
Novel DYRK1A Inhibitor Rescues Learning and Memory Deficits in a Mouse Model of Down Syndrome

 , ,
, ,  ,
,
Abstract
:1. Introduction
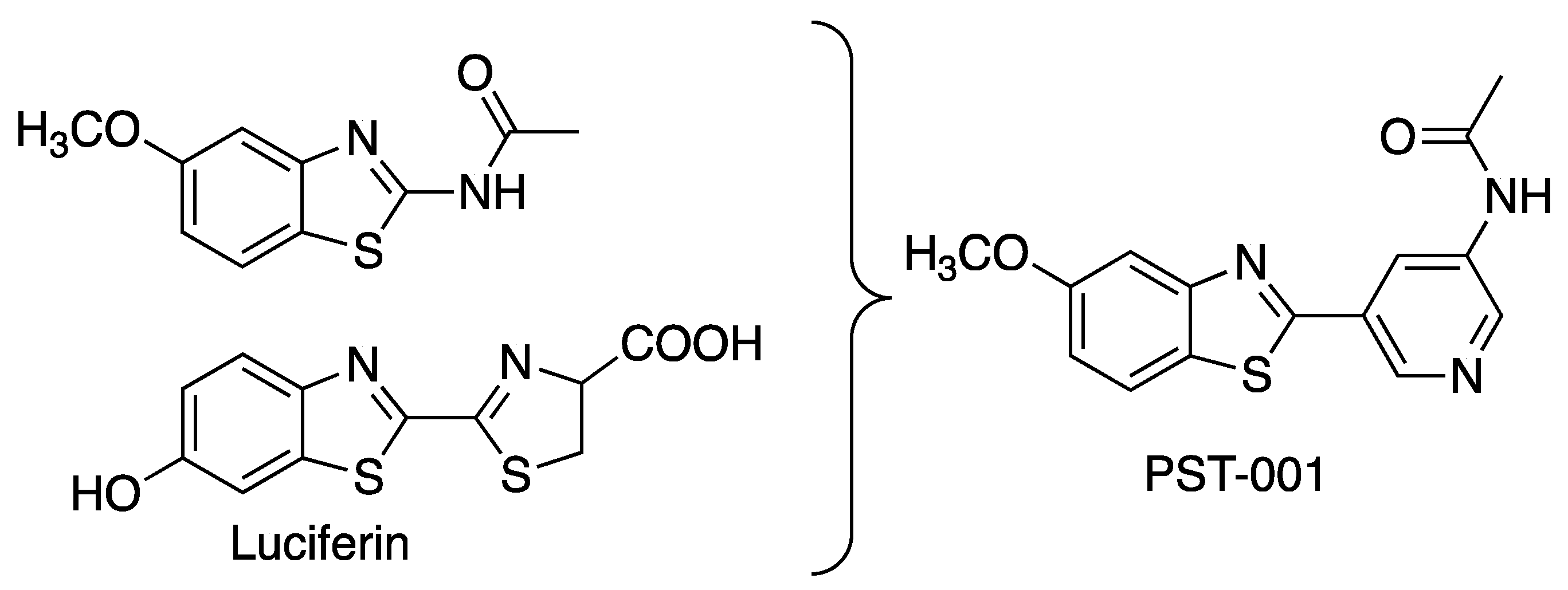
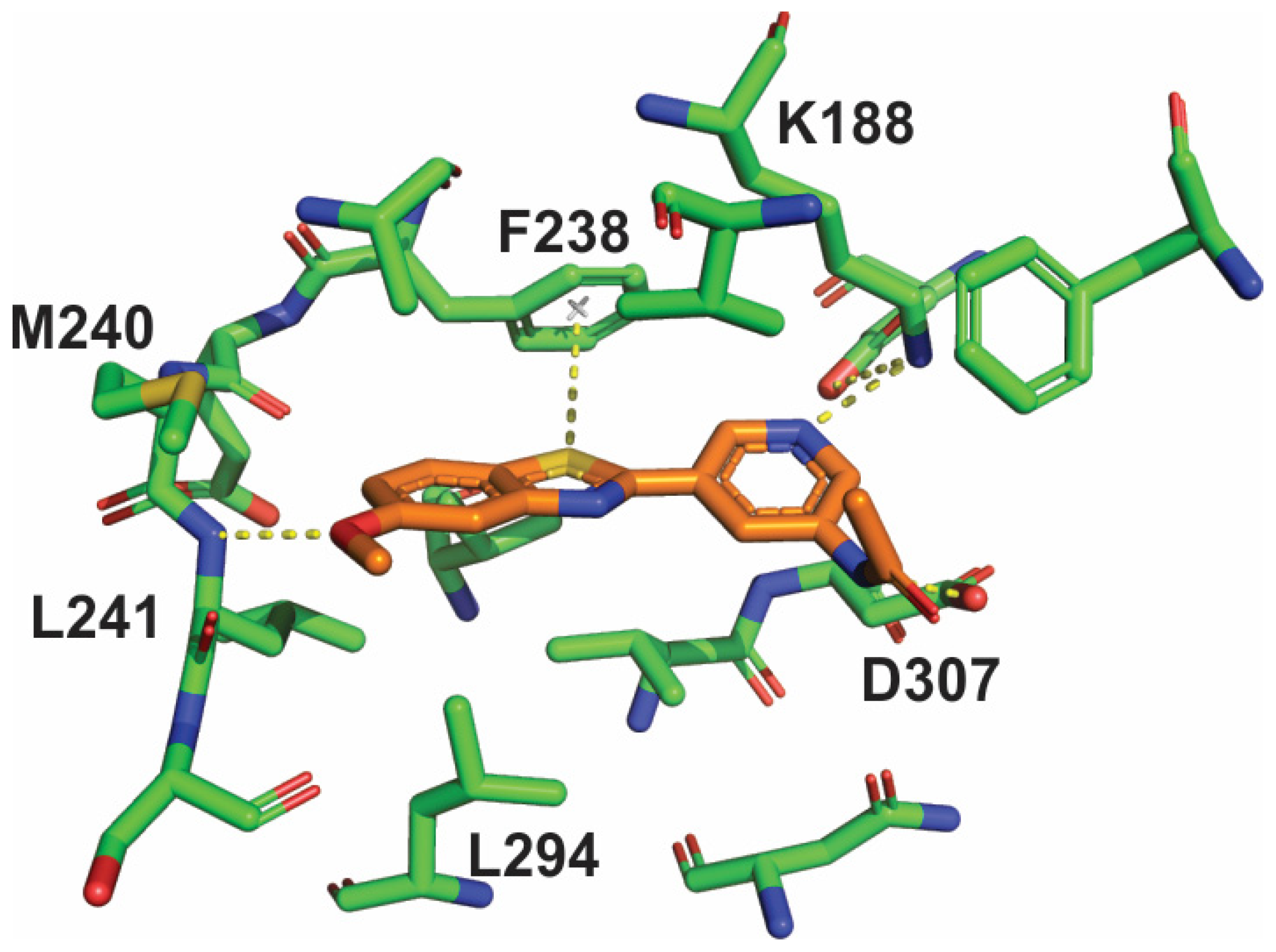
2. Results
3. Discussion
4. Materials and Methods
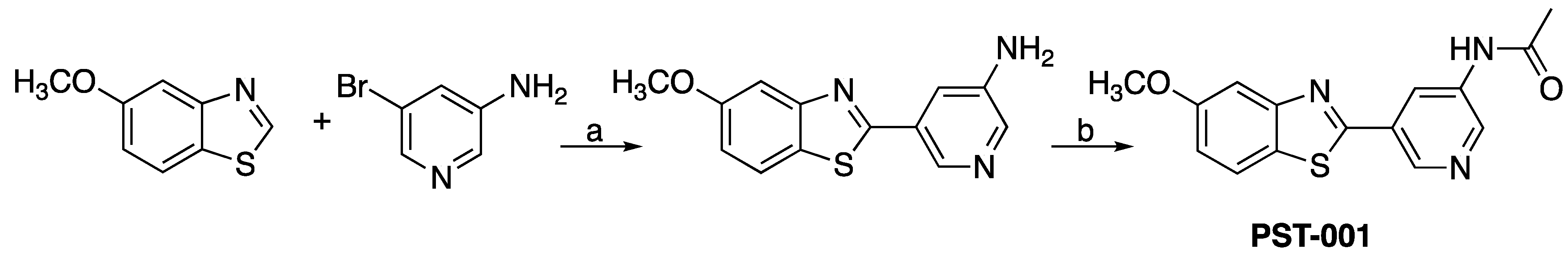
4.1. Chemical Preparation
- 5-(5-Methoxybenzo[d]thiazol-2-yl)pyridin-3-amine (1). 5-Methoxybenzothiazole (6.34 g, 38.4 mmol), 3-amino-5-bromopyridine (7.41 g, 42.8 mmol), cesium carbonate (12.5 g, 38.4 mmol), copper(I)bromide (1.12 g) and Pd(OAc)2 (0.56 g, 2.50 mmol) were suspended in dry DMF (200 mL) under argon. P(t-Bu)3 (1.00 g, 4.94 mmol) dissolved in 10 mL dry DMF was added. The reaction mixture was heated at 150 °C for 1.5 hrs, cooled to room temperature and poured into EtOAc (100 mL). The organic phase was washed with water (100 mL) and the aqueous phase extracted with EtOAc (2 × 100 mL). The combined organic phase was washed with water, dried (MgSO4), filtered and concentrated. Flash chromatography (Heptane: EtOAc 80: 20–50: 50–EtOAc) afforded 4.09 g (41%) of the title compound as a pale yellow solid. Purity >95% (HPLC). 1H NMR (300 MHz, DMSO-d6) δ 8.39 (s, 1H), 8.08 (s, 1H), 8.01 (d, J = 8.8, 1H), 7.73–7.49 (m, 2H), 7.11 (dd, J = 8.8, 2.5, 1H), 5.71 (s, 2H), 3.87 (s, 3H). MS (pos): 258 (M+H), HR (M+H): 258.0695 (observed), 258.0701 (calculated).
- N-(5-(5-Methoxybenzo[d]thiazol-2-yl)pyridin-3-yl)acetamide (PST-001) [70]. To a suspension of 5-(5-methoxybenzo[d]thiazol-2-yl)pyridin-3-amine (1.29 g, 5.00 mmol) in DCM (25 mL) was added pyridine (10 mL), followed by acetic anhydride (0.95 mL, 10.0 mmol). The reaction mixture was stirred at room temperature overnight, poured into water (100 mL) and the aqueous phase extracted with CHCl3: MeOH (90:10) (3 × 100 mL). The combined organic extract was dried (Na2SO4), filtered and concentrated. The crude material was treated with EtOAc (75 mL), sonicated for 2 min, and filtered. Drying allowed the isolation of 1.30 g (73%) of the title compound as a beige solid from 1.54 g substrate. Purity 99.5% (HPLC). 1H NMR (400 MHz, DMSO-d6) δ 10.41 (s, 1H), 8.88 (s, 1H), 8.80 (s, 2H), 8.03 (d, J = 8.8, 1H), 7.66 (d, J = 2.3, 1H), 7.13 (dd, J = 8.8, 2.4, 1H), 3.87 (s, 3H), 2.13 (s, 3H). 13C NMR (150 MHz, DMSO-d6) δ 169.8, 165.8, 159.5, 155.2, 142.8, 142.2, 136.8, 129.3, 126.7, 123.5, 123.2, 116.3, 106.1, 56.0, 24.4. IR (ATR, cm−1): 3301, 1680, 1603, 1551, 1503. MS (pos): 322 (M+Na), HR (M+H): 300.0801 (observed), 300.0807 (calculated).
4.2. DYRK1A Protein Production and Crystallization
4.3. Structure Solution and Refinement
4.4. IC50 Determination
4.5. Kinase Profile of Benzothiazolylpyridine Derivatives
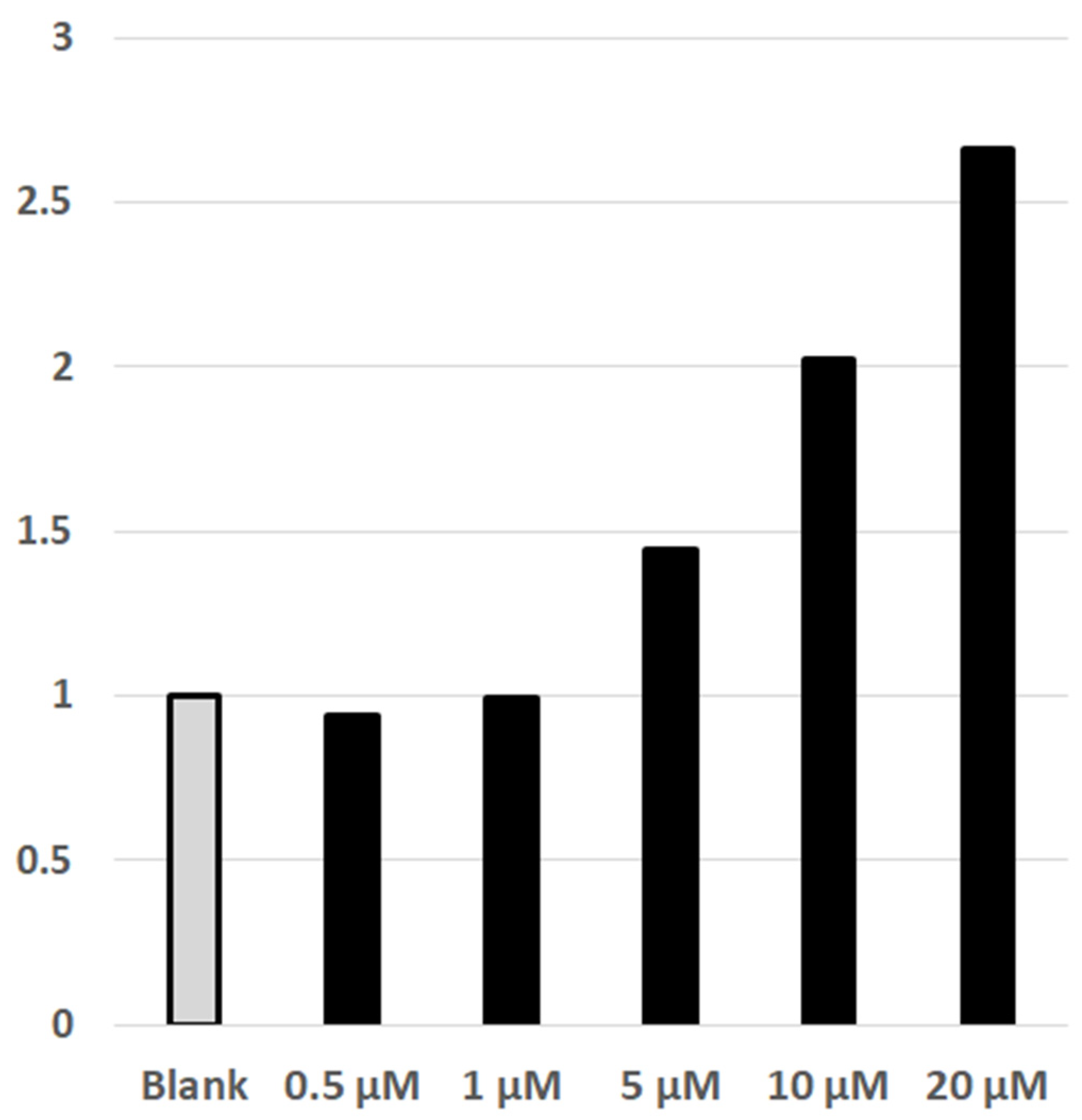
4.6. Luciferin/Luciferase Detected NFAT-Calcineurin Assay of Cellular Effect of DYRK1A
4.7. Rat IV Pharmacokinetics
4.8. Mouse PO Pharmacokinetics
4.9. Bioanalysis
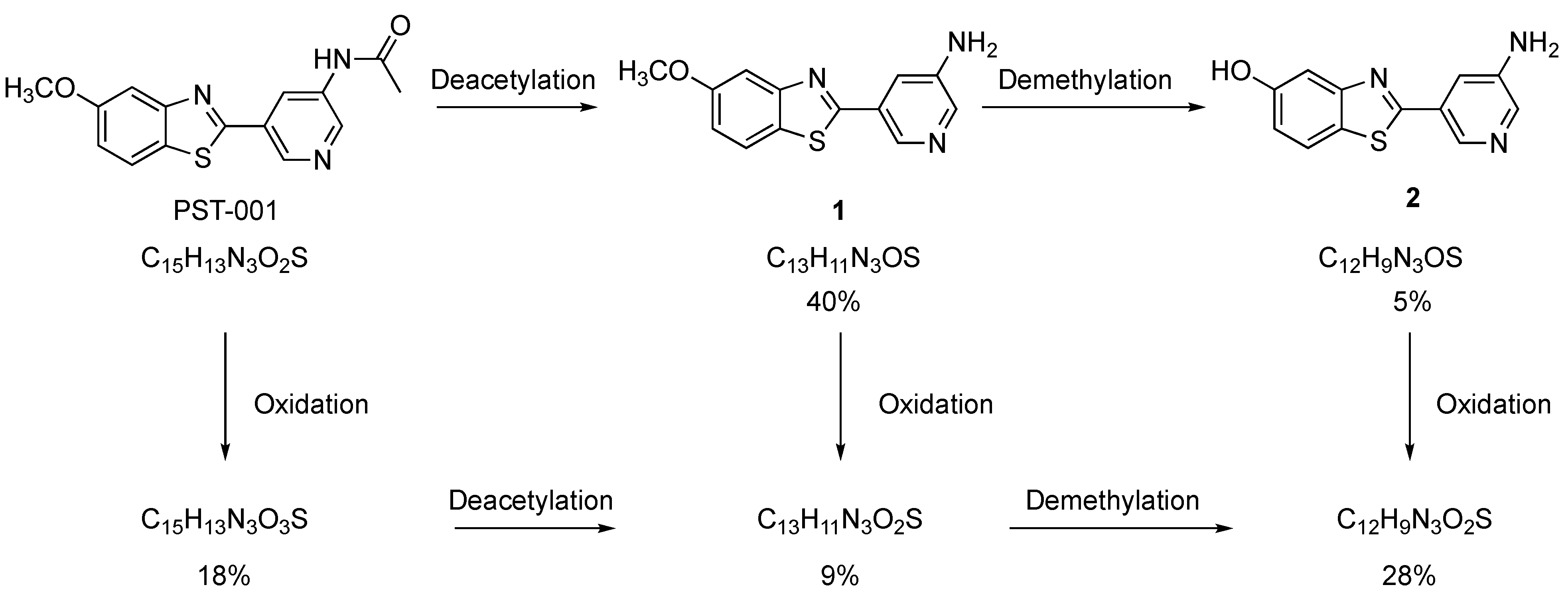
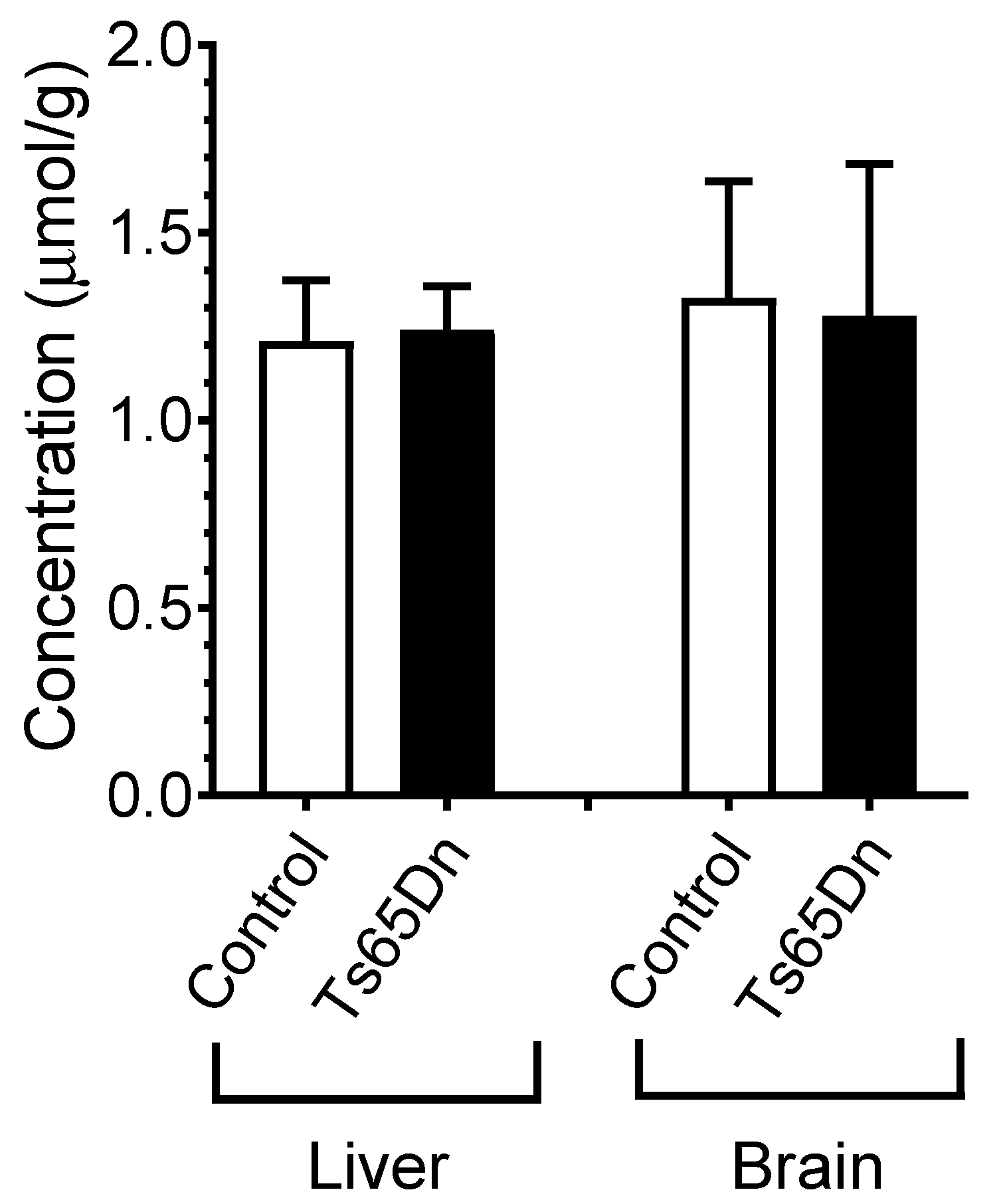
4.10. Ts65Ds Model of Cognition. Determination of Blood, Liver, and Brain PST-001 Concentrations
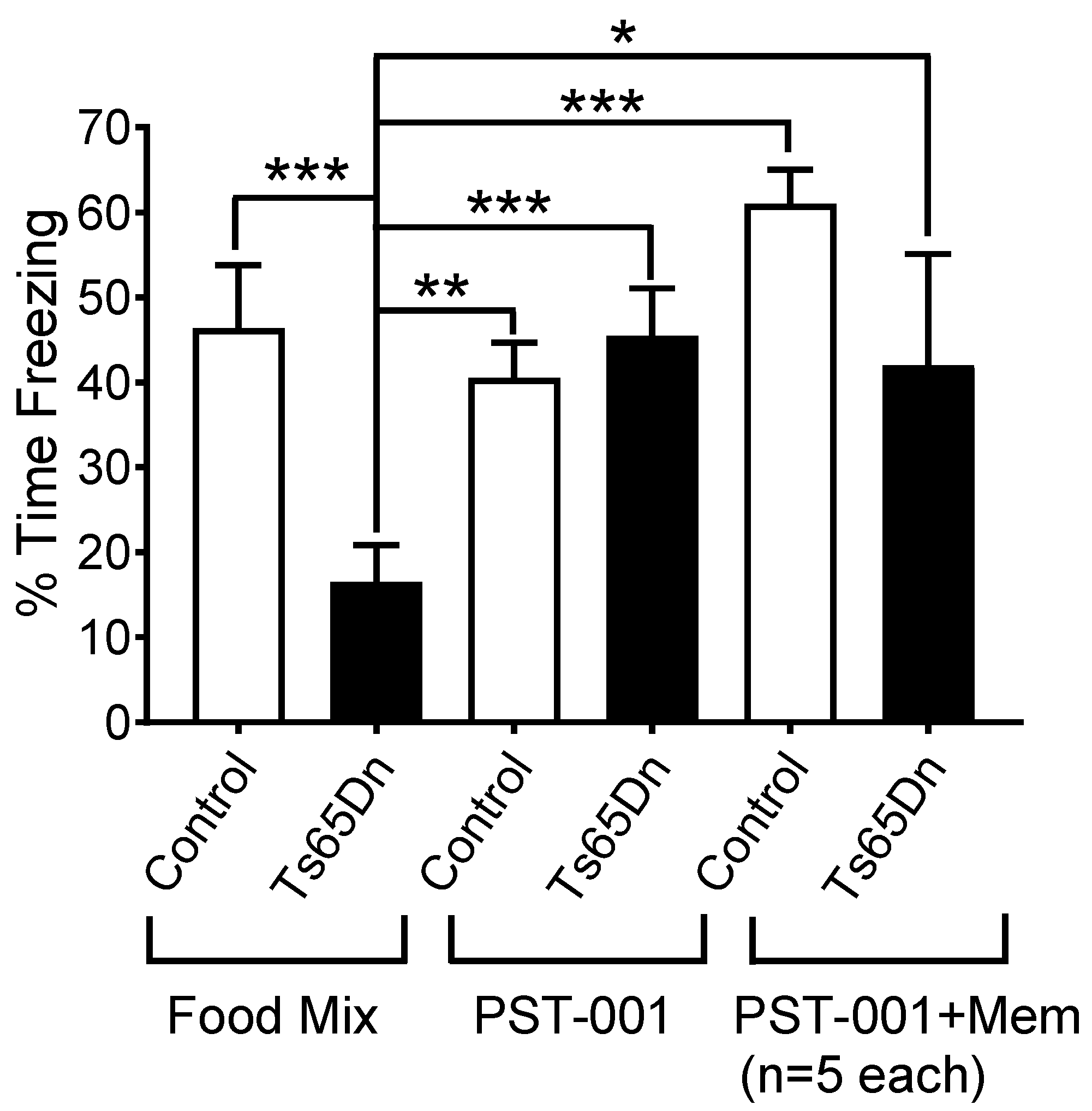
4.11. Ts65Ds Model of Cognition. Contextual Fear Conditioning
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Down, J.L.H. Observations on an Ethnic Classification of Idiots. Lond. Hosp. Rep. 1866, 3, 259–262. [Google Scholar]
- Antonarakis, S.E.; Epstein, C.J. The challenge of Down syndrome. Trends Mol. Med. 2006, 12, 473–479. [Google Scholar] [CrossRef]
- Lejeune, J.; Gautier, M.; Turpin, R. Study of somatic chromosomes from 9 mongoloid children. C. R. Hebd. Seances Acad. Sci. 1959, 248, 1721–1722. [Google Scholar]
- Tejedor, F.; Zhu, X.R.; Kaltenbach, E.; Ackermann, A.; Baumann, A.; Canal, I.; Heisenberg, M.; Fischbach, K.F.; Pongs, O. minibrain: A new protein kinase family involved in postembryonic neurogenesis in Drosophila. Neuron 1995, 14, 287–301. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.; Antonarakis, S.E. Localisation of a human homologue of the Drosophila mnb and rat Dyrk genes to chromosome 21q22.2. Hum. Genet. 1997, 99, 262–265. [Google Scholar] [CrossRef]
- Lyle, R.; Bena, F.; Gagos, S.; Gehrig, C.; Lopez, G.; Schinzel, A.; Lespinasse, J.; Bottani, A.; Dahoun, S.; Taine, L.; et al. Genotype-phenotype correlations in Down syndrome identified by array CGH in 30 cases of partial trisomy and partial monosomy chromosome 21. Eur. J. Hum. Genet. 2009, 17, 454–466. [Google Scholar] [CrossRef] [Green Version]
- Korenberg, J.R.; Chen, X.N.; Schipper, R.; Sun, Z.; Gonsky, R.; Gerwehr, S.; Carpenter, N.; Daumer, C.; Dignan, P.; Disteche, C. Down syndrome phenotypes: The consequences of chromosomal imbalance. Proc. Natl. Acad. Sci. USA 1994, 91, 4997–5001. [Google Scholar] [CrossRef] [Green Version]
- Ahn, K.J.; Jeong, H.K.; Choi, H.S.; Ryoo, S.R.; Kim, Y.J.; Goo, J.S.; Choi, S.Y.; Han, J.S.; Ha, I.; Song, W.J. DYRK1A BAC transgenic mice show altered synaptic plasticity with learning and memory defects. Neurobiol. Dis. 2006, 22, 463–472. [Google Scholar] [CrossRef]
- Dowjat, W.K.; Adayev, T.; Kuchna, I.; Nowicki, K.; Palminiello, S.; Hwang, Y.W.; Wegiel, J. Trisomy-driven overexpression of DYRK1A kinase in the brain of subjects with Down syndrome. Neurosci. Lett. 2007, 413, 77–81. [Google Scholar] [CrossRef] [Green Version]
- Guimera, J.; Casas, C.; Estivill, X.; Pritchard, M. Human minibrain homologue (MNBH/DYRK1): Characterization, alternative splicing, differential tissue expression, and overexpression in Down syndrome. Genomics 1999, 57, 407–418. [Google Scholar] [CrossRef]
- Moller, R.S.; Kubart, S.; Hoeltzenbein, M.; Heye, B.; Vogel, I.; Hansen, C.P.; Menzel, C.; Ullmann, R.; Tommerup, N.; Ropers, H.H.; et al. Truncation of the Down syndrome candidate gene DYRK1A in two unrelated patients with microcephaly. Am. J. Hum. Genet. 2008, 82, 1165–1170. [Google Scholar] [CrossRef] [Green Version]
- Altafaj, X.; Dierssen, M.; Baamonde, C.; Marti, E.; Visa, J.; Guimera, J.; Oset, M.; Gonzalez, J.R.; Florez, J.; Fillat, C.; et al. Neurodevelopmental delay, motor abnormalities and cognitive deficits in transgenic mice overexpressing Dyrk1A (minibrain), a murine model of Down’s syndrome. Hum. Mol. Genet. 2001, 10, 1915–1923. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, F.; Liang, Z.; Wegiel, J.; Hwang, Y.W.; Iqbal, K.; Grundke-Iqbal, I.; Ramakrishna, N.; Gong, C.X. Overexpression of Dyrk1A contributes to neurofibrillary degeneration in Down syndrome. FASEB J. 2008, 22, 3224–3233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feki, A.; Hibaoui, Y. DYRK1A Protein, A Promising Therapeutic Target to Improve Cognitive Deficits in Down Syndrome. Brain Sci. 2018, 8, 187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, O.H.; Cho, H.J.; Han, E.; Hong, T.I.; Ariyasiri, K.; Choi, J.H.; Hwang, K.S.; Jeong, Y.M.; Yang, S.Y.; Yu, K.; et al. Zebrafish knockout of Down syndrome gene, DYRK1A, shows social impairments relevant to autism. Mol. Autism 2017, 8, 50. [Google Scholar] [CrossRef]
- Gupta, M.; Dhanasekaran, A.R.; Gardiner, K.J. Mouse models of Down syndrome: Gene content and consequences. Mamm. Genome 2016, 27, 538–555. [Google Scholar] [CrossRef]
- Reeves, R.H.; Irving, N.G.; Moran, T.H.; Wohn, A.; Kitt, C.; Sisodia, S.S.; Schmidt, C.; Bronson, R.T.; Davisson, M.T. A mouse model for Down syndrome exhibits learning and behaviour deficits. Nat. Genet. 1995, 11, 177–184. [Google Scholar] [CrossRef]
- Sago, H.; Carlson, E.J.; Smith, D.J.; Kilbridge, J.; Rubin, E.M.; Mobley, W.C.; Epstein, C.J.; Huang, T.T. Ts1Cje, a partial trisomy 16 mouse model for Down syndrome, exhibits learning and behavioral abnormalities. Proc. Natl. Acad. Sci. USA 1998, 95, 6256–6261. [Google Scholar] [CrossRef] [Green Version]
- Jarhad, D.B.; Mashelkar, K.K.; Kim, H.R.; Noh, M.; Jeong, L.S. Dual-Specificity Tyrosine Phosphorylation-Regulated Kinase 1A (DYRK1A) Inhibitors as Potential Therapeutics. J. Med. Chem. 2018, 61, 9791–9810. [Google Scholar] [CrossRef]
- Nguyen, T.L.; Fruit, C.; Hérault, Y.; Meijer, L.; Besson, T. Dual-specificity tyrosine phosphorylation-regulated kinase 1A (DYRK1A) inhibitors: A survey of recent patent literature. Expert Opin. Ther. Pat. 2017, 27, 1183–1199. [Google Scholar] [CrossRef]
- Gockler, N.; Jofre, G.; Papadopoulos, C.; Soppa, U.; Tejedor, F.J.; Becker, W. Harmine specifically inhibits protein kinase DYRK1A and interferes with neurite formation. FEBS J. 2009, 276, 6324–6337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adayev, T.; Wegiel, J.; Hwang, Y.W. Harmine is an ATP-competitive inhibitor for dual-specificity tyrosine phosphorylation-regulated kinase 1A (Dyrk1A). Arch. Biochem. Biophys. 2011, 507, 212–218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De la Torre, R.; De Sola, S.; Pons, M.; Duchon, A.; de Lagran, M.M.; Farre, M.; Fito, M.; Benejam, B.; Langohr, K.; Rodriguez, J.; et al. Epigallocatechin-3-gallate, a DYRK1A inhibitor, rescues cognitive deficits in Down syndrome mouse models and in humans. Mol. Nutr. Food Res. 2014, 58, 278–288. [Google Scholar] [CrossRef] [PubMed]
- Guedj, F.; Sebrie, C.; Rivals, I.; Ledru, A.; Paly, E.; Bizot, J.C.; Smith, D.; Rubin, E.; Gillet, B.; Arbones, M.; et al. Green tea polyphenols rescue of brain defects induced by overexpression of DYRK1A. PLoS ONE 2009, 4, e4606. [Google Scholar] [CrossRef] [Green Version]
- Ogawa, Y.; Nonaka, Y.; Goto, T.; Ohnishi, E.; Hiramatsu, T.; Kii, I.; Yoshida, M.; Ikura, T.; Onogi, H.; Shibuya, H.; et al. Development of a novel selective inhibitor of the Down syndrome-related kinase Dyrk1A. Nat. Commun. 2010, 1, 86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rothweiler, U.; Eriksson, J.; Stensen, W.; Leeson, F.; Engh, R.A.; Svendsen, J.S. Luciferin and derivatives as a DYRK selective scaffold for the design of protein kinase inhibitors. Eur. J. Med. Chem. 2015, 94, 140–148. [Google Scholar] [CrossRef]
- Rothweiler, U.; Stensen, W.; Brandsdal, B.O.; Isaksson, J.; Leeson, F.A.; Engh, R.A.; Svendsen, J.S. Probing the ATP-Binding Pocket of Protein Kinase DYRK1A with Benzothiazole Fragment Molecules. J. Med. Chem. 2016, 59, 9814–9824. [Google Scholar] [CrossRef] [PubMed]
- Leblond, B.; Casagrande, A.-S.; Désiré, L.; Foucourt, A.; Besson, T. DYRK1 Inhibitors and Uses Thereof. WO Patent 2013/026806 A1, 28 February 2013. [Google Scholar]
- Foucourt, A.; Hedou, D.; Dubouilh-Benard, C.; Girard, A.; Taverne, T.; Casagrande, A.S.; Desire, L.; Leblond, B.; Besson, T. Design and synthesis of thiazolo[5, 4-f]quinazolines as DYRK1A inhibitors, part II. Molecules 2014, 19, 15411–15439. [Google Scholar] [CrossRef] [Green Version]
- Coutadeur, S.; Benyamine, H.; Delalonde, L.; de Oliveira, C.; Leblond, B.; Foucourt, A.; Besson, T.; Casagrande, A.S.; Taverne, T.; Girard, A.; et al. A novel DYRK1A (dual specificity tyrosine phosphorylation-regulated kinase 1A) inhibitor for the treatment of Alzheimer’s disease: Effect on Tau and amyloid pathologies in vitro. J. Neurochem. 2015, 133, 440–451. [Google Scholar] [CrossRef]
- Sander, T.; Freyss, J.; von Korff, M.; Rufener, C. DataWarrior: An Open-Source Program For Chemistry Aware Data Visualization And Analysis. J. Chem. Inf. Modeling 2015, 55, 460–473. [Google Scholar] [CrossRef]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef]
- Graczyk, P.P. Gini Coefficient: A New Way To Express Selectivity of Kinase Inhibitors against a Family of Kinases. J. Med. Chem. 2007, 50, 5773–5779. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, C.; Frank, D.; Will, R.; Jaschinski, C.; Frauen, R.; Katus, H.A.; Frey, N. DYRK1A is a novel negative regulator of cardiomyocyte hypertrophy. J. Biol. Chem. 2009, 284, 17320–17327. [Google Scholar] [CrossRef] [Green Version]
- Arron, J.R.; Winslow, M.M.; Polleri, A.; Chang, C.P.; Wu, H.; Gao, X.; Neilson, J.R.; Chen, L.; Heit, J.J.; Kim, S.K.; et al. NFAT dysregulation by increased dosage of DSCR1 and DYRK1A on chromosome 21. Nature 2006, 441, 595–600. [Google Scholar] [CrossRef]
- Olsen, L.; Rydberg, P.; Rod, T.H.; Ryde, U. Prediction of Activation Energies for Hydrogen Abstraction by Cytochrome P450. J. Med. Chem. 2006, 49, 6489–6499. [Google Scholar] [CrossRef] [PubMed]
- de Bruyn Kops, C.; Stork, C.; Sicho, M.; Kochev, N.; Svozil, D.; Jeliazkova, N.; Kirchmair, J. GLORY: Generator of the Structures of Likely Cytochrome P450 Metabolites Based on Predicted Sites of Metabolism. Front. Chem. 2019, 7, 1–15. [Google Scholar] [CrossRef]
- Stork, C.; Embruch, G.; Šícho, M.; de Bruyn Kops, C.; Chen, Y.; Svozil, D.; Kirchmair, J. NERDD: A web portal providing access to in silico tools for drug discovery. Bioinformatics 2019, 36, 1291–1292. [Google Scholar] [CrossRef]
- Ackley, D.C.; Rockich, K.T.; Baker, T.R. Metabolic Stability Assessed by Liver Microsomes and Hepatocytes. In Optimization in Drug Discovery; Yan, Z., Caldwell, G.W., Eds.; Humana Press: Totowa, NJ, USA, 2004; pp. 151–162. [Google Scholar]
- Costa, A.C.; Scott-McKean, J.J.; Stasko, M.R. Acute injections of the NMDA receptor antagonist memantine rescue performance deficits of the Ts65Dn mouse model of Down syndrome on a fear conditioning test. Neuropsychopharmacology 2008, 33, 1624–1632. [Google Scholar] [CrossRef] [Green Version]
- Arbones, M.L.; Thomazeau, A.; Nakano-Kobayashi, A.; Hagiwara, M.; Delabar, J.M. DYRK1A and cognition: A lifelong relationship. Pharmacol. Ther. 2019, 194, 199–221. [Google Scholar] [CrossRef]
- Wegiel, J.; Gong, C.X.; Hwang, Y.W. The role of DYRK1A in neurodegenerative diseases. FEBS J. 2011, 278, 236–245. [Google Scholar] [CrossRef]
- Souchet, B.; Duchon, A.; Gu, Y.; Dairou, J.; Chevalier, C.; Daubigney, F.; Nalesso, V.; Creau, N.; Yu, Y.; Janel, N.; et al. Prenatal treatment with EGCG enriched green tea extract rescues GAD67 related developmental and cognitive defects in Down syndrome mouse models. Sci. Rep. 2019, 9, 3914. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pathak, A.; Rohilla, A.; Gupta, T.; Akhtar, M.J.; Haider, M.R.; Sharma, K.; Haider, K.; Yar, M.S. DYRK1A kinase inhibition with emphasis on neurodegeneration: A comprehensive evolution story-cum-perspective. Eur. J. Med. Chem. 2018, 158, 559–592. [Google Scholar] [CrossRef]
- Ruiz-Mejias, M.; Martinez de Lagran, M.; Mattia, M.; Castano-Prat, P.; Perez-Mendez, L.; Ciria-Suarez, L.; Gener, T.; Sancristobal, B.; Garcia-Ojalvo, J.; Gruart, A.; et al. Overexpression of Dyrk1A, a Down Syndrome Candidate, Decreases Excitability and Impairs Gamma Oscillations in the Prefrontal Cortex. J. Neurosci. 2016, 36, 3648–3659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duchon, A.; Herault, Y. DYRK1A, a Dosage-Sensitive Gene Involved in Neurodevelopmental Disorders, Is a Target for Drug Development in Down Syndrome. Front. Behav. Neurosci. 2016, 10, e54285. [Google Scholar] [CrossRef] [Green Version]
- Becker, W.; Soppa, U.; Tejedor, F.J. DYRK1A: A potential drug target for multiple Down syndrome neuropathologies. CNS Neurol. Disord. Drug Targets 2014, 13, 26–33. [Google Scholar] [CrossRef] [PubMed]
- Laguna, A.; Barallobre, M.J.; Marchena, M.A.; Mateus, C.; Ramirez, E.; Martinez-Cue, C.; Delabar, J.M.; Castelo-Branco, M.; de la Villa, P.; Arbones, M.L. Triplication of DYRK1A causes retinal structural and functional alterations in Down syndrome. Hum. Mol. Genet. 2013, 22, 2775–2784. [Google Scholar] [CrossRef] [Green Version]
- Courcet, J.B.; Faivre, L.; Malzac, P.; Masurel-Paulet, A.; Lopez, E.; Callier, P.; Lambert, L.; Lemesle, M.; Thevenon, J.; Gigot, N.; et al. The DYRK1A gene is a cause of syndromic intellectual disability with severe microcephaly and epilepsy. J. Med. Genet. 2012, 49, 731–736. [Google Scholar] [CrossRef]
- Ryu, Y.S.; Park, S.Y.; Jung, M.S.; Yoon, S.H.; Kwen, M.Y.; Lee, S.Y.; Choi, S.H.; Radnaabazar, C.; Kim, M.K.; Kim, H.; et al. Dyrk1A-mediated phosphorylation of Presenilin 1: A functional link between Down syndrome and Alzheimer’s disease. J. Neurochem. 2010, 115, 574–584. [Google Scholar] [CrossRef]
- Ryoo, S.R.; Jeong, H.K.; Radnaabazar, C.; Yoo, J.J.; Cho, H.J.; Lee, H.W.; Kim, I.S.; Cheon, Y.H.; Ahn, Y.S.; Chung, S.H.; et al. DYRK1A-mediated hyperphosphorylation of Tau. A functional link between Down syndrome and Alzheimer disease. J. Biol. Chem. 2007, 282, 34850–34857. [Google Scholar] [CrossRef] [Green Version]
- Kimura, R.; Kamino, K.; Yamamoto, M.; Nuripa, A.; Kida, T.; Kazui, H.; Hashimoto, R.; Tanaka, T.; Kudo, T.; Yamagata, H.; et al. The DYRK1A gene, encoded in chromosome 21 Down syndrome critical region, bridges between beta-amyloid production and tau phosphorylation in Alzheimer disease. Hum. Mol. Genet. 2007, 16, 15–23. [Google Scholar] [CrossRef] [Green Version]
- Dierssen, M.; de Lagran, M.M. DYRK1A (dual-specificity tyrosine-phosphorylated and -regulated kinase 1A): A gene with dosage effect during development and neurogenesis. Sci. World J. 2006, 6, 1911–1922. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kentrup, H.; Joost, H.G.; Heimann, G.; Becker, W. Minibrain/DYRK1A gene: Candidate gene for mental retardation in Down’s syndrome? Klin Padiatr 2000, 212, 60–63. [Google Scholar] [CrossRef] [PubMed]
- Barallobre, M.J.; Perier, C.; Bove, J.; Laguna, A.; Delabar, J.M.; Vila, M.; Arbones, M.L. DYRK1A promotes dopaminergic neuron survival in the developing brain and in a mouse model of Parkinson’s disease. Cell Death Dis. 2014, 5, e1289. [Google Scholar] [CrossRef] [PubMed]
- Jones, E.L.; Aarsland, D.; Londos, E.; Ballard, C. A pilot study examining associations between DYRK1A and alpha-synuclein dementias. Neurodegener. Dis. 2012, 10, 229–231. [Google Scholar] [CrossRef]
- Kim, E.J.; Sung, J.Y.; Lee, H.J.; Rhim, H.; Hasegawa, M.; Iwatsubo, T.; Min do, S.; Kim, J.; Paik, S.R.; Chung, K.C. Dyrk1A phosphorylates alpha-synuclein and enhances intracellular inclusion formation. J. Biol. Chem. 2006, 281, 33250–33257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferrer, I.; Barrachina, M.; Puig, B. Glycogen synthase kinase-3 is associated with neuronal and glial hyperphosphorylated tau deposits in Alzheimer’s disease, Pick’s disease, progressive supranuclear palsy and corticobasal degeneration. Acta Neuropathol. 2002, 104, 583–591. [Google Scholar] [CrossRef]
- Ferrer, I.; Barrachina, M.; Puig, B.; Martínez de Lagrán, M.; Martí, E.; Avila, J.; Dierssen, M. Constitutive Dyrk1A is abnormally expressed in Alzheimer disease, Down syndrome, Pick disease, and related transgenic models. Neurobiol. Dis. 2005, 20, 392–400. [Google Scholar] [CrossRef]
- Pozo, N.; Zahonero, C.; Fernandez, P.; Linares, J.M.; Ayuso, A.; Hagiwara, M.; Perez, A.; Ricoy, J.R.; Hernandez-Lain, A.; Sepulveda, J.M.; et al. Inhibition of DYRK1A destabilizes EGFR and reduces EGFR-dependent glioblastoma growth. J. Clin. Investig. 2013, 123, 2475–2487. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.L.; Ding, K.; Hu, X.; Wu, L.W.; Zhou, D.M.; Rao, M.J.; Lin, N.M.; Zhang, C. DYRK1A inhibition suppresses STAT3/EGFR/Met signalling and sensitizes EGFR wild-type NSCLC cells to AZD9291. J. Cell. Mol. Med. 2019, 23, 7427–7437. [Google Scholar] [CrossRef] [Green Version]
- Fernandez-Martinez, P.; Zahonero, C.; Sanchez-Gomez, P. DYRK1A: The double-edged kinase as a protagonist in cell growth and tumorigenesis. Mol. Cell. Oncol. 2015, 2, e970048. [Google Scholar] [CrossRef] [Green Version]
- Rachdi, L.; Kariyawasam, D.; Aiello, V.; Herault, Y.; Janel, N.; Delabar, J.M.; Polak, M.; Scharfmann, R. Dyrk1A induces pancreatic beta cell mass expansion and improves glucose tolerance. Cell Cycle 2014, 13, 2221–2229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de la Torre, R.; de Sola, S.; Hernandez, G.; Farré, M.; Pujol, J.; Rodriguez, J.; Espadaler, J.M.; Langohr, K.; Cuenca-Royo, A.; Principe, A.; et al. Safety and efficacy of cognitive training plus epigallocatechin-3-gallate in young adults with Down’s syndrome (TESDAD): A double-blind, randomised, placebo-controlled, phase 2 trial. Lancet Neurol. 2016, 15, 801–810. [Google Scholar] [CrossRef]
- Shen, W.; Taylor, B.; Jin, Q.; Nguyen-Tran, V.; Meeusen, S.; Zhang, Y.Q.; Kamireddy, A.; Swafford, A.; Powers, A.F.; Walker, J.; et al. Inhibition of DYRK1A and GSK3B induces human beta-cell proliferation. Nat. Commun. 2015, 6, 8372. [Google Scholar] [CrossRef] [PubMed]
- Stringer, M.; Goodlett, C.R.; Roper, R.J. Targeting trisomic treatments: Optimizing Dyrk1a inhibition to improve Down syndrome deficits. Mol. Genet. Genomic. Med. 2017, 5, 451–465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tahtouh, T.; Elkins, J.M.; Filippakopoulos, P.; Soundararajan, M.; Burgy, G.; Durieu, E.; Cochet, C.; Schmid, R.S.; Lo, D.C.; Delhommel, F.; et al. Selectivity, cocrystal structures, and neuroprotective properties of leucettines, a family of protein kinase inhibitors derived from the marine sponge alkaloid leucettamine B. J. Med. Chem. 2012, 55, 9312–9330. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, M.; Estivill, X.; de la Luna, S. DYRK1A accumulates in splicing speckles through a novel targeting signal and induces speckle disassembly. J. Cell Sci. 2003, 116, 3099–3107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, H.; Wang, K.; Chen, S.; Sun, Q.; Zhang, Y.; Chen, L.; Sun, X. NFATc1 phosphorylation by DYRK1A increases its protein stability. PLoS ONE 2017, 12, e0172985. [Google Scholar] [CrossRef] [Green Version]
- Svendsen, J.S.; Stensen, W.; Porter, R.A. Benzothiazole Derivatives as DYRK1 Inhibitors. WO 2018/069468 A1, 23 February 2021. [Google Scholar]
- Alexeeva, M.; Aberg, E.; Engh, R.A.; Rothweiler, U. The structure of a dual-specificity tyrosine phosphorylation-regulated kinase 1A-PKC412 complex reveals disulfide-bridge formation with the anomalous catalytic loop HRD(HCD) cysteine. Acta Crystallogr. Sect. D Biol. Crystallogr. 2015, 71, 1207–1215. [Google Scholar] [CrossRef]
- Krug, M.; Weiss, M.S.; Heinemann, U.; Mueller, U. XDSAPP: A graphical user interface for the convenient processing of diffraction data using XDS. J. Appl. Crystallogr. 2012, 45, 568–572. [Google Scholar] [CrossRef]
- Kabsch, W. XDS. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010, 66, 125–132. [Google Scholar] [CrossRef] [Green Version]
- McCoy, A.J.; Grosse-Kunstleve, R.W.; Adams, P.D.; Winn, M.D.; Storoni, L.C.; Read, R.J. Phaser crystallographic software. J. Appl. Crystallogr. 2007, 40, 658–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adams, P.D.; Afonine, P.V.; Bunkóczi, G.; Chen, V.B.; Davis, I.W.; Echols, N.; Headd, J.J.; Hung, L.W.; Kapral, G.J.; Grosse-Kunstleve, R.W.; et al. PHENIX: A comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010, 66, 213–221. [Google Scholar] [CrossRef] [Green Version]
- Winn, M.D.; Ballard, C.C.; Cowtan, K.D.; Dodson, E.J.; Emsley, P.; Evans, P.R.; Keegan, R.M.; Krissinel, E.B.; Leslie, A.G.; McCoy, A.; et al. Overview of the CCP4 suite and current developments. Acta Crystallogr. Sect. D Biol. Crystallogr. 2011, 67, 235–242. [Google Scholar] [CrossRef] [Green Version]
- Murshudov, G.N.; Skubák, P.; Lebedev, A.A.; Pannu, N.S.; Steiner, R.A.; Nicholls, R.A.; Winn, M.D.; Long, F.; Vagin, A.A. REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr. Sect. D Biol. Crystallogr. 2011, 67, 355–367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schüttelkopf, A.W.; van Aalten, D.M. PRODRG: A tool for high-throughput crystallography of protein-ligand complexes. Acta Crystallogr. Sect. D Biol. Crystallogr. 2004, 60, 1355–1363. [Google Scholar] [CrossRef] [Green Version]
- Hastie, C.J.; McLauchlan, H.J.; Cohen, P. Assay of protein kinases using radiolabeled ATP: A protocol. Nat. Protoc. 2006, 1, 968–971. [Google Scholar] [CrossRef] [PubMed]
- Bain, J.; Plater, L.; Elliott, M.; Shapiro, N.; Hastie, C.J.; McLauchlan, H.; Klevernic, I.; Arthur, J.S.; Alessi, D.R.; Cohen, P. The selectivity of protein kinase inhibitors: A further update. Biochem. J. 2007, 408, 297–315. [Google Scholar] [CrossRef] [Green Version]
- Czarna, A.; Wang, J.; Zelencova, D.; Liu, Y.; Deng, X.; Choi, H.G.; Zhang, T.; Zhou, W.; Chang, J.W.; Kildalsen, H.; et al. Novel Scaffolds for Dual Specificity Tyrosine-Phosphorylation-Regulated Kinase (DYRK1A) Inhibitors. J. Med. Chem. 2018, 61, 7560–7572. [Google Scholar] [CrossRef] [Green Version]
- Victorino, D.B.; Bederman, I.R.; Costa, A.C.S. Pharmacokinetic Properties of Memantine after a Single Intraperitoneal Administration and Multiple Oral Doses in Euploid Mice and in the Ts65Dn Mouse Model of Down’s Syndrome. Basic Clin. Pharmacol. Toxicol. 2017, 121, 382–389. [Google Scholar] [CrossRef] [Green Version]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Kinase | % Remaining Activity | |
|---|---|---|
| 100 μM | 1 μM | |
| DYRK1A | 3 | 8 |
| DYRK2 | 4 | 21 |
| DYRK3 | 5 | 20 |
| Aurora A | 136 | 133 |
| Aurora B | 24 | 106 |
| GSK3β | 100 | 107 |
| ERK8 | 28 | 89 |
| CLK2 | 5 | 22 |
| PK Parameter | Administration Route and Measured Tissue | ||||
|---|---|---|---|---|---|
| IV (Plasma) (Rat) | IV Plasma (Mouse) | IV Brain (Mouse) | PO Plasma (Mouse) | PO Brain (Mouse) | |
| PST-001 dose | 1 mg/kg | 1 mg/kg | 1 mg/kg | 5 mg/kg | 5 mg/kg |
| t½ (h) | 0.72 | 0.15 | 0.15 | NQ | 0.27 |
| Tmax (h) | 0.03 | 0.08 | 0.08 | 1.00 | 0.25 |
| Cmax (ng/mL) | 647 | 362 | 867 | 76 | 153 |
| AUCall (h ∗ ng/mL) | 158 | 84 | 200 | 89 | 189 |
| CL (mL/h/kg) | 6281 | 11865 | 4989 | ||
| Vd (mL/kg) | 2387 | 1388 | 553 | ||
| f | 21.2% | ||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stensen, W.; Rothweiler, U.; Engh, R.A.; Stasko, M.R.; Bederman, I.; Costa, A.C.S.; Fugelli, A.; Svendsen, J.S.M. Novel DYRK1A Inhibitor Rescues Learning and Memory Deficits in a Mouse Model of Down Syndrome. Pharmaceuticals 2021, 14, 1170. https://doi.org/10.3390/ph14111170
Stensen W, Rothweiler U, Engh RA, Stasko MR, Bederman I, Costa ACS, Fugelli A, Svendsen JSM. Novel DYRK1A Inhibitor Rescues Learning and Memory Deficits in a Mouse Model of Down Syndrome. Pharmaceuticals. 2021; 14(11):1170. https://doi.org/10.3390/ph14111170
Chicago/Turabian StyleStensen, Wenche, Ulli Rothweiler, Richard Alan Engh, Melissa R. Stasko, Ilya Bederman, Alberto C. S. Costa, Anders Fugelli, and John S. Mjøen Svendsen. 2021. "Novel DYRK1A Inhibitor Rescues Learning and Memory Deficits in a Mouse Model of Down Syndrome" Pharmaceuticals 14, no. 11: 1170. https://doi.org/10.3390/ph14111170
APA StyleStensen, W., Rothweiler, U., Engh, R. A., Stasko, M. R., Bederman, I., Costa, A. C. S., Fugelli, A., & Svendsen, J. S. M. (2021). Novel DYRK1A Inhibitor Rescues Learning and Memory Deficits in a Mouse Model of Down Syndrome. Pharmaceuticals, 14(11), 1170. https://doi.org/10.3390/ph14111170