
Investigation of Thiocarbamates as Potential Inhibitors of the SARS-CoV-2 Mpro

 , , , ,
, , , ,  , and
, and 
{kind=link}
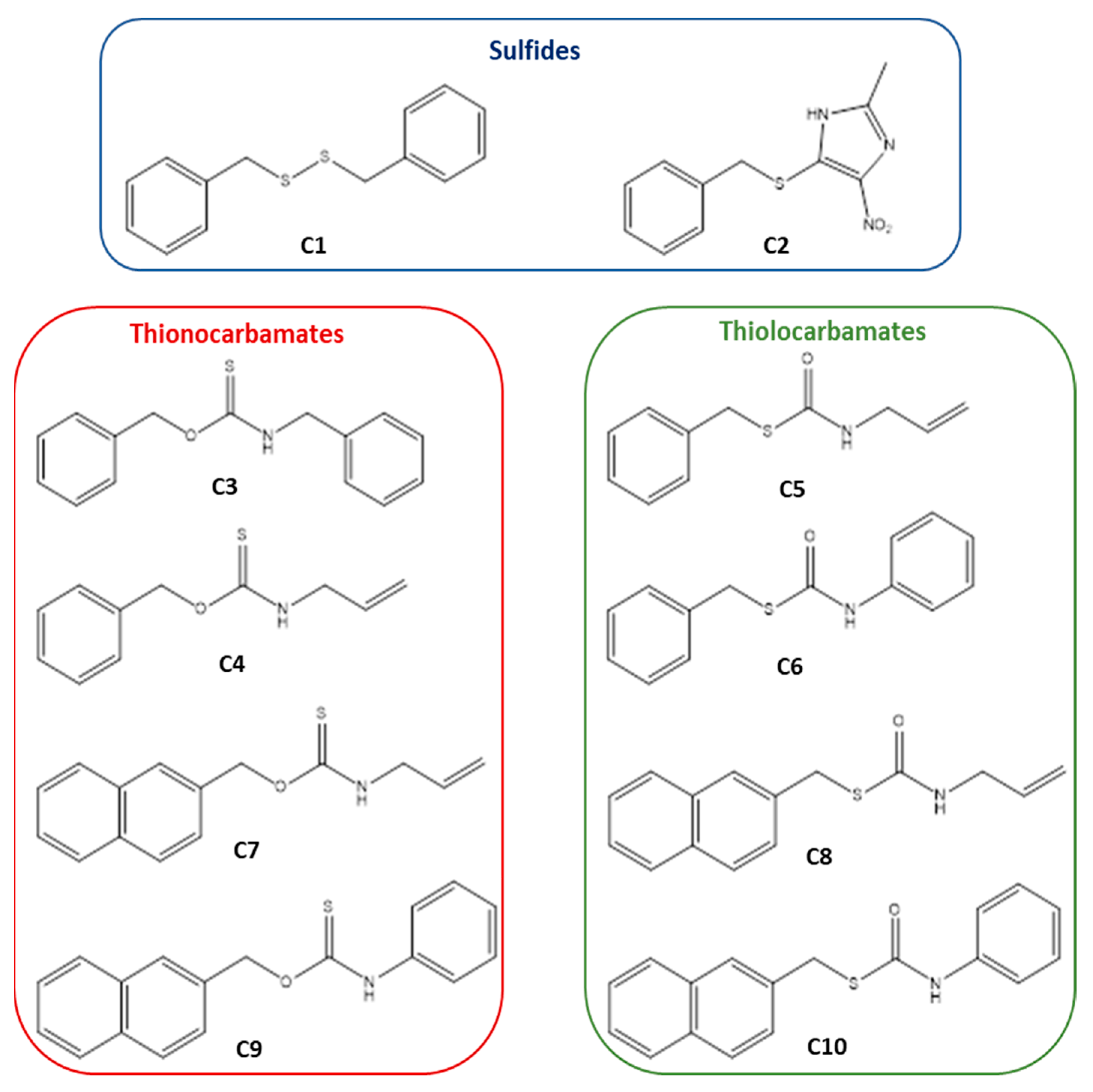{kind=link}
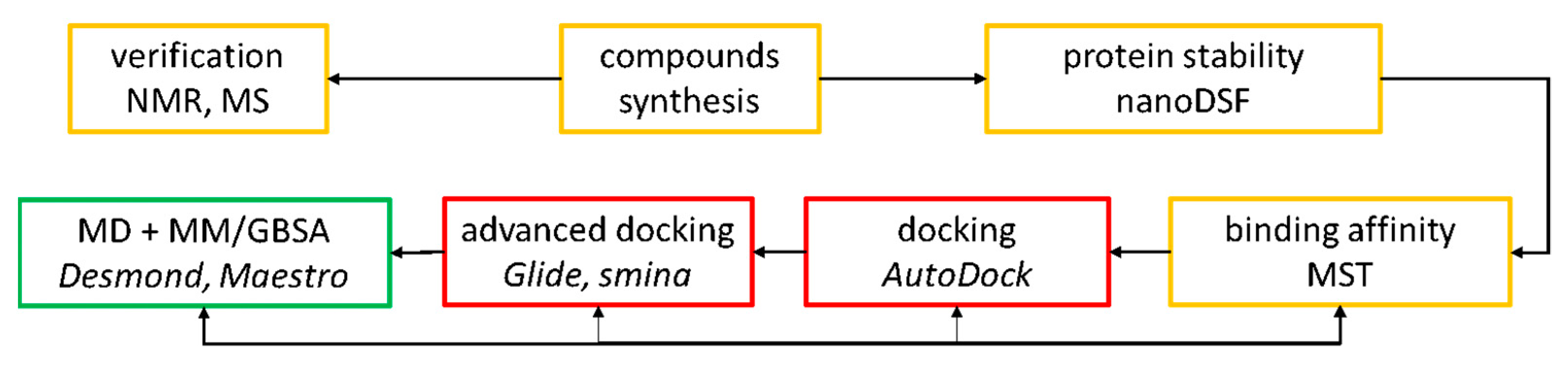{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. Thermal Stability Analysis
2.2. The Binding Affinity of the Compounds to the SARS-CoV-2 Mpro Protein
2.3. Molecular Docking of Sulfur-Containing Compounds
3. Discussion
4. Materials and Methods
4.1. Protein Preparation
4.2. Compounds Preparation
4.3. Thermal Stability Analysis
4.3.1. The Influence of DMSO Concentration on the SARS-CoV-2 Mpro Protein Stability
4.3.2. Thermal Stability of the SARS-CoV-2 Mpro with the Examined Compounds
4.4. The Binding Affinity of the Compounds to the SARS-CoV-2 Mpro Protein
4.4.1. The Intrinsic Fluorescence of the Compounds
4.4.2. Binding Affinity Measurement-MST Experiment
4.5. Molecular Docking of Sulfur-Containing Compounds
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Liu, X.H.; Zhang, X.; Lu, Z.H.; Zhu, Y.S.; Wang, T. Potential molecular targets of nonstructural proteins for the development of antiviral drugs against SARS-CoV-2 infection. Biomed. Pharmacother. 2021, 133, 111035. [Google Scholar] [CrossRef] [PubMed]
- Bzówka, M.; Mitusińska, K.; Raczyńska, A.; Samol, A.; Tuszyński, J.A.; Góra, A. Structural and evolutionary analysis indicate that the SARS-CoV-2 Mpro is a challenging target for small-molecule inhibitor design. Int. J. Mol. Sci. 2020, 21, 3099. [Google Scholar] [CrossRef] [PubMed]
- Alves, V.M.; Bobrowski, T.; Melo-Filho, C.C.; Korn, D.; Auerbach, S.; Schmitt, C.; Muratov, E.N.; Tropsha, A. QSAR modeling of SARS-CoV Mpro inhibitors identifies Sufugolix, Cenicriviroc, Proglumetacin, and other drugs as candidates for repurposing against SARS-CoV-2. Mol. Inform. 2021, 40, 2000113. [Google Scholar] [CrossRef]
- Li, Y.; Xie, Z.; Lin, W.; Cai, W.; Wen, C.; Guan, Y.; Mo, X.; Wang, J.; Wang, Y.; Peng, P.; et al. Efficacy and safety of Lopinavir/Ritonavir or Arbidol in adult patients with mild/moderate COVID-19: An exploratory randomized controlled trial. Med 2020, 1, 105–113. [Google Scholar] [CrossRef] [PubMed]
- Gasmi, M.A.; Peana, M.; Noor, S.; Lysiuk, R.; Menzel, A.; Gasmi Benahmed, A.; Bjørklund, G. Chloroquine and hydroxychloroquine in the treatment of COVID-19: The never-ending story. Appl. Microbiol. Biotechnol. 2021, 105, 1333–1343. [Google Scholar] [CrossRef]
- Wadaa-Allah, A.; Emhamed, M.S.; Sadeq, M.A.; Ben Hadj Dahman, N.; Ullah, I.; Farrag, N.S.; Negida, A. Efficacy of the current investigational drugs for the treatment of COVID-19: A scoping review. Ann. Med. 2021, 53, 318–334. [Google Scholar] [CrossRef] [PubMed]
- World Health Organisation. Novel Coronavirus (2019-nCoV) Situation Reports; “Solidarity” Clinical Trial for COVID-19 Treatments. Geneva, Switzerland, 2020. Available online: https://www.who.int/docs/default-source/coronaviruse/situation-reports/20200131-sitrep-11-ncov.pdf?sfvrsn=de7c0f7_4 (accessed on 22 May 2021).
- Jiménez-Alberto, A.; Ribas-Aparicio, R.M.; Aparicio-Ozores, G.; Castelán-Vega, J.A. Virtual screening of approved drugs as potential SARS-CoV-2 main protease inhibitors. Comput. Biol. Chem. 2020, 88, 107325. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Li, X.; Huang, Y.Y.; Wu, Y.; Liu, R.; Zhou, L.; Lin, Y.; Wu, D.; Zhang, L.; Liu, H.; et al. Identify potent SARS-CoV-2 main protease inhibitors via accelerated free energy perturbation-based virtual screening of existing drugs. Proc. Natl. Acad. Sci. USA 2020, 117, 27381–27387. [Google Scholar] [CrossRef]
- Fischer, A.; Sellner, M.; Neranjan, S.; Smieško, M.; Lill, M.A. Potential Inhibitors for Novel Coronavirus Protease Identified by Virtual Screening of 606 Million Compounds. Int. J. Mol. Sci. 2020, 21, 3626. [Google Scholar] [CrossRef] [PubMed]
- Gorgulla, C.; Padmanabha Das, K.M.; Leigh, K.E.; Cespugli, M.; Fischer, P.D.; Wang, Z.F.; Tesseyre, G.; Pandita, S.; Shnapir, A.; Calderaio, A.; et al. A multi-pronged approach targeting SARS-CoV-2 proteins using ultra-large virtual screening. iScience 2021, 24, 102021. [Google Scholar] [CrossRef]
- Teli, D.M.; Shah, M.B.; Chhabria, M.T. In silico screening of natural compounds as potential Iihibitors of SARS-CoV-2 main protease and spike RBD: Targets for COVID-19. Front. Mol. Biosci. 2021, 7, 599079. [Google Scholar] [CrossRef] [PubMed]
- Majumder, R.; Mandal, M. Screening of plant-based natural compounds as a potential COVID-19 main protease inhibitor: An in silico docking and molecular dynamics simulation approach. J. Biomol. Struct. Dyn. 2020, 1–16. [Google Scholar] [CrossRef]
- Mazzini, S.; Musso, L.; Dallavalle, S.; Artali, R. Putative SARS-CoV-2 Mpro inhibitors from an in-house library of natural and natureinspired products: A virtual screening and molecular docking study. Molecules 2020, 25, 3745. [Google Scholar] [CrossRef] [PubMed]
- Joshi, T.; Joshi, T.; Sharma, P.; Mathpal, S.; Pundir, H.; Bhatt, V.; Chandra, S. In silico screening of natural compounds against COVID-19 by targeting Mpro and ACE2 using molecular docking. Eur. Rev. Med. Pharmacol. Sci. 2020, 24, 4529–4536. [Google Scholar] [PubMed]
- Wang, L.; Bao, B.B.; Song, G.Q.; Chen, C.; Zhang, X.M.; Lu, W.; Wang, Z.; Cai, Y.; Li, S.; Fu, S.; et al. Discovery of unsymmetrical aromatic disulfides as novel inhibitors of SARS-CoV main protease: Chemical synthesis, biological evaluation, molecular docking and 3D-QSAR study. Eur. J. Med. Chem. 2017, 137, 450–461. [Google Scholar] [CrossRef]
- Günther, S.; Reinke, P.Y.A.; Fernández-García, Y.; Lieske1, J.; Lane1, T.J.; Ginn, H.M.; Koua1, F.H.M.; Ehrt, C.; Ewert, W.; Oberthuer, D.; et al. X-ray screening identifies active site and allosteric inhibitors of SARS-CoV-2 main protease. Science 2021, 372, 642–646. [Google Scholar] [CrossRef]
- Guo, H.; Sun, B.; Gao, H.; Chen, X.; Liu, S.; Yao, X.; Liu, X.; Che, Y. Diketopiperazines from the Cordyceps-colonizing fungus Epicoccum nigrum. J. Nat. Prod. 2009, 72, 2115–2119. [Google Scholar] [CrossRef] [PubMed]
- Choudhury, C. Fragment tailoring strategy to design novel chemical entities as potential binders of novel corona virus main protease. J. Biomol. Struct. Dyn. 2020, 39, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Yang, J. A review of the latest research on Mpro targeting SARS-COV inhibitors. RSC Med. Chem. 2021, 12, 1026–1036. [Google Scholar] [CrossRef] [PubMed]
- ClinicalTrials.gov. Study of PF-07321332 in Helthy Participants. 24 May 2021. Available online: https://clinicaltrials.gov/ct2/show/study/NCT04756531 (accessed on 1 May 2021).
- Halford, B. Pfizer unveils its oral CARS-CoV-2 inhibitor. Chem. Eng. News 2021, 99, 7. [Google Scholar] [CrossRef]
- Guo, S.; Xie, H.; Lei, Y.; Liu, B.; Zhang, L.; Xub, Y.; Zuo, Z. Discovery of novel inhibitors against main protease (Mpro) of SARS-CoV-2 via virtual screening and biochemical evaluation. Bioorg. Chem. 2021, 110, 104767. [Google Scholar] [CrossRef]
- Wang, J.M.; Ding, G.Z.; Fang, L.; Dai, J.G.; Yu, S.S.; Wang, Y.H.; Chen, X.G.; Ma, S.G.; Jing, Q.; Xu, S.; et al. Thiodiketopiperazines Produced by the Endophytic Fungus Epicoccum nigrum. J. Nat. Prod. 2010, 73, 1240–1249. [Google Scholar] [CrossRef]
- Gil, C.; Ginex, T.; Maestro, I.; Nozal, V.; Barrado-Gil, L.; Cuesta-Geijo, M.A.; Urquiza, J.; Ramírez, D.; Alonso, C.; Campillo, N.E.; et al. COVID-19: Drug Targets and Potential Treatments. J. Med. Chem. 2020, 63, 12359–12386. [Google Scholar] [CrossRef]
- Ma, C.; Wang, J. Dipyridamole, chloroquine, montelukast sodium, candesartan, oxytetracycline, and atazanavir are not SARS-CoV-2 main protease inhibitors. Proc. Natl. Acad. Sci. USA 2021, 118, e2024420118. [Google Scholar] [CrossRef] [PubMed]
- Jin, Z.; Du, X.; Xu, Y.; Deng, Y.; Liu, M.; Zhao, Y.; Zhang, B.; Li, X.; Zhang, L.; Peng, C.; et al. Structure of Mpro from SARS-CoV-2 and discovery of its inhibitors. Nature 2020, 582, 289–293. [Google Scholar] [CrossRef] [PubMed]
- Mahdi, M.; Mótyán, J.A.; Szojka, Z.I.; Golda, M.; Miczi, M.; Tőzsér, J. Analysis of the efficacy of HIV protease inhibitors against SARS-CoV-2’s main protease. Virol. J. 2020, 17, 190. [Google Scholar] [CrossRef]
- Abuo-Rahma, G.E.-D.A.; Mohamed, M.F.A.; Ibrahim, T.S.; Shoman, M.E.; Samir, E.; Abd El-Baky, R.M. Potential repurposed SARS-CoV-2 (COVID-19) infection drugs. RSC Adv. 2020, 10, 26895–26916. [Google Scholar] [CrossRef]
- Amaro, R.E.; Baudry, J.; Chodera, J.; Demir, O.; McCammon, J.A.; Miao, Y.; Smith, J.C. Ensemble Docking in Drug Discovery. Biophys. J. 2018, 114, 2271–2278. [Google Scholar] [CrossRef] [PubMed]
- Kenny, P.W. The nature of ligand efficiency. J. Cheminform. 2019, 11, 8. [Google Scholar] [CrossRef]
- Li, J.; Abel, R.; Zhu, K.; Cao, Y.; Zhao, S.; Friesner, R.A. The VSGB 2.0 model: A next generation energy model for high resolution protein structure modeling. Proteins Struct. Funct. Bioinform. 2011, 79, 2794–2812. [Google Scholar] [CrossRef] [PubMed]
- Bissantz, C.; Kuhn, B.; Stahl, M. A medicinal chemist’s guide to molecular interacions. J. Med. Chem. 2010, 53, 5061–5084. [Google Scholar] [CrossRef] [PubMed]
- Gao, C.; Park, M.S.; Stern, H.A. Accounting for Ligand Conformational Restriction in Calculations of Protein-Ligand Binding Affinities. Biophys. J. 2010, 98, 901–910. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Choudhuri, K.; Pramanik, M.; Mal, P. Direct C–S Bond Functionalization of Benzyl Mercaptan. Eur. J. Org. Chem. 2020, 25, 3906–3913. [Google Scholar] [CrossRef]
- Kasprzycka, A.; Ptaszek-Budniok, A.; Szeja, W. Simple and efficient method for the protection of hydroxyl groups as 4-methoxybenzyl ethers. Synth. Commun. 2014, 44, 2276–2284. [Google Scholar] [CrossRef]
- Komor, R.; Kasprzycka, A.; Pastuch-Gawołek, G.; Szeja, W. Simple synthesis of glycosylthiols and thioglycosides by rearrangement of O-glycosyl thionocarbamates. Carbohydr. Res. 2014, 396, 37–42. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef]
- Allouche, A.R. Gabedit—A graphical user interface for computational chemistry softwares. J. Comput. Chem. 2011, 32, 174–182. [Google Scholar] [CrossRef] [PubMed]
- Anandakrishnan, R.; Aguilar, B.; Onufriev, A.V. H++ 3.0: Automating pK prediction and the preparation of biomolecular structures for atomistic molecular modeling and simulations. Nucleic Acids Res. 2012, 40, 537–541. [Google Scholar] [CrossRef]
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open Babel: An open chemical toolbox. J. Cheminform. 2011, 3, 1–14. [Google Scholar] [CrossRef]
- Halgren, T.A.; Murphy, R.B.; Friesner, R.A.; Beard, H.S.; Frye, L.L.; Pollard, W.T.; Banks, J.L. Glide: A new approach for rapid, accurate docking and scoring. 2. Enrichment factors in database screening. J. Med. Chem. 2004, 47, 1750–1759. [Google Scholar] [CrossRef]
- Koes, D.R.; Baumgartner, M.P.; Camacho, C.J. Lessons learned in empirical scoring with smina from the CSAR 2011 benchmarking exercise. J. Chem. Inf. Model. 2013, 53, 1893–1904. [Google Scholar] [CrossRef] [PubMed]
- Sastry, G.M.; Adzhigirey, M.; Day, T.; Annabhimoju, R.; Sherman, W. Protein and ligand preparation: Parameters, protocols, and influence on virtual screening enrichments. J. Comput.-Aided Mol. Des. 2013, 27, 221–234. [Google Scholar] [CrossRef] [PubMed]
- Schrödinger Release 2019-3: Maestro; Schrödinger, LLC: New York, NY, USA, 2019.
- Schrödinger Release 2019-3: LigPrep; Schrödinger, LLC: New York, NY, USA, 2019.
- Bowers, K.J.; Chow, E.; Xu, H.; Dror, R.O.; Eastwood, M.P.; Gregersen, B.A.; Klepeis, J.L.; Kolossvary, I.; Moraes, M.A.; Sacerdoti, F.D.; et al. Scalable Algorithms for Molecular Dynamics Simulations on Commodity Clusters. In Proceedings of the 2006 ACM/IEEE Conference on Supercomputing, Tampa, FL, USA, 11–17 November 2006; pp. 43–56. [Google Scholar]
- Shaw, D.E.; Grossman, J.P.; Bank, J.A.; Batson, B.; Butts, J.A.; Chao, J.C.; Deneroff, M.M.; Dror, R.O.; Even, A.; Fenton, C.H.; et al. Anton 2: Raising the Bar for Performance and Programmability in a Special-Purpose Molecular Dynamics Supercomputer. In Proceedings of the International Conference for High Performance Computing, Networking, Storage and Analysis, New Orleans, LA, USA, 16–21 November 2014; pp. 41–53. [Google Scholar]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Papaj, K.; Spychalska, P.; Hopko, K.; Kapica, P.; Fisher, A.; Lill, M.A.; Bagrowska, W.; Nowak, J.; Szleper, K.; Smieško, M.; et al. Investigation of Thiocarbamates as Potential Inhibitors of the SARS-CoV-2 Mpro. Pharmaceuticals 2021, 14, 1153. https://doi.org/10.3390/ph14111153
Papaj K, Spychalska P, Hopko K, Kapica P, Fisher A, Lill MA, Bagrowska W, Nowak J, Szleper K, Smieško M, et al. Investigation of Thiocarbamates as Potential Inhibitors of the SARS-CoV-2 Mpro. Pharmaceuticals. 2021; 14(11):1153. https://doi.org/10.3390/ph14111153
Chicago/Turabian StylePapaj, Katarzyna, Patrycja Spychalska, Katarzyna Hopko, Patryk Kapica, Andre Fisher, Markus A. Lill, Weronika Bagrowska, Jakub Nowak, Katarzyna Szleper, Martin Smieško, and et al. 2021. "Investigation of Thiocarbamates as Potential Inhibitors of the SARS-CoV-2 Mpro" Pharmaceuticals 14, no. 11: 1153. https://doi.org/10.3390/ph14111153
APA StylePapaj, K., Spychalska, P., Hopko, K., Kapica, P., Fisher, A., Lill, M. A., Bagrowska, W., Nowak, J., Szleper, K., Smieško, M., Kasprzycka, A., & Góra, A. (2021). Investigation of Thiocarbamates as Potential Inhibitors of the SARS-CoV-2 Mpro. Pharmaceuticals, 14(11), 1153. https://doi.org/10.3390/ph14111153