
Late Na+ Current Is [Ca2+]i-Dependent in Canine Ventricular Myocytes

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
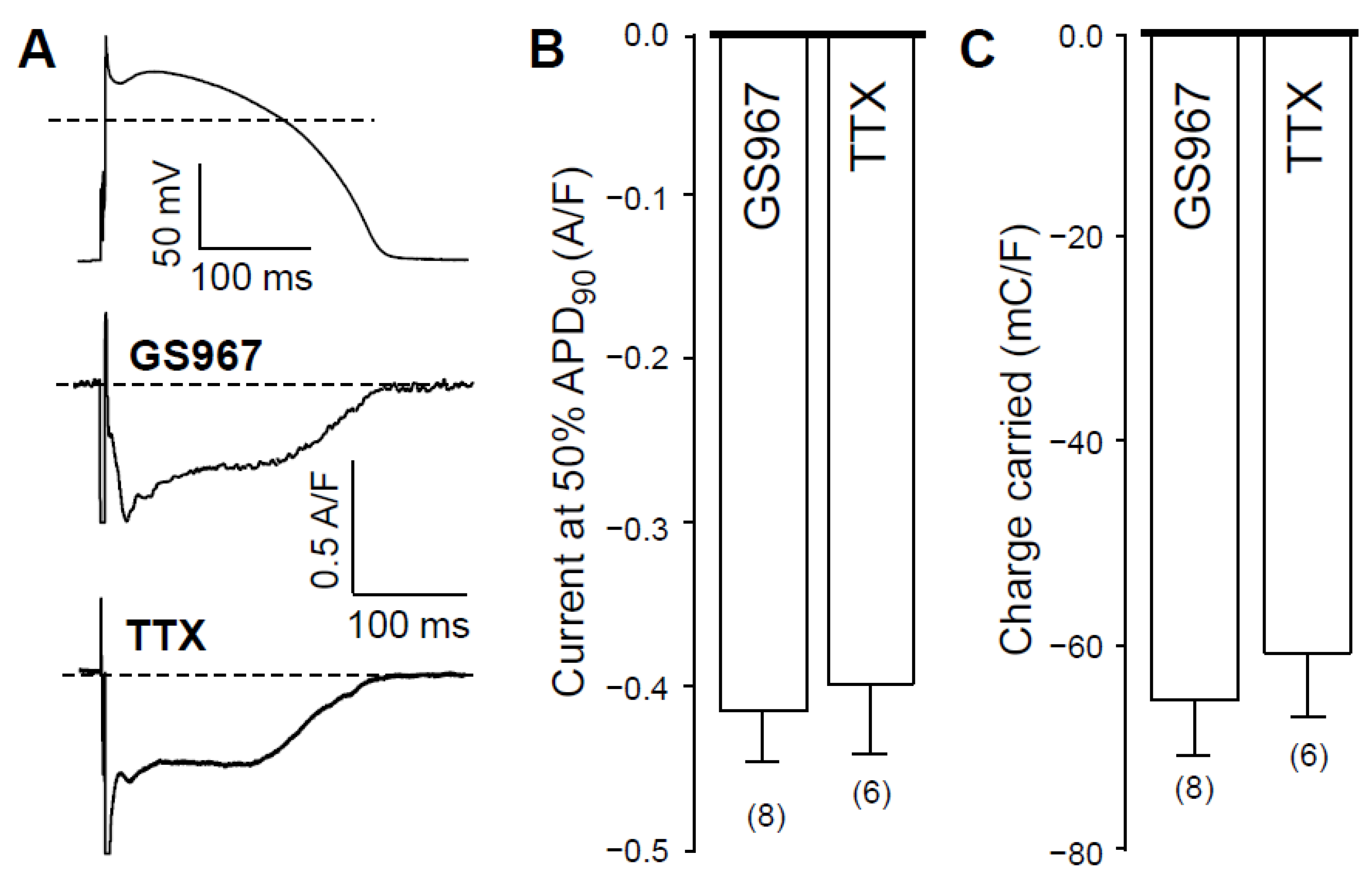
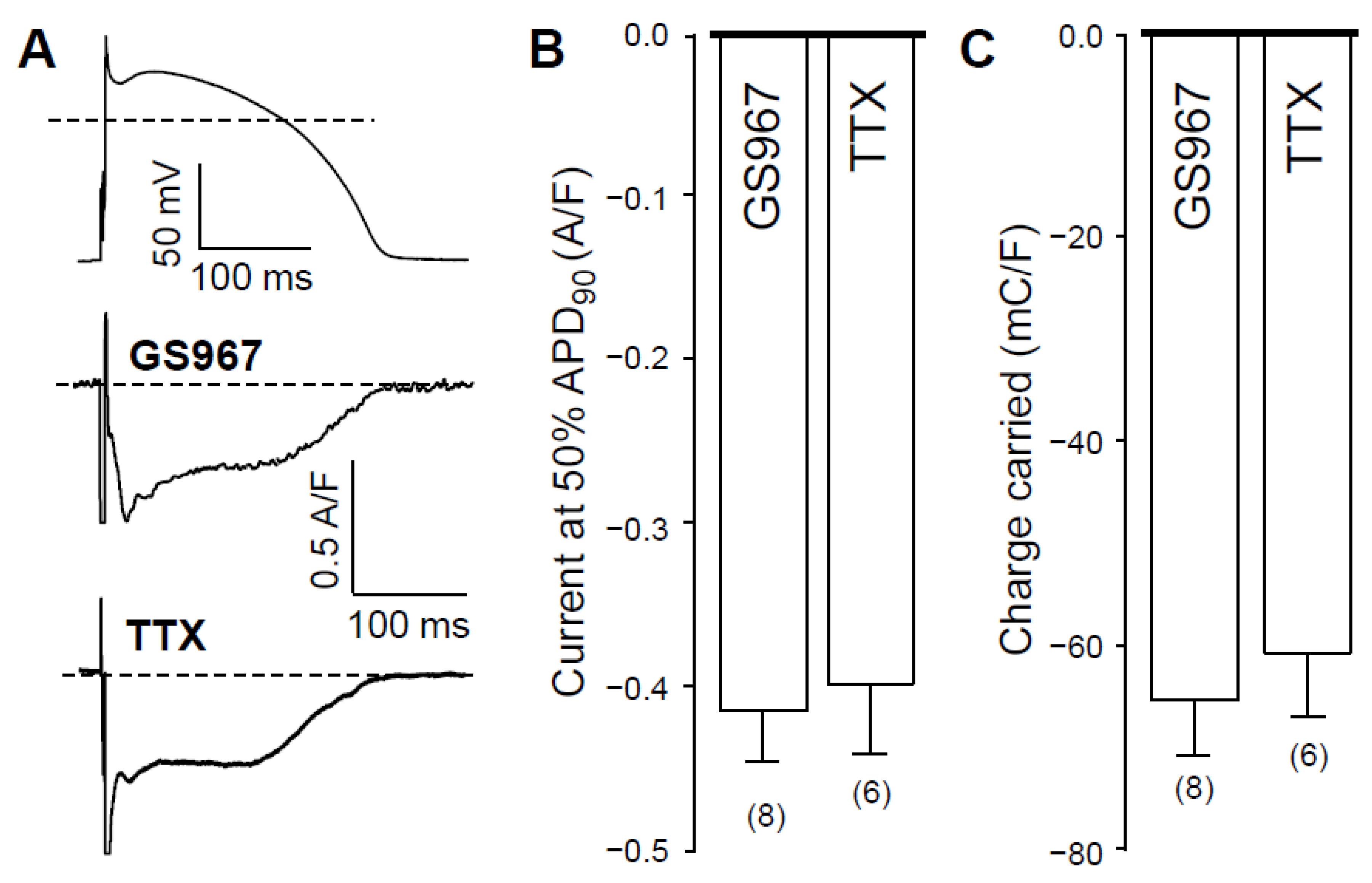
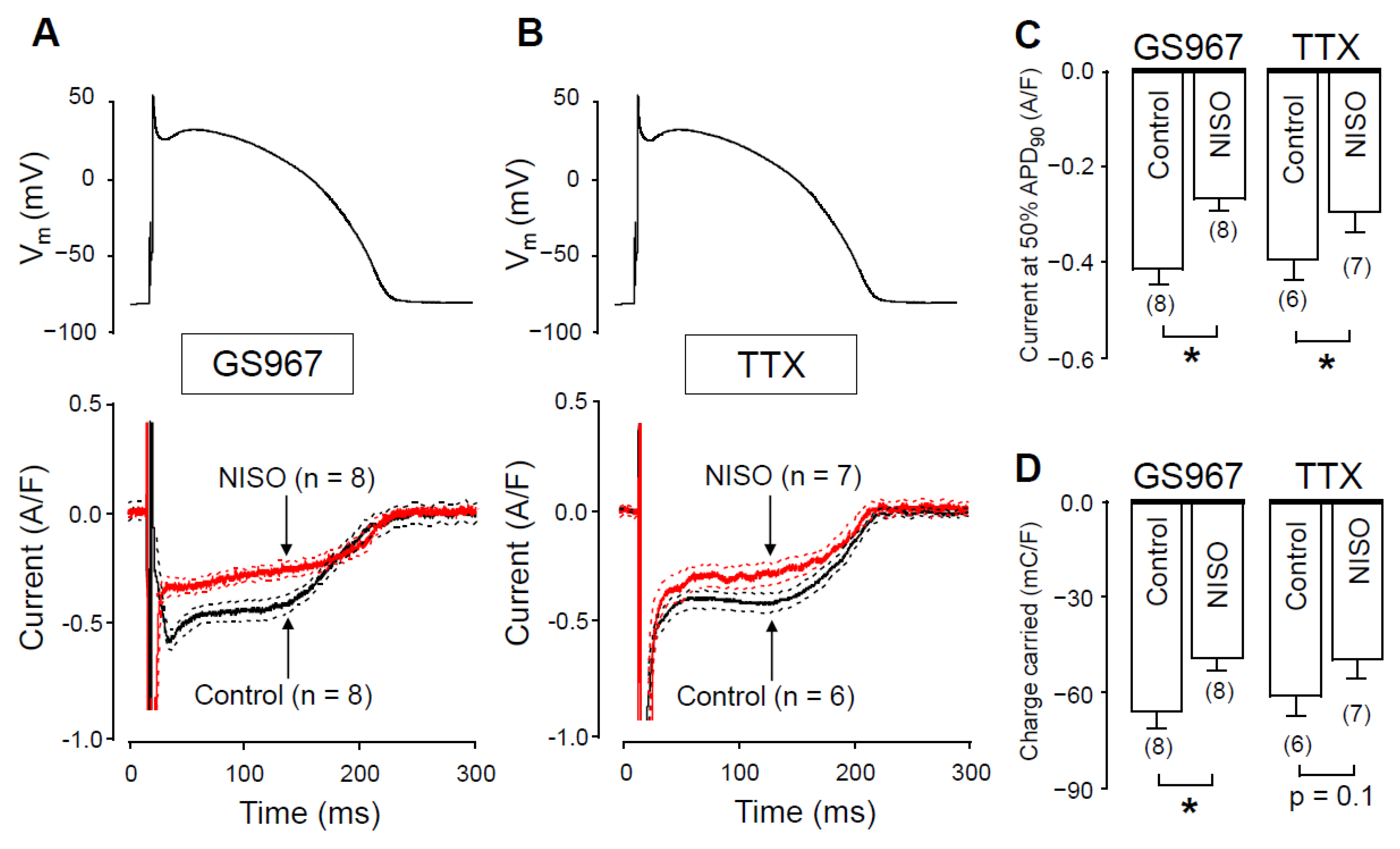
2.1. Effects of GS967 and TTX on INaL under Action Potential Voltage Clamp Conditions
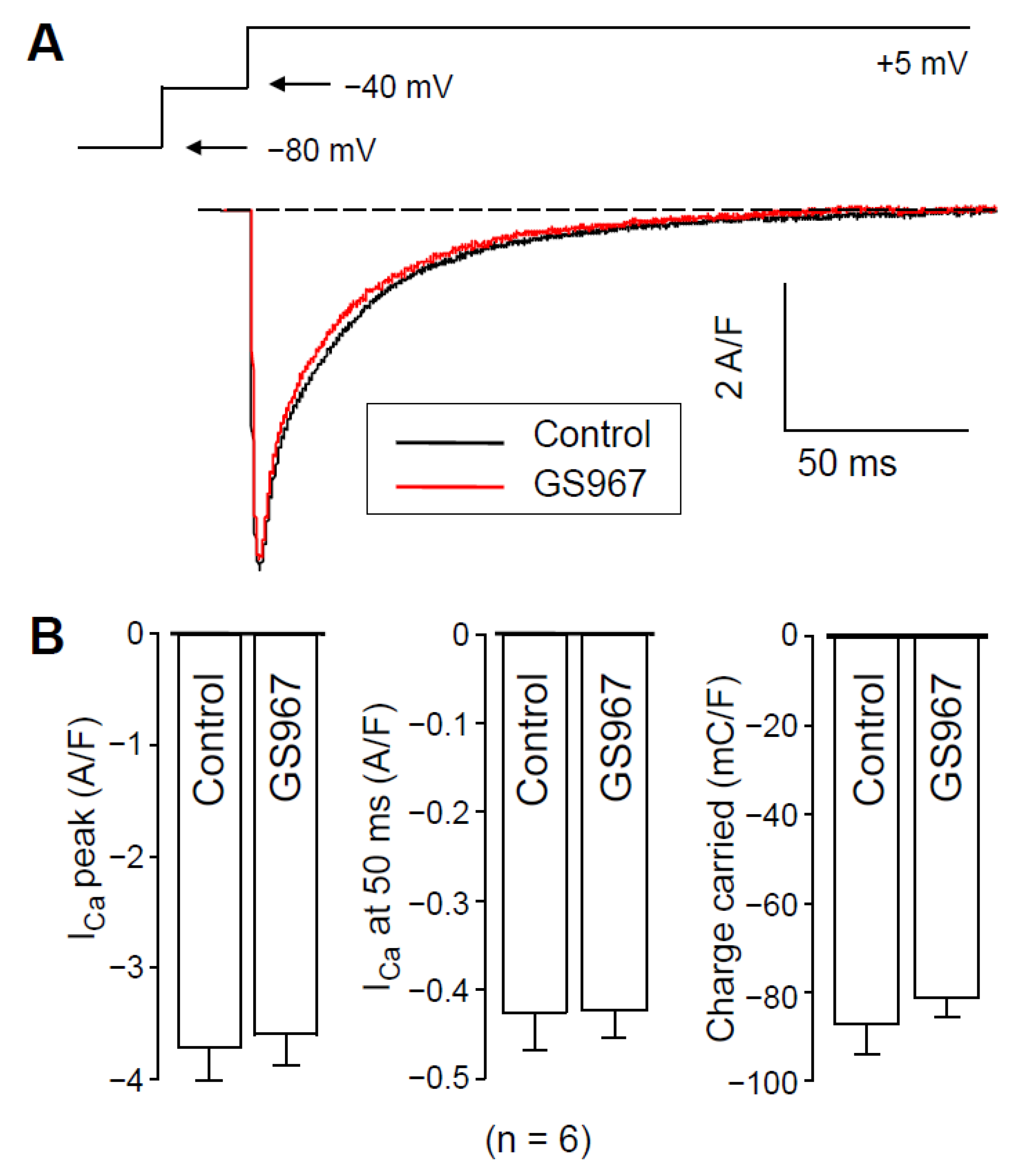
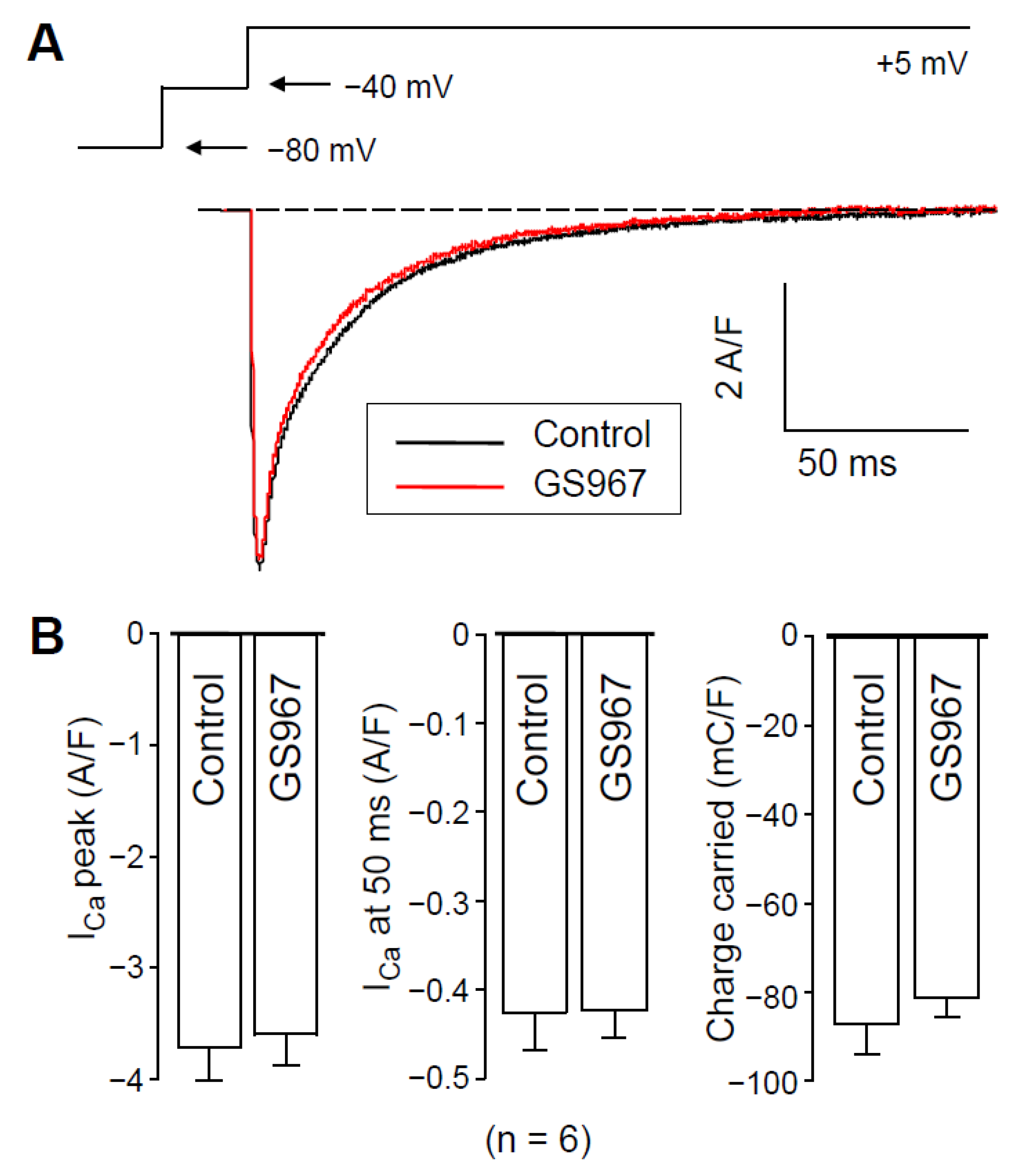
2.2. Effect of GS967 on ICa under Conventional Voltage Clamp Conditions
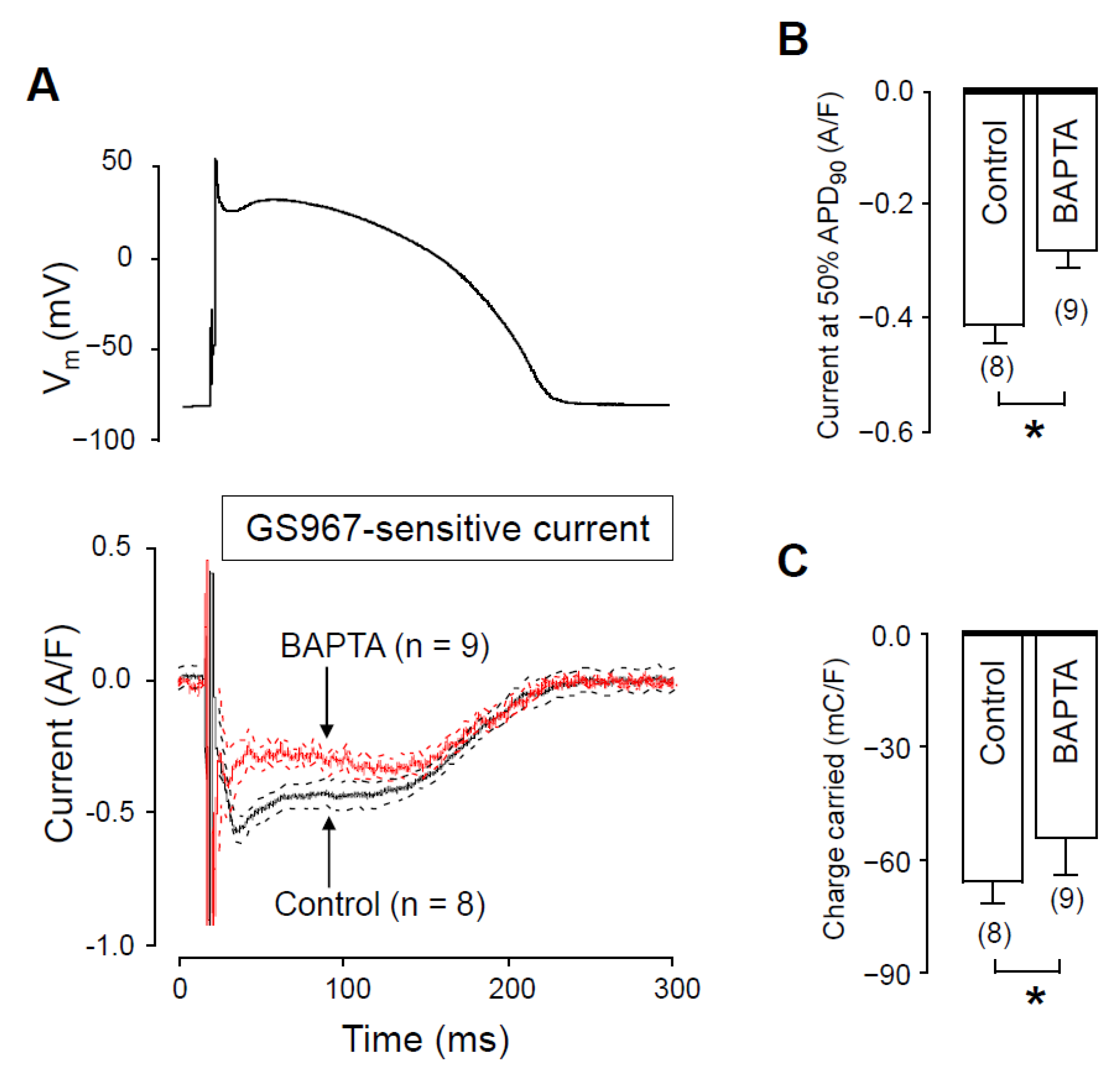
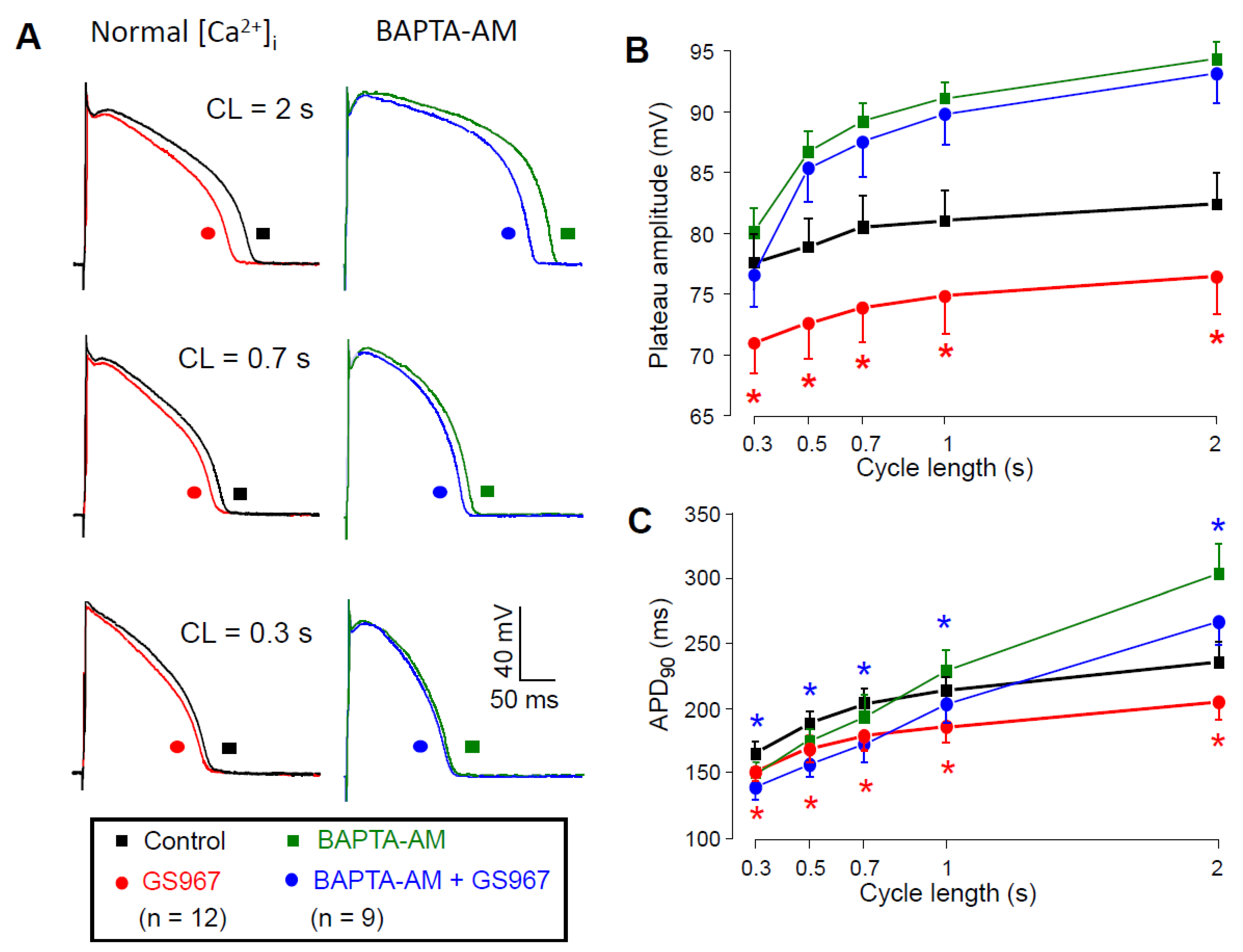
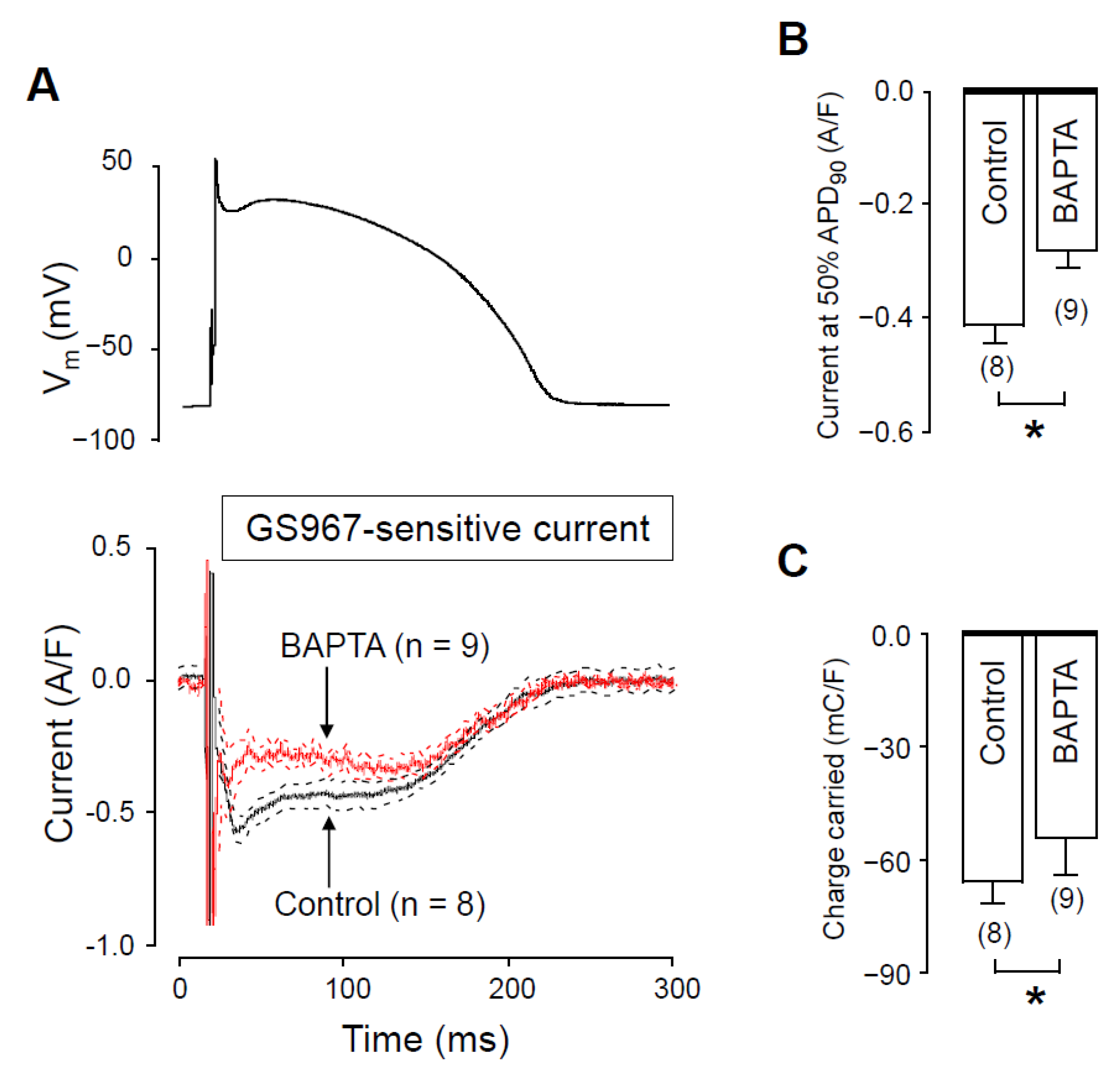
2.3. Effects of GS967 on INaL in the Presence of Intracellular BAPTA
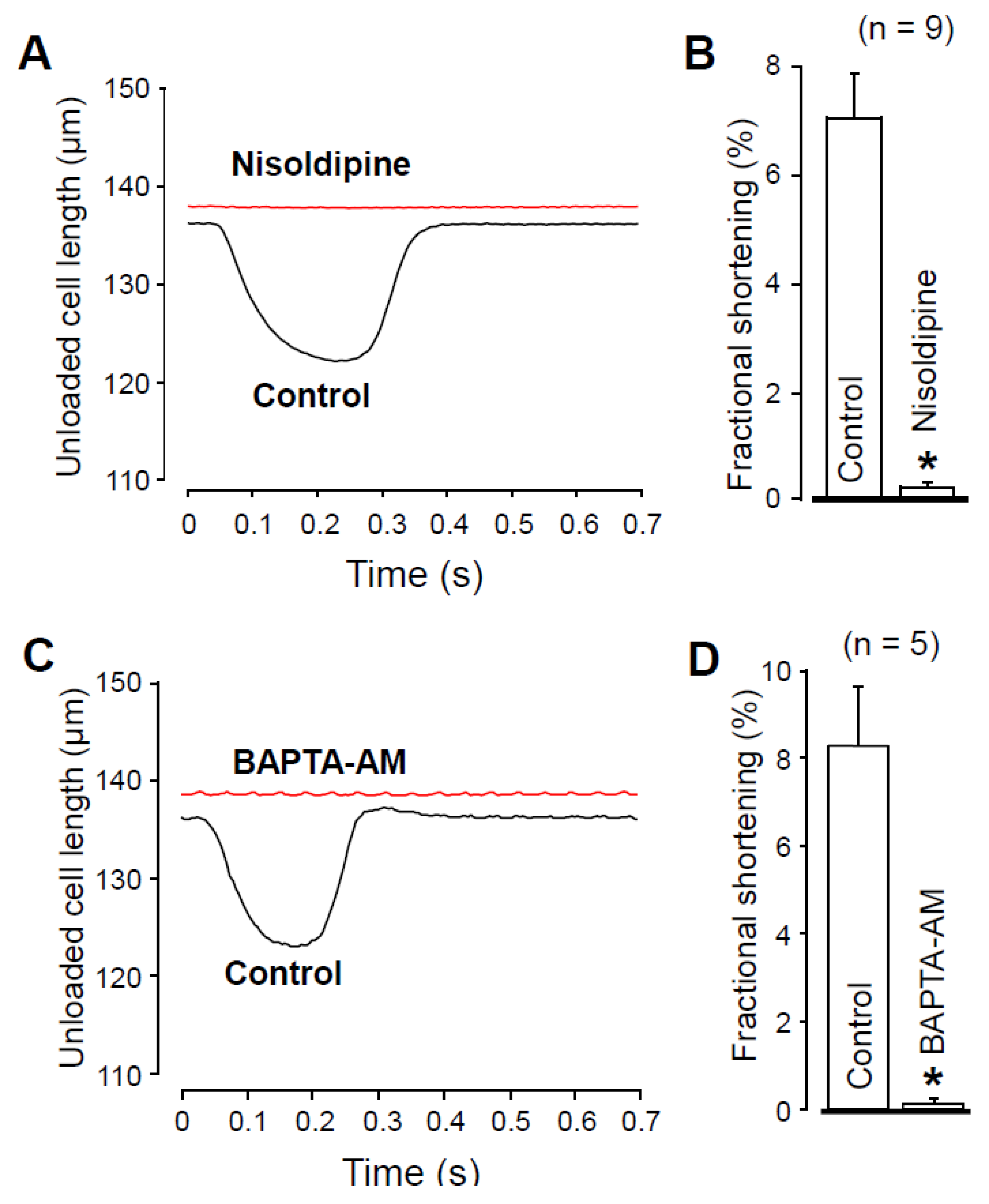
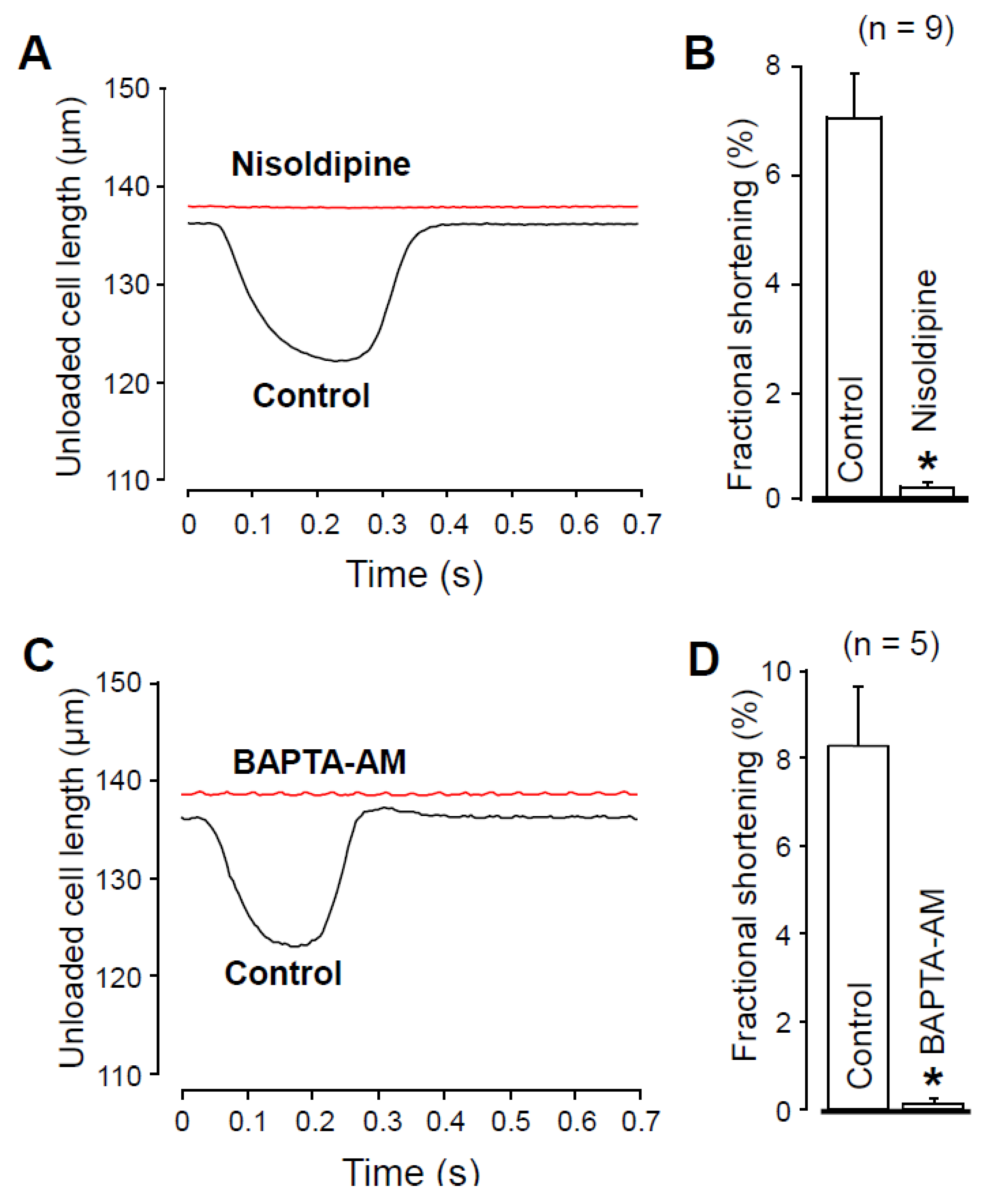
2.4. Effects of Nisoldipine and BAPTA-AM on Unloaded Cell Shortening
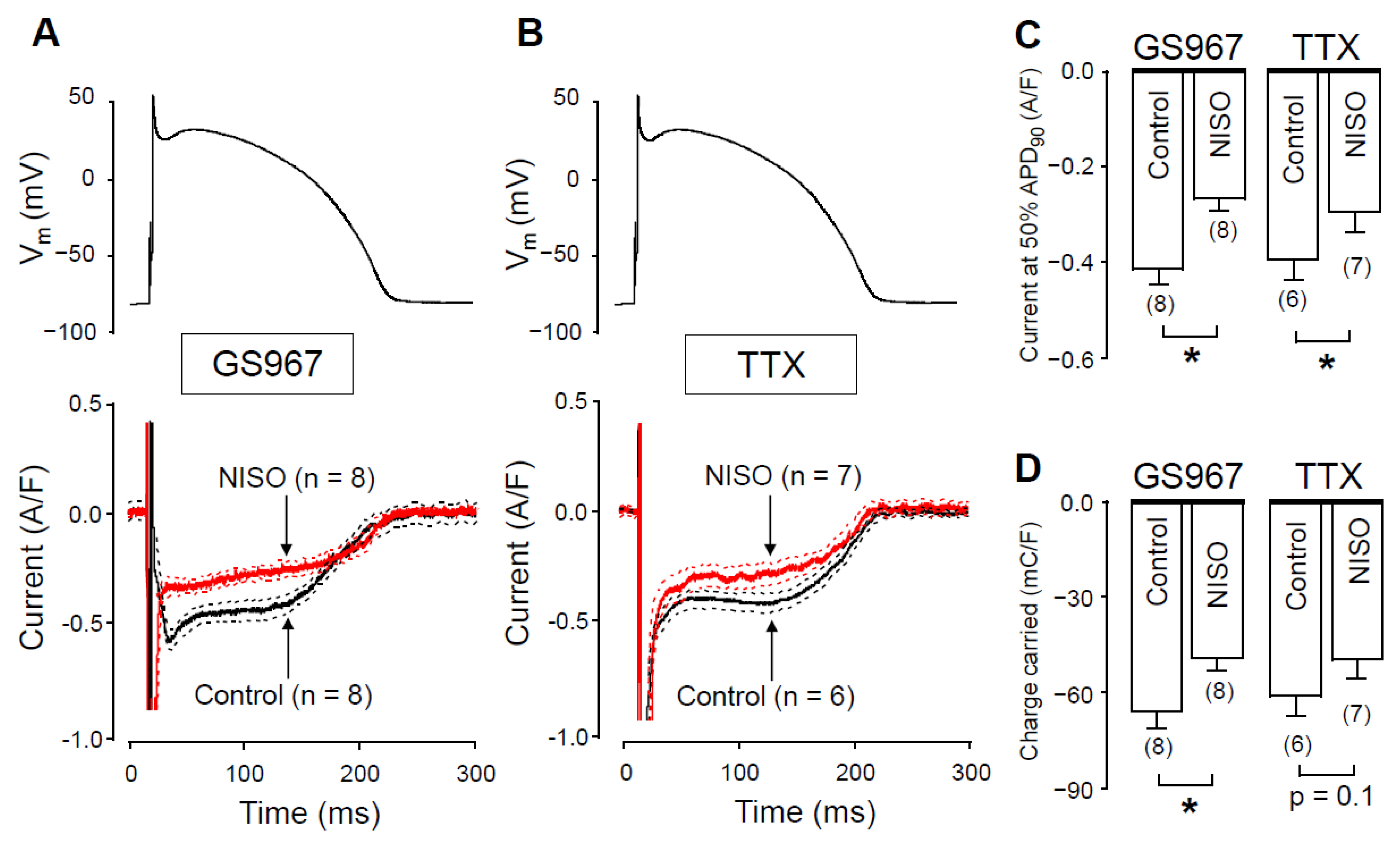
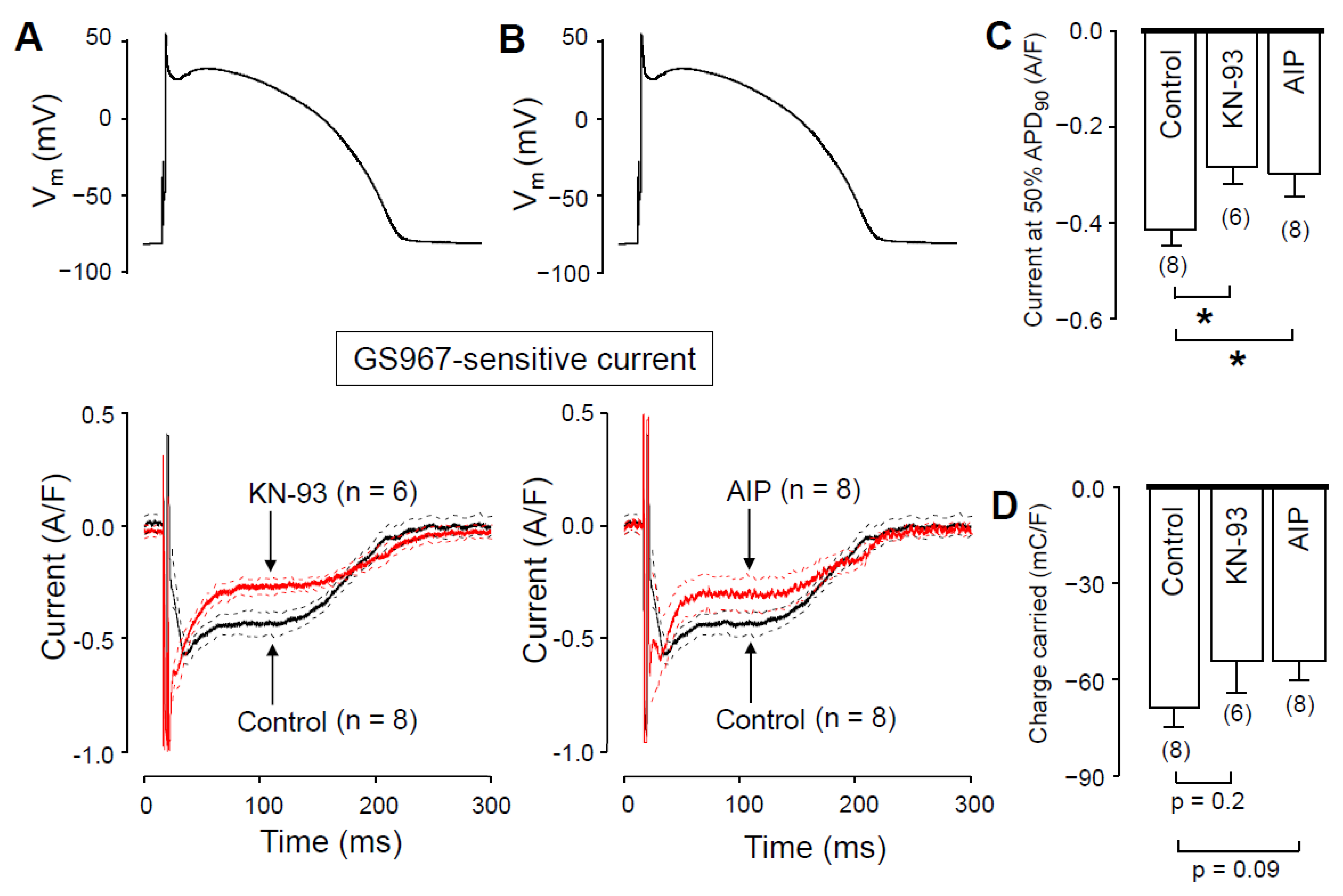
2.5. The Role of CaMKII in Regulation of INaL
2.6. Effect of GS967 on Action Potential Morphology
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Isolation of Cardiomyocytes
4.3. Electrophysiology
4.4. Action Potential Voltage Clamp
4.5. Conventional Voltage Clamp
4.6. Recording of Action Potentials
4.7. Recording of Unloaded Cell Shortening
4.8. Statistics
5. Conclusions
- INaL depends on the magnitude of [Ca2+]i in canine ventricular cells.
- The [Ca2+]i-dependence of INaL is mediated by the Ca2+-dependent activation of CaMKII.
- INaL is augmented by the baseline CaMKII activity.
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Song, Y.; Belardinelli, L. Basal late sodium current is a significant contributor to the duration of action potential of guinea pig ventricular myocytes. Physiol. Rep. 2017, 5, e13295. [Google Scholar] [CrossRef] [PubMed]
- Maltsev, V.A.; Undrovinas, A.I. A multi-modal composition of the late Na+ current in human ventricular cardiomyocytes. Cardiovasc. Res. 2006, 69, 116–127. [Google Scholar] [CrossRef] [Green Version]
- Noble, D.; Noble, P.J. Late sodium current in the pathophysiology of cardiovascular disease: Consequences of sodium-calcium overload. Heart 2006, 92 (Suppl. 4), 1–5. [Google Scholar] [CrossRef] [PubMed]
- Attwell, D.; Cohen, I.S.; Eisner, D.A.; Ohba, M.; Ojeda, C. The steady state TTX-sensitive (“window”) sodium current in cardiac Purkinje fibres. Pflügers Arch. 1979, 379, 137–142. [Google Scholar] [CrossRef] [PubMed]
- Chadda, K.R.; Jeevaratnam, K.; Lei, M.; Huang, C.L.H. Sodium channel biophysics, late sodium current and genetic arrhythmic syndromes. Pflügers Arch. 2017, 469, 629–641. [Google Scholar] [CrossRef] [Green Version]
- Undrovinas, A.I.; Maltsev, V.A.; Kyle, J.W.; Silverman, N.; Sabbah, H.N. Gating of the late Na+ channel in normal and failing human myocardium. J. Mol. Cell. Cardiol. 2002, 34, 1477–1489. [Google Scholar] [CrossRef]
- Clancy, C.E.; Tateyama, M.; Liu, H.; Wehrens, X.H.; Kass, R.S. Non-equilibrium gating in cardiac sodium cahannels: An original mechanism of arrhythmia. Circulation 2003, 107, 2233–2237. [Google Scholar] [CrossRef] [Green Version]
- Biet, M.; Barajas-Martínez, H.; Ton, A.T.; Delabre, J.F.; Morin, N.; Dumaine, R. About half of the late sodium current in cardiac myocytes from dog ventricle is due to non-cardiac-type Na+ channels. J. Mol. Cell. Cardiol 2012, 53, 593–598. [Google Scholar] [CrossRef] [PubMed]
- Coraboeuf, E.; Deroubaix, E.; Coulombe, A. Effect of tetrodotoxin on action potentials of the conducting system in the dog heart. Am. J. Physiol. 1979, 236, H561–H567. [Google Scholar] [CrossRef] [PubMed]
- Carmeliet, E. Slow inactivation of sodium current and voltage-dependent block by tetrodotoxin in rabbit cardiac Purkinje fibers. Biomed. Biochim. Acta 1986, 45, S163–S166. [Google Scholar]
- Carmeliet, E. Voltage-dependent block by tetrodotoxin of the sodium channel in rabbit cardiac Purkinje fibers. Biophys. J. 1987, 51, 109–114. [Google Scholar] [CrossRef] [Green Version]
- Horvath, B.; Bers, D.M. The late sodium current in heart failure: Pathophysiology and clinical relevance. ESC Heart Fail. 2014, 1, 26–40. [Google Scholar] [CrossRef]
- Valdivia, C.R.; Chu, W.W.; Pu, J.; Foell, J.D.; Haworth, R.A.; Wolff, M.R.; Kamp, T.J.; Makielski, J.C. Increased late sodium current in myocytes from a canine heart failure model and from failing human heart. J. Mol. Cell. Cardiol. 2005, 38, 475–483. [Google Scholar] [CrossRef] [PubMed]
- Maltsev, V.A.; Silverman, N.; Sabbah, H.N.; Undrovinas, A.I. Chronic heart failure slows late sodium current in human and canine ventricular myocytes: Implications for repolarization variability. Eur. J. Heart Fail. 2007, 9, 219–227. [Google Scholar] [CrossRef] [Green Version]
- Zaza, A.; Rocchetti, M. The late Na+ current–Origin and pathophysiological relevance. Cardiovasc. Drugs 2013, 27, 61–68. [Google Scholar] [CrossRef] [Green Version]
- Shyrock, J.C.; Song, Y.; Rajamani, S.; Antzelecitch, C.; Belardinelli, L. The antiarrhythmogenic consequences of increasing late INa in the cardiomyocyte. Cardiovasc. Res. 2013, 99, 600–611. [Google Scholar] [CrossRef] [Green Version]
- Yu, S.; Li, G.; Huang, C.L.H.; Lei, M.; Wu, L. Late sodium current associated cardiac electrophysiological and mechanical dysfunction. Pflügers Arch. 2018, 470, 461–469. [Google Scholar] [CrossRef]
- Hegyi, B.; Bányász, T.; Izu, L.T.; Belardinelli, L.; Bers, D.M.; Chen-Izu, Y. β-adrenergic regulation of late Na+ current during cardiac action potential is mediated by both PKA and CaMKII. J. Mol. Cell. Cardiol. 2018, 123, 168–179. [Google Scholar] [CrossRef]
- Fu, C.; Hao, J.; Zeng, M.; Song, Y.; Jiang, W.; Zhang, P.; Luo, A.; Cao, Z.; Belardinelli, L.; Ma, J. Modulation of late sodium current by Ca2+–calmodulin-dependent protein kinase II, protein kinase C and Ca2+ during hypoxia in rabbit ventricular myocytes. Exp. Physiol. 2017, 102.7, 818–834. [Google Scholar] [CrossRef] [PubMed]
- Hashambhoy, Y.; Winslow, R.L.; Greenstein, J.L. CaMKII-dependent activation of late INa contributes to cellular arrhythmia in a model of the cardiac myocyte. Conf. Proc. IEEE Eng. Med. Biol. Soc. 2011, 2011, 4665–4668. [Google Scholar]
- Wagner, S.; Dybkova, N.; Rasenack, E.C.L.; Jacobshagen, C.; Fabritz, L.; Kirchhof, P.; Maier, S.K.G.; Zhang, T.; Hasenfuss, G.; Brown, J.H.; et al. Ca2+/calmodulin-dependent protein kinase II regulates cardiac Na+ channels. J. Clin. Investig. 2006, 116, 3127–3138. [Google Scholar] [CrossRef]
- Howard, T.; Greer-Short, A.; Satroplus, T.; Patel, N.; Nassal, D.; Mohler, P.J.; Hund, T.J. CaMKII-dependent late Na+ current increases electrical dispersion and arrhythmia in ischemia-reperfusion. Am. J. Physiol. Heart Circ. Physiol. 2018, 315, H794–H801. [Google Scholar] [CrossRef] [Green Version]
- Toischer, K.; Hartmann, N.; Wagner, S.; Fischer, T.H.; Herting, J.; Danner, B.C.; Sag, C.M.; Hund, T.J.; Mohler, P.J.; Belardinelli, L.; et al. Role of late sodium current as a potential arrhythmogenic mechanism in the progression of pressure-induced heart disease. J. Mol. Cell. Cardiol. 2013, 61, 111–122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horvath, B.; Banyasz, T.; Jian, Z.; Hegyi, B.; Kistamas, K.; Nanasi, P.P.; Izu, L.T.; Chen-Izu, Y. Dynamics of the late Na+ current during cardiac action potential and its contribution to afterdepolarizations. J. Mol. Cell. Cardiol. 2013, 64, 59–68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hegyi, B.; Bossuyt, J.; Griffiths, L.G.; Shimkunas, R.; Coulibaly, Z.; Jian, Z.; Grimsrud, K.N.; Sondergaard, C.S.; Ginsburg, K.S.; Chiamvimonvat, N.; et al. Complex electrophysiological remodeling in postinfarction ischemic heart failure. Proc. Natl. Acad. Sci. USA 2018, 115, E3036–E3044. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szabo, G.; Szentandrassy, N.; Biro, T.; Toth, B.I.; Czifra, G.; Magyar, J.; Banyasz, T.; Varro, A.; Kovacs, L.; Nanasi, P.P. Asymmetrical distribution of ion channels in canine and human left-ventricular wall: Epicardium versus midmyocardium. Pflug. Arch. 2005, 450, 307–316. [Google Scholar] [CrossRef]
- Szentadrassy, N.; Banyasz, T.; Biro, T.; Szabo, G.; Toth, B.I.; Magyar, J.; Lazar, J.; Varro, A.; Kovacs, L.; Nanasi, P.P. Apico-basal inhomogeneity in distribution of ion channels in canine and human ventricular myocardium. Cardiovasc. Res. 2005, 65, 851–860. [Google Scholar] [CrossRef] [Green Version]
- Jost, N.; Acsai, K.; Horvath, B.; Banyasz, T.; Baczko, I.; Bitay, M.; Bogáts, G.; Nánási, P.P. Contribution of IKr and IK1 to ventricular repolarization in canine and human myocytes: Is there any influence of duration? Basic Res. Cardiol. 2009, 104, 33–41. [Google Scholar] [CrossRef]
- Jost, N.; Virág, L.; Comtois, P.; Ordög, B.; Szuts, V.; Seprényi, G.; Bitay, M.; Kohajda, Z.; Koncz, I.; Nagy, N.; et al. Ionic mechanisms limiting cardiac repolarization reserve in humans compared to dogs. J. Physiol. 2013, 591, 4189–4206. [Google Scholar] [CrossRef] [Green Version]
- Horváth, B.; Hézső, T.; Szentandrássy, N.; Kistamás, K.; Árpádffy-Lovas, T.; Varga, R.; Gazdag, P.; Veress, R.; Dienes, C.; Baranyai, D.; et al. Late sodium current in human, canine and guinea pig ventricular myocardium. J. Mol. Cell. Cardiol. 2020, 139, 14–23. [Google Scholar] [CrossRef] [Green Version]
- Zygmunt, A.C.; Eddelstone, G.T.; Thomas, G.P.; Nesterenko, V.V.; Antzelevitch, C. Larger late sodium conductance in M cells contributes to electrical heterogeneity in canine ventricle. Am. J. Physiol. Heart Circ. Physiol. 2001, 281, H689–H697. [Google Scholar] [CrossRef] [PubMed]
- Belardinelli, L.; Liu, G.; Smith-Maxwell, C.; Wang, W.-Q.; El-Bizri, N.; Hirakawa, R.; Karpinski, S.; Li, C.H.; Hu, L.; Li, X.-J.; et al. A novel, potent, and selective inhibitor of cardiac late sodium current suppresses experimental arrhythmias. J. Pharm. Exp. 2013, 344, 23–32. [Google Scholar] [CrossRef] [PubMed]
- Bossu, A.; Houtman, M.J.C.; Meijborg, V.M.F.; Varkevisser, R.; Beekman, H.D.M.; Dunnink, A.; de Bakker, J.M.T.; Mollova, N.; Rajamani, S.; Belardinelli, L.; et al. Selective late sodium current inhibitor GS-458967 suppresses torsades de pointes by mostly affecting perpetuation but not initiation of the arrhythmia. Br. J. Pharm. 2018, 175, 2470–2482. [Google Scholar] [CrossRef] [Green Version]
- Bonatti, R.; Silva, A.F.G.; Batatinha, J.A.P.; Sobrado, L.F.; Machado, A.D.; Varone, B.B.; Nearing, B.D.; Belardinelli, L.; Verrier, R.L. Selective late sodium current blockade with GS-458967 markedly reduces ischemia-induced atrial and ventricular repolarization alternans and ECG heterogeneity. Heart Rhythm 2014, 11, 827–1835. [Google Scholar] [CrossRef]
- Bányász, T.; Horváth, B.; Virág, L.; Bárándi, L.; Szentandrássy, N.; Harmati, G.; Magyar, J.; Marangoni, S.; Zaza, A.; Varró, A.; et al. Reverse rate dependency is an intrinsic property of canine cardiac preparations. Cardiovasc. Res. 2009, 84, 237–244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bárándi, L.; Virág, L.; Jost, N.; Horváth, Z.; Koncz, I.; Papp, R.; Harmati, G.; Horváth, B.; Szentandrássy, N.; Bányász, T.; et al. Reverse rate-dependent changes are determined by baseline action potential duration in mammalian and human ventricular preparations. Basic Res. Cardiol. 2010, 105, 315–323. [Google Scholar] [CrossRef]
- Zaza, A.; Varro, A. Rate-dependent modulation of repolarization: Biology or math? Eur. Heart J. 2006, 27, 412. [Google Scholar]
- Maltsev, V.A.; Reznikov, V.; Undrovinas, N.A.; Sabbah, H.N.; Undrovinas, A. Modulation of the late sodium current by Ca2+, calmodulin, and CaMKII in normal and failing dog cardiomyocytes: Similarities and differences. Am. J. Physiol. Heart Circ. Physiol. 2008, 294, H1597–H1608. [Google Scholar] [CrossRef] [Green Version]
- Wingo, T.L.; Shah, V.N.; Anderson, M.E.; Lybrand, T.P.; Chazin, W.J.; Balser, J.R. An EF-hand in the sodium channel couples intracellular calcium to cardiac excitability. Nat. Struct. Mol. Biol. 2004, 11, 219–225. [Google Scholar] [CrossRef]
- Tan, H.L.; Kupershmidt, S.; Zhang, R.; Stepanovic, S.; Roden, D.M.; Wilde, A.A.; Anderson, M.E.; Balser, J.R. A calcium sensor in the sodium channel modulates cardiac excitability. Nature 2002, 415, 442–447. [Google Scholar] [CrossRef]
- Kim, J.; Ghosh, S.; Liu, H.; Tateyama, M.; Kass, R.S.; Pitt, G.S. Calmodulin mediates Ca2+ sensitivity of sodium channels. J. Biol. Chem. 2004, 279, 45004–45012. [Google Scholar] [CrossRef] [Green Version]
- Sarhan, M.F.; Tung, C.C.; Van Petegem, F.; Ahern, C.A. Crystallographic basis for calcium regulation of sodium channels. Proc. Natl. Acad. Sci. USA 2012, 109, 3558–3563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erickson, J.R.; Patel, R.; Ferguson, A.; Bossuyt, J.; Bers, D.M. Fluorescence resonance energy transfer-based sensor Camui provides new insight into mechanisms of calcium/calmodulin-dependent protein kinase II activation in intact cardiomyocytes. Circ. Res. 2011, 109, 729–738. [Google Scholar] [CrossRef] [PubMed]
- Wood, B.M.; Simon, M.; Galice, S.; Alim, C.C.; Ferrero, M.; Pinna, N.N.; Bers, D.M.; Bossuyt, J. Cardiac CaMKII activation promotes rapid translocation to its extra-dyadic targets. J. Mol. Cell. Cardiol. 2018, 125, 18–28. [Google Scholar] [CrossRef] [PubMed]
- Lu, Q.; Harris, V.A.; Sun, X.; Hou, Y.; Black, S.M. Ca2+/calmodulin-dependent protein kinase II contributes to hypoxic ischemic cell death in neonatal hippocampal slice cultures. PLoS ONE 2013, 8, e70750. [Google Scholar] [CrossRef]
- Wong, M.H.; Samal, A.B.; Lee, M.; Vlach, J.; Novikov, N.; Niedziela-Majka, A.; Feng, J.Y.; Koltun, D.O.; Brendza, K.M.; Kwon, H.J.; et al. The KN-93 molecule inhibits calcium/calmodulin-dependent protein kinase II (CaMKII) activity by binding to Ca2+/CaM. J. Mol. Biol. 2019, 431, 1440–1459. [Google Scholar] [CrossRef]
- Horváth, B.; Váczi, K.; Hegyi, B.; Gönczi, M.; Dienes, B.; Kistamás, K.; Bányász, T.; Magyar, J.; Baczkó, I.; Varró, A.; et al. Sarcolemmal Ca2+-entry through L-type Ca2+ channels controls the profile of Ca2+-activated Cl- current in canine ventricular myocytes. J. Mol. Cell Cardiol. 2016, 97, 125–139. [Google Scholar] [CrossRef] [Green Version]
- Banyasz, T.; Fulop, L.; Magyar, J.; Szentandrassy, N.; Varro, A.; Nanasi, P.P. Endocardial versus epicardial differences in L-type calcium current in canine ventricular myocytes studied by action potential voltage clamp. Cardiovasc. Res. 2003, 58, 66–75. [Google Scholar] [CrossRef] [Green Version]
- Bányász, T.; Magyar, J.; Szentandrássy, N.; Horváth, B.; Birinyi, P.; Szentmiklósi, J.; Nánási, P.P. Action potential clamp fingerprints of K+ currents in canine cardiomyocytes: Their role in ventricular repolarization. Acta Physiol. Scand. 2007, 190, 189–198. [Google Scholar] [CrossRef]
- Szentandrássy, N.; Kistamás, K.; Hegyi, B.; Horváth, B.; Ruzsnavszky, F.; Váczi, K.; Magyar, J.; Bányász, T.; Varró, A.; Nánási, P.P. Contribution of ion currents to beat-to-beat variability of action potential duration in canine ventricular myocytes. Pflügers Arch. 2015, 467, 1431–1443. [Google Scholar] [CrossRef] [Green Version]
- Horváth, B.; Szentandrássy, N.; Veress, R.; Almássy, J.; Magyar, J.; Bányász, T.; Tóth, A.; Papp, Z.; Nánási, P.P. Frequency-dependent effects of omecamtiv mecarbil on cell shortening of isolated canine ventricular cardiomyocytes. Naunyn Schmiedeberg Arch. Pharm. 2017, 390, 1239–1246. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kiss, D.; Horváth, B.; Hézső, T.; Dienes, C.; Kovács, Z.; Topal, L.; Szentandrássy, N.; Almássy, J.; Prorok, J.; Virág, L.; et al. Late Na+ Current Is [Ca2+]i-Dependent in Canine Ventricular Myocytes. Pharmaceuticals 2021, 14, 1142. https://doi.org/10.3390/ph14111142
Kiss D, Horváth B, Hézső T, Dienes C, Kovács Z, Topal L, Szentandrássy N, Almássy J, Prorok J, Virág L, et al. Late Na+ Current Is [Ca2+]i-Dependent in Canine Ventricular Myocytes. Pharmaceuticals. 2021; 14(11):1142. https://doi.org/10.3390/ph14111142
Chicago/Turabian StyleKiss, Dénes, Balázs Horváth, Tamás Hézső, Csaba Dienes, Zsigmond Kovács, Leila Topal, Norbert Szentandrássy, János Almássy, János Prorok, László Virág, and et al. 2021. "Late Na+ Current Is [Ca2+]i-Dependent in Canine Ventricular Myocytes" Pharmaceuticals 14, no. 11: 1142. https://doi.org/10.3390/ph14111142
APA StyleKiss, D., Horváth, B., Hézső, T., Dienes, C., Kovács, Z., Topal, L., Szentandrássy, N., Almássy, J., Prorok, J., Virág, L., Bányász, T., Varró, A., Nánási, P. P., & Magyar, J. (2021). Late Na+ Current Is [Ca2+]i-Dependent in Canine Ventricular Myocytes. Pharmaceuticals, 14(11), 1142. https://doi.org/10.3390/ph14111142