significance in cancer treatment. Front. Immunol. 2017, 8, 70. [Google Scholar] [CrossRef] [Green Version]
- Khan, M.A.; Tania, M.; Wei, C.; Mei, Z.; Fu, S.; Cheng, J.; Xu, J.; Fu, J. Thymoquinone inhibits cancer metastasis by downregulating TWIST1 expression to reduce epithelial to mesenchymal transition. Oncotarget 2015, 6, 19580. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.H.; Yang, L.; Liu, X.J.; Zhan, Y.; Pan, Y.X.; Wang, X.Z.; Luo, J.M. Association between methylation of tumor suppressor gene SOCS1 and acute myeloid leukemia. Oncol. Rep. 2018, 40, 1008–1016. [Google Scholar] [CrossRef] [PubMed]
- Chougule, R.A.; Kazi, J.U.; Rönnstrand, L. FYN expression potentiates FLT3-ITD induced STAT5 signaling in acute myeloid leukemia. Oncotarget 2016, 7, 9964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Larrue, C.; Heydt, Q.; Saland, E.; Boutzen, H.; Kaoma, T.; Sarry, J.-E.; Joffre, C.; Récher, C. Oncogenic KIT mutations induce STAT3-dependent autophagy to support cell proliferation in acute myeloid leukemia. Oncogenesis 2019, 8, 39. [Google Scholar] [CrossRef]
- Venugopal, S.; Bar-Natan, M.; Mascarenhas, J.O. JAKs to STATs: A tantalizing therapeutic target in acute myeloid leukemia. Blood Rev. 2020, 40, 100634. [Google Scholar] [CrossRef]
- Liu, K.; Tian, T.; Zheng, Y.; Zhou, L.; Dai, C.; Wang, M.; Lin, S.; Deng, Y.; Hao, Q.; Zhai, Z. Scutellarin inhibits proliferation and invasion of hepatocellular carcinoma cells via down-regulation of JAK2/STAT3 pathway. J. Cell. Mol. Med. 2019, 23, 3040–3044. [Google Scholar] [CrossRef] [Green Version]
- Zhu, W.-Q.; Wang, J.; Guo, X.-F.; Liu, Z.; Dong, W.-G. Thymoquinone inhibits proliferation in gastric cancer via the STAT3 pathway in vivo and in vitro. World J. Gastroenterol. 2016, 22, 4149. [Google Scholar] [CrossRef] [PubMed]
- Dalgıç, C.T.; Kaymaz, B.T.; Özkan, M.C.; Dalmızrak, A.; Şahin, F.; Saydam, G. Investigating the role of JAK/STAT pathway on dasatinib-induced apoptosis for CML cell model K562. Clin. Lymphoma Myeloma Leuk. 2015, 15, S161–S166. [Google Scholar] [CrossRef]
- Li, Y.; Sun, W.; Han, N.; Zou, Y.; Yin, D. Curcumin inhibits proliferation, migration, invasion and promotes apoptosis of retinoblastoma cell lines through modulation of miR-99a and JAK/STAT pathway. BMC Cancer 2018, 18, 1230. [Google Scholar] [CrossRef] [Green Version]
- Rummelt, C.; Gorantla, S.P.; Meggendorfer, M.; Charlet, A.; Endres, C.; Döhner, K.; Heidel, F.H.; Fischer, T.; Haferlach, T.; Duyster, J. Activating JAK-mutations confer resistance to FLT3 kinase inhibitors in FLT3-ITD positive AML in vitro and in vivo. Leukemia 2020, 35, 2017–2029. [Google Scholar] [CrossRef]
- Dumas, P.-Y.; Naudin, C.; Martin-Lannerée, S.; Izac, B.; Casetti, L.; Mansier, O.; Rousseau, B.; Artus, A.; Dufossée, M.; Giese, A. Hematopoietic niche drives FLT3-ITD acute myeloid leukemia resistance to quizartinib via STAT5-and hypoxia-dependent upregulation of AXL. Haematologica 2019, 104, 2017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cook, A.M.; Li, L.; Ho, Y.; Lin, A.; Li, L.; Stein, A.; Forman, S.; Perrotti, D.; Jove, R.; Bhatia, R. Role of altered growth factor receptor-mediated JAK2 signaling in growth and maintenance of human acute myeloid leukemia stem cells. Blood 2014, 123, 2826–2837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, J.-H.; Jang, Y.-S.; Lee, H.J.; Lee, C.-Y.; Shin, D.Y.; Oh, S.H. Inhibition of STAT3 signaling induces apoptosis and suppresses growth of lung cancer: Good and bad. Lab. Anim. Res. 2019, 35, 30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Jamal, H.A.N.; Johan, M.F.; Jusoh, S.A.M.; Ismail, I.; Taib, W.R.W. Re-expression of bone marrow proteoglycan-2 by 5-azacytidine is associated with STAT3 inactivation and sensitivity response to imatinib in resistant CML cells. Asian Pac. J. Cancer Prev. 2018, 19, 1585. [Google Scholar]
- Almajali, B.; Al-Jamal, H.A.N.; Taib, W.R.W.; Ismail, I.; Johan, M.F.; Doolaanea, A.A.; Ibrahim, W.N. Thymoquinone, as a Novel Therapeutic Candidate of Cancers. Pharmaceuticals 2021, 14, 369. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2− ΔΔCT method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Hodroj, M.H.; Jardaly, A.; Abi Raad, S.; Zouein, A.; Rizk, S. Andrographolide potentiates the antitumor effect of topotecan in acute myeloid leukemia cells through an intrinsic apoptotic pathway. Cancer Manag. Res. 2018, 10, 1079. [Google Scholar] [CrossRef] [Green Version]
- Musalli, M.G.; Hassan, M.A.; Sheikh, R.A.; Kalantan, A.A.; Halwani, M.A.; Zeyadi, M.; Hosawi, S.; Alhosin, M. Thymoquinone induces cell proliferation inhibition and apoptosis in acute myeloid leukemia cells: Role of apoptosis-related WT1 and BCL2 genes. Eur. J. Cell Sci. 2019, 1, 2–9. [Google Scholar] [CrossRef]
- Quentmeier, H.; Reinhardt, J.; Zaborski, M.; Drexler, H. FLT3 mutations in acute myeloid leukemia cell lines. Leukemia 2003, 17, 120–124. [Google Scholar] [CrossRef] [Green Version]
- Gupta, M.; Han, J.J.; Stenson, M.; Maurer, M.; Wellik, L.; Hu, G.; Ziesmer, S.; Dogan, A.; Witzig, T.E. Elevated serum IL-10 levels in diffuse large B-cell lymphoma: A mechanism of aberrant JAK2 activation. Blood 2012, 119, 2844–2853. [Google Scholar] [CrossRef]
- Bellon, M.; Moles, R.; Chaib-Mezrag, H.; Pancewicz, J.; Nicot, C. JAG1 overexpression contributes to Notch1 signaling and the migration of HTLV-1-transformed ATL cells. J. Hematol. Oncol. 2018, 11, 119. [Google Scholar] [CrossRef]
- Ai, L.; Mu, S.; Sun, C.; Fan, F.; Yan, H.; Qin, Y.; Cui, G.; Wang, Y.; Guo, T.; Mei, H. Myeloid-derived suppressor cells endow stem-like qualities to multiple myeloma cells by inducing piRNA-823 expression and DNMT3B activation. Mol. Cancer 2019, 18, 88. [Google Scholar] [CrossRef]
- Zhang, T.J.; Zhou, J.D.; Yang, D.Q.; Wang, Y.X.; Wen, X.M.; Guo, H.; Yang, L.; Lian, X.Y.; Lin, J.; Qian, J. TET2 expression is a potential prognostic and predictive biomarker in cytogenetically normal acute myeloid leukemia. J. Cell. Physiol. 2018, 233, 5838–5846. [Google Scholar] [CrossRef]
- Ayatollahi, H.; Sadeghian, M.H.; Naderi, M.; Jafarian, A.H.; Shams, S.F.; Motamedirad, N.; Sheikhi, M.; Bahrami, A.; Shakeri, S. Quantitative assessment of Wilms tumor 1 expression by real-time quantitative polymerase chain reaction in patients with acute myeloblastic leukemia. J. Res. Med. Sci. 2017, 22, 54. [Google Scholar] [PubMed]
- Relles, D.; Chipitsyna, G.I.; Gong, Q.; Yeo, C.J.; Arafat, H.A. Thymoquinone promotes pancreatic cancer cell death and reduction of tumor size through combined inhibition of histone deacetylation and induction of histone acetylation. Adv. Prev. Med. 2016, 2016, 1407840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamal, A.; Nazari, M.; Yaseen, M.; Iqbal, M.A.; Ahamed, M.B.K.; Majid, A.S.A.; Bhatti, H.N. Green synthesis of selenium-N-heterocyclic carbene compounds: Evaluation of antimicrobial and anticancer potential. Bioorg. Chem. 2019, 90, 103042. [Google Scholar] [CrossRef]
- Habib, A.; Nazari, M.; Iqbal, M.A.; Bhatti, H.N.; Ahmed, M.K.; Majid, A.A. Unsymmetrically substituted benzimidazolium based Silver (I)-N-heterocyclic carbene complexes: Synthesis, characterization and in vitro anticancer study against human breast cancer and colon cancer. J. Saudi Chem. Soc. 2019, 23, 795–808. [Google Scholar] [CrossRef]
- Badroon, N.; Abdul Majid, N.; Al-Suede, F.S.R.; Nazari, V.M.; Giribabu, N.; Abdul Majid, A.M.S.; Eid, E.E.; Alshawsh, M.A. Cardamonin exerts antitumor effect on human hepatocellular carcinoma xenografts in athymic nude mice through inhibiting NF-κβ pathway. Biomedicines 2020, 8, 586. [Google Scholar] [CrossRef]
- Nazari, M.; Mahmood, S.; Raman, S. Comparative docking studies of rosmarinic acid and sinesitin to inhibit HSP70. Int. J. Eng. Technol. Sci. 2019, 6, 115–119. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cells | CpG-1 | CpG-2 | CpG-3 | CpG-4 | Mean ± SD |
|---|---|---|---|---|---|
| Untreated MV4-11 cells | 12 | 9 | 8 | 8 | 9.3 ± 1.8 |
| TQ-treated MV4-11 cells | 8 | 8 | 8 | 6 | 7.5 ± 1.0 |
| 5-Aza-treated MV4-11 cells | 13 | 8 | 0 | 0 | 5.2 ± 2.1 |
| Unmethylated DNA control | 6 | 0 | 0 | 0 | 1.5 ± 0.4 |
| Methylated DNA control | 95 | 88 | 100 | 84 | 91.8 ± 15.7 |
| Unmethylated Bisulfite-unconverted DNA control | 0 | 0 | 0 | 0 | 0 |
| Cells | CpG-1 | CpG-2 | CpG-3 | Mean ± SD |
|---|---|---|---|---|
| Untreated MV4-11 cells | 6 | 0 | 8 | 4.7 ± 1.1 |
| TQ-treated MV4-11 cells | 6 | 0 | 0 | 2.0 ± 0.7 |
| 5-Aza-treated MV4-11 cells | 0 | 0 | 0 | 0 |
| Unmethylated DNA control | 0 | 0 | 0 | 0 |
| Methylated DNA control | 89 | 96 | 100 | 95.0 ± 15.5 |
| Unmethylated Bisulfite- unconverted DNA control | 0 | 0 | 0 | 0 |
| SOCS-1 | CpG-1 | CpG-2 | CpG-3 | CpG-4 | CpG-5 | CpG-6 | CpG-7 | Mean ± SD |
|---|---|---|---|---|---|---|---|---|
| Untreated MV4-11 cells | 0 | 0 | 7 | 0 | 0 | 7 | 0 | 2.0 ± 0.4 |
| TQ-treated MV4-11 cells | 0 | 0 | 5 | 7 | 0 | 0 | 0 | 1.7 ± 0.9 |
| 5-Aza-treated MV4-11 cells | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Unmethylated DNA control | 5 | 0 | 8 | 6 | 0 | 6 | 0 | 3.6 ± 1.3 |
| Methylated DNA control | 82 | 86 | 100 | 89 | 93 | 100 | 79 | 89.9 ± 22.3 |
| Unmethylated Bisulfite-unconverted DNA control | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| FBE (Kcal/mol) | Ki | |||
|---|---|---|---|---|
| TQ | 5-Aza | TQ | 5-Aza | |
| JAK2 | −5.99 | −6.37 | 40.79 μM | 21.51 μM |
| STAT3 | −6.68 | −6.52 | 12.76 μM | 16.68 μM |
| STAT5 | −6.04 | −6.0 | 37.64 μM | 40.21 μM |
| Genes | Accession Numbers | Primer Sequence (5′-3′) | Reference |
|---|---|---|---|
| SHP-1 | NC_000012.12 | Forward: GCCTGGACTGTGACATTGAC Reverse: ATGTTCCCGTACTCCGACTC | [43] |
| SOCS-1 | NC_000016.10 | Forward: GACGCCTGCGGATTCTAC Reverse: AGCGGCCGGCCTGAAAG | [54] |
| SOCS-3 | NC_000017.11 | Forward: GACCAGCGCCACTTCTTCAC Reverse: CTGGATGCGCAGGTTCTTG | [55] |
| FLT3-ITD | NC_000013.11 | Forward: ACGCTTGGAAGCAGGAGAT Reverse: CACAAGGCTGCCCTCTAGTT | [56] |
| JAK2 | NC_000009.12 | Forward: TGTCTTACCTCTTTGCTCAGTGGCG Reverse: CAATGACATTTTCTCGCTCGACAGC | [57] |
| STAT3 | NC_000017.11 | Forward: GATTGACCAGCAGTATAGCCGCTTC Reverse: CTGCAGTCTGTAGAAGGCGTG | [58] |
| STAT5A | NC_000017.11 | Forward: GAAGCTGAACGTGCACATGAATC Reverse: GTAGGGACAGAGTCTTCACCTGG | [45] |
| STAT5B | NC_000017.13 | Forward: AGTTTGATTCTCAGGAAAGAATGT Reverse: TCCATCAACAGCTTTAGCAGT | [45] |
| DNMT1 | NC_000019.10 | Forward: TAT CCG AGG AGG GCT ACC Reverse: TAA GCA TGA GCA CCG TTCT | [59] |
| DNMT3A | NC_000002.12 | Forward: GGA GGA CCGAAA GGA CGG A Reverse: CCC CATTGG GTA ATA GCTCTG AG | [59] |
| DNMT3B | NC_000020.11 | Forward: GAG ATC AGA GGC CGA AGA T Reverse: CTG TCA AGT CCT GTG TGTAG | [59] |
| TET2 | NC_000004.12 | Forward: ACGCTTGGAAGCAGGAGAT Reverse: CACAAGGCTGCCCTCTAGTT | [60] |
| WT1 | NC_000011.10 | Forward: CAGGCTGCAAAAGAGATATTTTAAGCT Reverse: GAAGTCACACTGGTATGGTTTCTCA | [61] |
| β-actin | NC_000071.7 | Forward: CTGGCACCCAGGACAATG Reverse: GCCGATCCACACGGAGTA | [62] |
| Assay Name | Analyzed Bisufite Sequence | Type of Primer | Primer Sequence (5′-3′) | Amplicon Length |
|---|---|---|---|---|
| SHP-1 | GAGATGTYGTGGGATYGTTTGGGTTYGTATGYGTGAAGTATTATTTGGGTTTGGAGTG | PCR-Forward | GGAGGGTTGAGTAAAAGTAGTTGG | 94 bp |
| PCR-Reverse | Biotin_ACACTCCAAACCCAAATAATACTT | |||
| Sequencing | TTGGTGGAGGAGGGA | |||
| SOCS-1 | GTYGAAGYGGTTTTYGTYGGAYGTTATYGYGGAAAGAGAAATTAAAAGTG GAGTTG | PCR-Forward | AGGGTTTAGAAGAGAGGGAAATA | 85 bp |
| PCR-Reverse | Biotin_CCCCCAACTCCACTTTTAATT | |||
| Sequencing | GAAGAGAGGGAAATAGG | |||
| SOCS-3 | GTYGGTATTTYGATTYGTTATTGTAGATGTAGTTATTAA TA | PCR-Forward | GGAGGTTTTGGGTTTGGTATTTAGTAA | 207 bp |
| PCR-Reverse | Biotin_AAACCCTTCCCCAAATCTCATAAAT | |||
| Sequencing | ATTTAGTAAAGTTGTGGTTTGAG |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Al-Rawashde, F.A.; Johan, M.F.; Taib, W.R.W.; Ismail, I.; Johari, S.A.T.T.; Almajali, B.; Al-wajeeh, A.S.; Nazari Vishkaei, M.; Al-Jamal, H.A.N. Thymoquinone Inhibits Growth of Acute Myeloid Leukemia Cells through Reversal SHP-1 and SOCS-3 Hypermethylation: In Vitro and In Silico Evaluation. Pharmaceuticals 2021, 14, 1287. https://doi.org/10.3390/ph14121287
Al-Rawashde FA, Johan MF, Taib WRW, Ismail I, Johari SATT, Almajali B, Al-wajeeh AS, Nazari Vishkaei M, Al-Jamal HAN. Thymoquinone Inhibits Growth of Acute Myeloid Leukemia Cells through Reversal SHP-1 and SOCS-3 Hypermethylation: In Vitro and In Silico Evaluation. Pharmaceuticals. 2021; 14(12):1287. https://doi.org/10.3390/ph14121287
Chicago/Turabian StyleAl-Rawashde, Futoon Abedrabbu, Muhammad Farid Johan, Wan Rohani Wan Taib, Imilia Ismail, Syed Ahmad Tajudin Tuan Johari, Belal Almajali, Abdullah Saleh Al-wajeeh, Mansoureh Nazari Vishkaei, and Hamid Ali Nagi Al-Jamal. 2021. "Thymoquinone Inhibits Growth of Acute Myeloid Leukemia Cells through Reversal SHP-1 and SOCS-3 Hypermethylation: In Vitro and In Silico Evaluation" Pharmaceuticals 14, no. 12: 1287. https://doi.org/10.3390/ph14121287
APA StyleAl-Rawashde, F. A., Johan, M. F., Taib, W. R. W., Ismail, I., Johari, S. A. T. T., Almajali, B., Al-wajeeh, A. S., Nazari Vishkaei, M., & Al-Jamal, H. A. N. (2021). Thymoquinone Inhibits Growth of Acute Myeloid Leukemia Cells through Reversal SHP-1 and SOCS-3 Hypermethylation: In Vitro and In Silico Evaluation. Pharmaceuticals, 14(12), 1287. https://doi.org/10.3390/ph14121287