
Towards Drug Repurposing in Cancer Cachexia: Potential Targets and Candidates

 ,
, 
 and
and 
Abstract
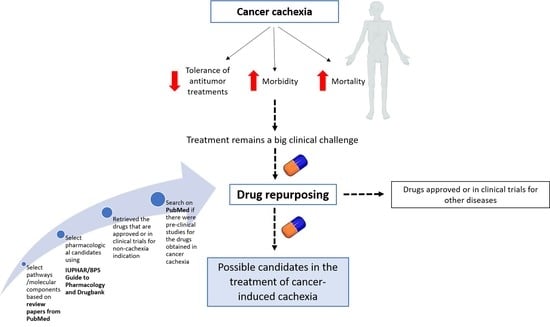
1. Introduction
2. Materials and Methods
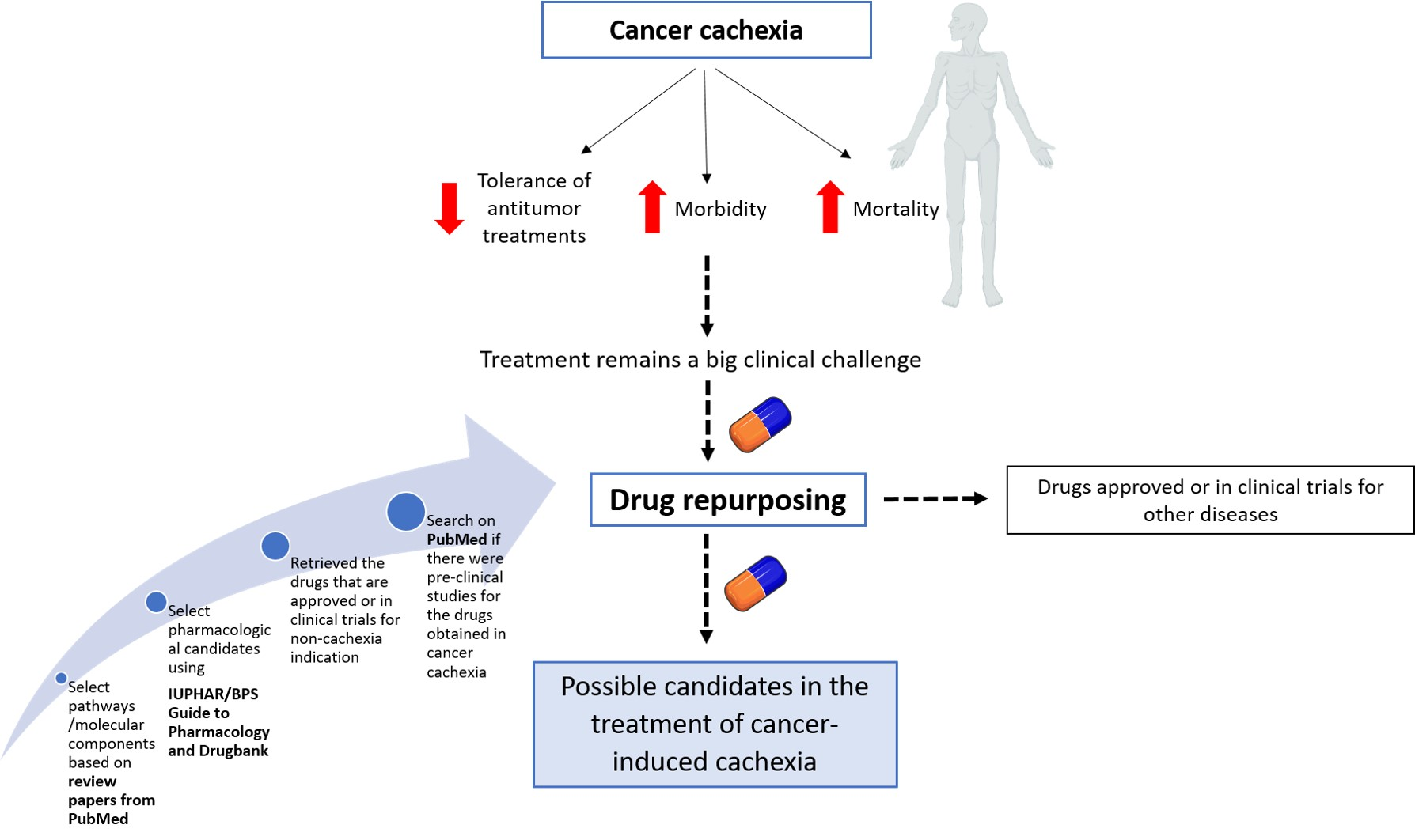
2.1. Selection of Phenotypes and Pathways/Molecular Components Involved in Cachexia Syndrome
2.2. Selection of Pharmacological Candidates
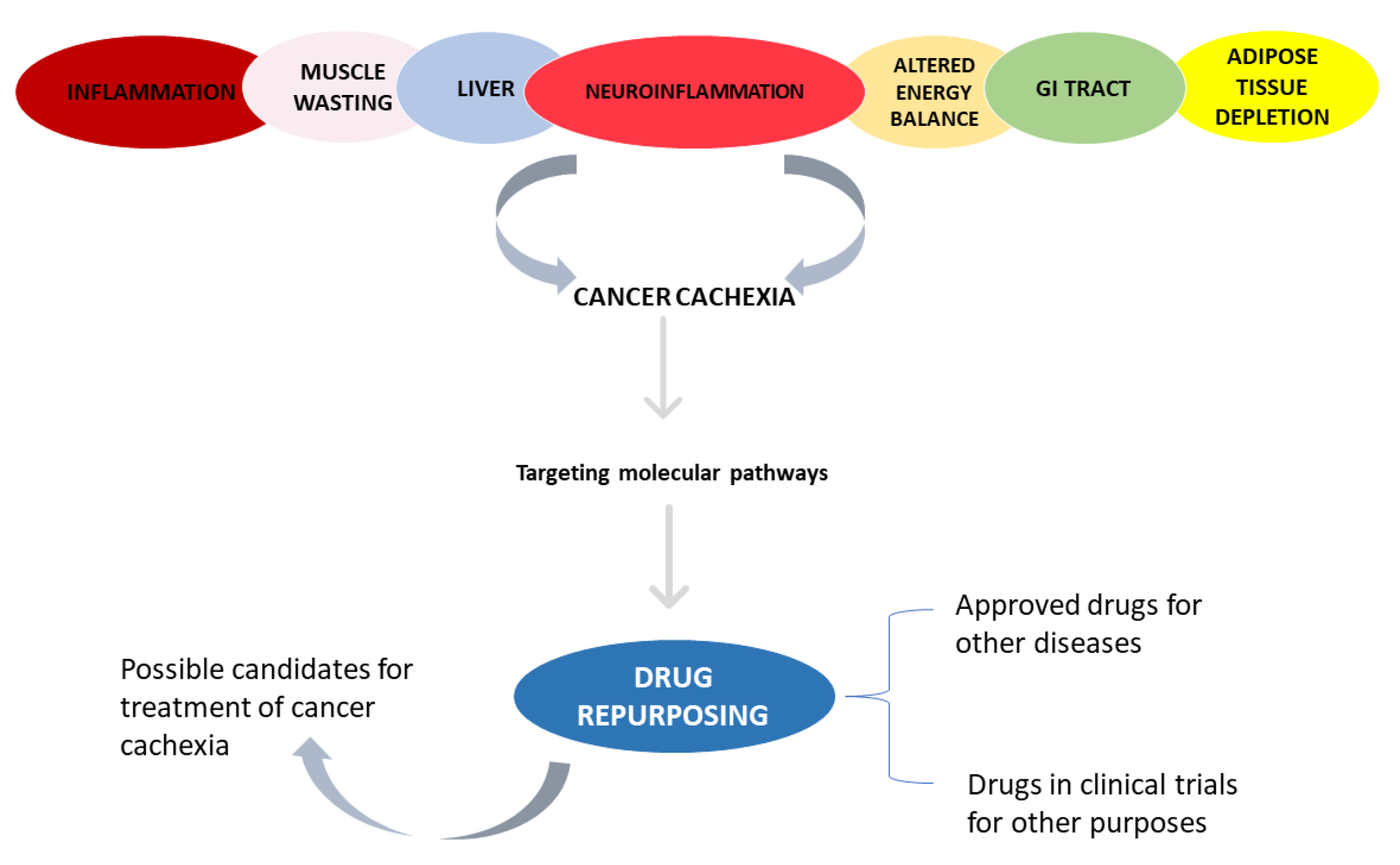
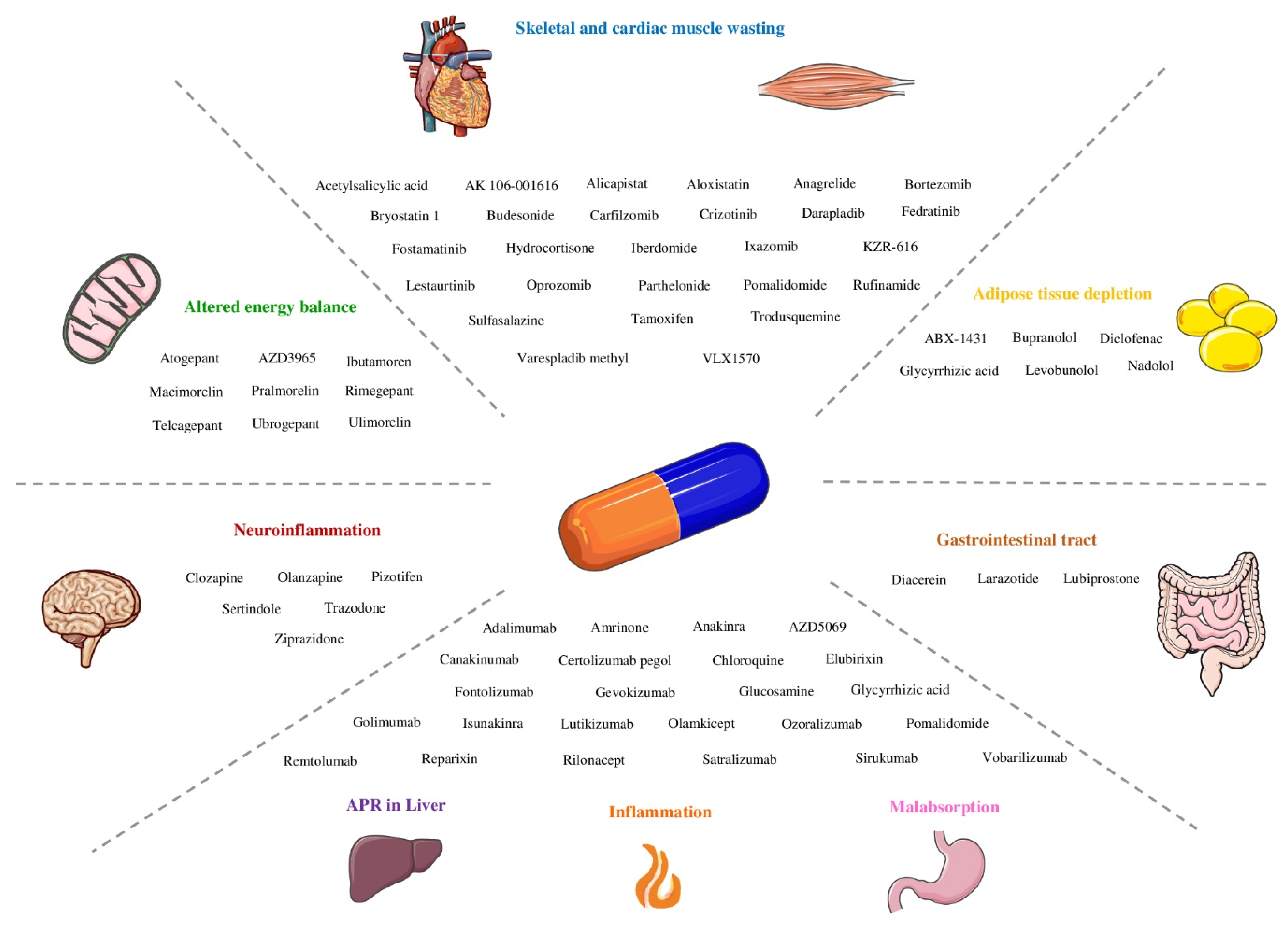
3. Results
3.1. Inflammation
3.1.1. TNF-α
3.1.2. IL-6
3.1.3. IL-1
3.2. Skeletal and Cardiac Muscle Wasting
3.2.1. Autophagy
3.2.2. Ubiquitin-Mediated Proteasome Degradation System (UPS)
3.2.3. Calcium-Activated Protease Calpains
3.2.4. Insulin Resistance
3.2.5. PIF
3.3. Adipose Tissue Depletion
3.3.1. Lipolysis
3.3.2. Inhibition of Lipogenesis
3.3.3. WAT Browning
3.4. Liver
3.5. Altered Energy Balance
3.5.1. Ghrelin Agonists
3.5.2. Inhibitor of Monocarboxylate Transporter 1 (MCT1)
3.5.3. Calcitonin Gene-Related Peptide (CGRP) Receptor Antagonist
3.6. Neuroinflammation
Serotonin Antagonists
3.7. Impaired Barrier Function and Malabsorption
3.7.1. Zonulin Inhibitor
3.7.2. ZO-1 and Claudins Agonist
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Da Silva, S.P.; Santos, J.M.O.; Costa e Silva, M.P.; Gil da Costa, R.M.; Medeiros, R. Cancer cachexia and its pathophysiology: Links with sarcopenia, anorexia and asthenia. J. Cachex Sarcopenia Muscle 2020, 11, 619–635. [Google Scholar] [CrossRef]
- Fearon, K.; Strasser, F.; Anker, S.D.; Bosaeus, I.; Bruera, E.; Fainsinger, R.L.; Jatoi, A.; Loprinzi, C.; MacDonald, N.; Mantovani, G.; et al. Definition and classification of cancer cachexia: An international consensus. Lancet Oncol. 2011, 12, 489–495. [Google Scholar] [CrossRef]
- Roeland, E.J.; Bohlke, K.; Baracos, V.E.; Bruera, E.; Del Fabbro, E.; Dixon, S.; Fallon, M.; Herrstedt, J.; Lau, H.; Platek, M.; et al. Management of Cancer Cachexia: ASCO Guideline. J. Clin. Oncol. 2020, 38, 2438–2453. [Google Scholar] [CrossRef]
- Nowak-Sliwinska, P.; Scapozza, L.; i Altaba, A.R. Drug repurposing in oncology: Compounds, pathways, phenotypes and computational approaches for colorectal cancer. Biochim. Biophys. Acta Bioenerg. 2019, 1871, 434–454. [Google Scholar] [CrossRef]
- Baracos, V.E.; Martin, L.; Korc, M.; Guttridge, D.C.; Fearon, K.C.H. Cancer-associated cachexia. Nat. Rev. Dis. Prim. 2018, 4, 17105. [Google Scholar] [CrossRef] [PubMed]
- Argiles, J.M.; Busquets, S.; Stemmler, B.; López-Soriano, F.J. Cancer cachexia: Understanding the molecular basis. Nat. Rev. Cancer 2014, 14, 754–762. [Google Scholar] [CrossRef]
- Porporato, P. Understanding cachexia as a cancer metabolism syndrome. Oncogenesis 2016, 5, e200. [Google Scholar] [CrossRef]
- Xue, H.; Li, J.; Xie, H.; Wang, Y. Review of Drug Repositioning Approaches and Resources. Int. J. Biol. Sci. 2018, 14, 1232–1244. [Google Scholar] [CrossRef] [PubMed]
- Serçinoğlu, O.; Sarica, P.O. In Silico Databases and Tools for Drug Repurposing. Chapter 24; In In Silico Drug Design; Roy, K., Ed.; Academic Press: Cambridge, MA, USA, 2019; pp. 703–742. [Google Scholar]
- Armstrong, J.F.; Faccenda, E.; Harding, S.D.; Pawson, A.J.; Southan, C.; Sharman, J.L.; Campo, B.; Cavanagh, D.R.; Alexander, S.; Davenport, A.P.; et al. The IUPHAR/BPS Guide to pharmacology in 2020: Extending immunopharmacology content and introducing the IUPHAR/MMV Guide to malaria pharmacology. Nucleic Acids Res. 2019, 48, D1006–D1021. [Google Scholar] [CrossRef]
- Wishart, D.S.; Feunang, Y.D.; Guo, A.C.; Lo, E.J.; Marcu, A.; Grant, J.R.; Sajed, T.; Johnson, D.; Li, C.; Sayeeda, Z.; et al. DrugBank 5.0: A major update to the DrugBank database for 2018. Nucleic Acids Res. 2017, 46, D1074–D1082. [Google Scholar] [CrossRef]
- Argilés, J.M.; López-Soriano, F.J.; Stemmler, B.; Busquets, S. Therapeutic strategies against cancer cachexia. Eur. J. Transl. Myol. 2019, 29, 7960. [Google Scholar] [CrossRef]
- Lu, X.; Hu, R.; Peng, L.; Liu, M.; Sun, Z. Efficacy and Safety of Adalimumab Biosimilars: Current Critical Clinical Data in Rheumatoid Arthritis. Front. Immunol. 2021, 12, 707. [Google Scholar] [CrossRef]
- Scheinfeld, N. Adalimumab: A review of side effects. Expert Opin. Drug Saf. 2005, 4, 637–641. [Google Scholar] [CrossRef]
- Saraceno, R.; Schipani, C.; Mazzotta, A.; Esposito, M.; DiRenzo, L.; DeLorenzo, A.; Chimenti, S. Effect of anti-tumor necrosis factor-α therapies on body mass index in patients with psoriasis. Pharmacol. Res. 2008, 57, 290–295. [Google Scholar] [CrossRef]
- Lee, A.; Scott, L.J. Certolizumab Pegol: A Review in Moderate to Severe Plaque Psoriasis. BioDrugs 2020, 34, 235–244. [Google Scholar] [CrossRef] [PubMed]
- Horton, S.; Walsh, C.; Emery, P. Certolizumab pegol for the treatment of rheumatoid arthritis. Expert Opin. Biol. Ther. 2011, 12, 235–249. [Google Scholar] [CrossRef]
- Goel, P.; Gerriets, V. Chloroquine; StatPearls Publishing LLC: Treasure Island, FL, USA, 2021. [Google Scholar]
- Gupta, A.; Preuss, C.V. Inamrinone; StatPearls Publishing LLC: Treasure Island, FL, USA, 2021. [Google Scholar]
- Jurasinski, C.V.; Kilpatrick, L.; Vary, T.C. Amrinone prevents muscle protein wasting during chronic sepsis. Am. J. Physiol. Metab. 1995, 268, E491–E500. [Google Scholar] [CrossRef]
- Lira, E.C.; Gonçalves, D.A.; Parreiras-E-Silva, L.T.; Zanon, N.M.; Kettelhut, I.C.; Navegantes, L.C. Phosphodiesterase-4 inhibition reduces proteolysis and atrogenes expression in rat skeletal muscles. Muscle Nerve 2011, 44, 371–381. [Google Scholar] [CrossRef]
- Burki, T.K. Pomalidomide for symptomatic Kaposi’s sarcoma. Lancet Oncol. 2016, 17, e526. [Google Scholar] [CrossRef]
- Ríos-Tamayo, R.; Martín-García, A.; Alarcón-Payer, C.; Sánchez-Rodríguez, D.; de la Guardia, A.; García Collado, C.G.; Jiménez Morales, A.; Jurado Chacón, M.; Cabeza Barrera, J. Pomalidomide in the treatment of multiple myeloma: Design, development and place in therapy. Drug Des. Dev. Ther. 2017, 11, 2399–2408. [Google Scholar] [CrossRef]
- Lacy, M.Q.; McCurdy, A.R. Pomalidomide. Blood 2013, 122, 2305–2309. [Google Scholar] [CrossRef]
- Li, J.-Y.; Cao, H.-Y.; Liu, P.; Cheng, G.-H.; Sun, M.-Y. Glycyrrhizic Acid in the Treatment of Liver Diseases: Literature Review. BioMed Res. Int. 2014, 2014, 872139. [Google Scholar] [CrossRef]
- Ming, L.J.; Yin, A.C.Y. Therapeutic Effects of Glycyrrhizic Acid. Nat. Prod. Commun. 2013, 8, 415–418. [Google Scholar] [CrossRef] [PubMed]
- Deng, Q.-P.; Wang, M.-J.; Zeng, X.; Chen, G.G.; Huang, R.-Y. Effects of Glycyrrhizin in a Mouse Model of Lung Adenocarcinoma. Cell. Physiol. Biochem. 2017, 41, 1383–1392. [Google Scholar] [CrossRef] [PubMed]
- Aletaha, D.; Bingham, C.O.; Karpouzas, G.A.; Takeuchi, T.; Thorne, C.; Bili, A.; Agarwal, P.; Hsu, B.; Rao, R.; Brown, K.; et al. Long-term safety and efficacy of sirukumab for patients with rheumatoid arthritis who previously received sirukumab in randomised controlled trials (SIRROUND-LTE). RMD Open 2021, 7, e001465. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, W.A.; Dasgupta, B.; Luqmani, R.; Unizony, S.H.; Blockmans, D.; Lai, Z.; Kurrasch, R.H.; Lazic, I.; Brown, K.; Rao, R. A Multicentre, Randomised, Double-Blind, Placebo-Controlled, Parallel-Group Study to Evaluate the Efficacy and Safety of Sirukumab in the Treatment of Giant Cell Arteritis. Rheumatol. Ther. 2020, 7, 793–810. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, T.; Tanaka, Y.; Yamanaka, H.; Harigai, M.; Nakano, T.; Akagi, K.; Ukyo, Y.; Hsu, B. Efficacy and safety of sirukumab in Japanese patients with moderate to severe rheumatoid arthritis inadequately controlled by disease modifying anti-rheumatic drugs: Subgroup analysis of a phase 3 study. Mod. Rheumatol. 2018, 28, 941–949. [Google Scholar] [CrossRef] [PubMed]
- Dhimolea, E. Canakinumab. mAbs 2010, 2, 3–13. [Google Scholar] [CrossRef] [PubMed]
- Pile, K.D.; Graham, G.G.; Mahler, S.M. Interleukin-1 (IL-1) Inhibitors: Anakinra, Rilonacept, and Canakinumab. In Compendium of Inflammatory Diseases; Parnham, M.J., Ed.; Springer: Basel, Switzerland, 2016; pp. 666–670. [Google Scholar] [CrossRef]
- McDermott, M. Rilonacept in the treatment of chronic inflammatory disorders. Drugs Today 2009, 45, 423–430. [Google Scholar] [CrossRef] [PubMed]
- Harnack, U.; Johnen, H.; Pecher, G. IL-1 receptor antagonist anakinra enhances tumour growth inhibition in mice receiving peptide vaccination and beta-(1–3), (1–6)-D-glucan. Anticancer Res. 2010, 30, 3959–3965. [Google Scholar] [PubMed]
- Rausch, V.; Sala, V.; Penna, F.; Porporato, P.E.; Ghigo, A. Understanding the common mechanisms of heart and skeletal muscle wasting in cancer cachexia. Oncogenesis 2021, 10, 1. [Google Scholar] [CrossRef]
- Siddiqui, J.A.; Pothuraju, R.; Jain, M.; Batra, S.K.; Nasser, M.W. Advances in cancer cachexia: Intersection between affected organs, mediators, and pharmacological interventions. Biochim. Biophys. Acta Bioenerg. 2020, 1873, 188359. [Google Scholar] [CrossRef]
- Sakuma, K.; Aoi, W.; Yamaguchi, A. Molecular mechanism of sarcopenia and cachexia: Recent research advances. Pflug. Arch. 2017, 469, 573–591. [Google Scholar] [CrossRef]
- Lim, S.M.; Hanif, E.A.M.; Chin, S.-F. Is targeting autophagy mechanism in cancer a good approach? The possible double-edge sword effect. Cell Biosci. 2021, 11, 56. [Google Scholar] [CrossRef]
- Talpaz, M.; Kiladjian, J.J. Fedratinib, a newly approved treatment for patients with myeloproliferative neoplasm-associated myelofibrosis. Leukemia 2021, 35, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Pardanani, A.; Harrison, C.; Cortes, J.E.; Cervantes, F.; Mesa, R.A.; Milligan, D.; Masszi, T.; Mishchenko, E.; Jourdan, E.; Vannucchi, A.M.; et al. Safety and Efficacy of Fedratinib in Patients With Primary or Secondary Myelofibrosis: A Randomized Clinical Trial. JAMA Oncol. 2015, 1, 643–651. [Google Scholar] [CrossRef] [PubMed]
- Sahu, A.; Prabhash, K.; Noronha, V.; Joshi, A.; Desai, S. Crizotinib: A comprehensive review. South Asian J. Cancer 2013, 2, 91–97. [Google Scholar] [CrossRef]
- Maringwa, J.; Kågedal, M.; Hamrén, U.W.; Martin, P.; Cox, E.; Hamrén, B. Pharmacokinetic-pharmacodynamic modeling of fostamatinib efficacy on ACR20 to support dose selection in patients with rheumatoid arthritis (RA). J. Clin. Pharmacol. 2015, 55, 328–335. [Google Scholar] [CrossRef] [PubMed]
- Bussel, J.; Arnold, D.M.; Grossbard, E.; Mayer, J.; Treliński, J.; Homenda, W.; Hellmann, A.; Windyga, J.; Sivcheva, L.; Khalafallah, A.A.; et al. Fostamatinib for the treatment of adult persistent and chronic immune thrombocytopenia: Results of two phase 3, randomized, placebo-controlled trials. Am. J. Hematol. 2018, 93, 921–930. [Google Scholar] [CrossRef] [PubMed]
- Glickman, M.; Ciechanover, A. The Ubiquitin-Proteasome Proteolytic Pathway: Destruction for the Sake of Construction. Physiol. Rev. 2002, 82, 373–428. [Google Scholar] [CrossRef]
- Fennell, D.A.; Chacko, A.; Mutti, L. BCL-2 family regulation by the 20S proteasome inhibitor bortezomib. Oncogene 2007, 27, 1189–1197. [Google Scholar] [CrossRef]
- Alexander, T.; Sarfert, R.; Klotsche, J.; Kühl, A.A.; Rubbert-Roth, A.; Lorenz, H.-M.; Rech, J.; Hoyer, B.F.; Cheng, Q.; Waka, A.; et al. The proteasome inhibitior bortezomib depletes plasma cells and ameliorates clinical manifestations of refractory systemic lupus erythematosus. Ann. Rheum. Dis. 2015, 74, 1474–1478. [Google Scholar] [CrossRef] [PubMed]
- Jakez-Ocampo, J.; Atisha-Fregoso, Y.; Llorente, L. Refractory Primary Sjögren Syndrome Successfully Treated with Bortezomib. J. Clin. Rheumatol. 2015, 21, 31–32. [Google Scholar] [CrossRef]
- Penna, F.; Bonetto, A.; Aversa, Z.; Minero, V.G.; Fanelli, F.R.; Costelli, P.; Muscaritoli, M. Effect of the specific proteasome inhibitor bortezomib on cancer-related muscle wasting. J. Cachex Sarcopenia Muscle 2015, 7, 345–354. [Google Scholar] [CrossRef] [PubMed]
- Groen, K.; van de Donk, N.W.C.J.; Stege, C.; Zweegman, S.; Nijhof, I. Carfilzomib for relapsed and refractory multiple myeloma. Cancer Manag. Res. 2019, 11, 2663–2675. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Li, C.; Peng, X.; Kang, Q.; Deng, D.; Zhang, L.; Zheng, Y.; Wang, C.; Qiao, Z.; Guo, D.; et al. Combined treatment of carfilzomib and z-VAD-fmk inhibits skeletal proteolysis and apoptosis and ameliorates cancer cachexia. Med. Oncol. 2015, 32. [Google Scholar] [CrossRef]
- Shirley, M. Ixazomib: First Global Approval. Drugs 2016, 76, 405–411. [Google Scholar] [CrossRef]
- Hughes, D.M.; Staron, A.; Sanchorawala, V. A pharmacist’s review of the treatment of systemic light chain amyloidosis. J. Oncol. Pharm. Pract. 2020, 27, 187–198. [Google Scholar] [CrossRef]
- Micheletto, M.L.J.; Hermes, T.D.A.; Bertassoli, B.M.; Petri, G.; Perez, M.M.; Fonseca, F.L.A.; Carvalho, A.A.S.; Feder, D. Ixazomib, an oral proteasome inhibitor, exhibits potential effect in dystrophin-deficient mdx mice. Int. J. Exp. Pathol. 2020, 102, 11–21. [Google Scholar] [CrossRef]
- Zeng, X.; Zhao, L.; Chen, S.; Li, X. Inhibition of mitochondrial and cytosolic calpain attenuates atrophy in myotubes co-cultured with colon carcinoma cells. Oncol. Lett. 2020, 21, 124. [Google Scholar] [CrossRef]
- Lin, X.-Y.; Chen, S.-Z. Calpain inhibitors ameliorate muscle wasting in a cachectic mouse model bearing CT26 colorectal adenocarcinoma. Oncol. Rep. 2017, 37, 1601–1610. [Google Scholar] [CrossRef] [PubMed]
- Barnoy, S.; Glaser, T.; Kosower, N.S. The calpain–calpastatin system and protein degradation in fusing myoblasts. Biochim. Biophys. Acta Bioenerg. 1998, 1402, 52–60. [Google Scholar] [CrossRef][Green Version]
- Petersen, M.; Shulman, G. Mechanisms of Insulin Action and Insulin Resistance. Physiol. Rev. 2018, 98, 2133–2223. [Google Scholar] [CrossRef]
- Salmeen, A.; Andersen, J.; Myers, M.P.; Tonks, N.K.; Barford, D. Molecular Basis for the Dephosphorylation of the Activation Segment of the Insulin Receptor by Protein Tyrosine Phosphatase 1B. Mol. Cell 2000, 6, 1401–1412. [Google Scholar] [CrossRef]
- Cariuk, P.; Lorite, M.; Todorov, P.; Field, W.; Wigmore, S.; Tisdale, M. Induction of cachexia in mice by a product isolated from the urine of cachectic cancer patients. Br. J. Cancer 1997, 76, 606–613. [Google Scholar] [CrossRef]
- Watchorn, T.M.; Waddell, I.D.; Dowidar, N.; Ross, J.A. Proteolysis-inducing factor regulates hepatic gene expression via the transcription factors NF-κΒ and STAT3. FASEB J. 2001, 15, 562–564. [Google Scholar] [CrossRef] [PubMed]
- Watchorn, T.M.; Waddell, I.; Ross, J.A. Proteolysis-inducing factor differentially influences transcriptional regulation in endothelial subtypes. Am. J. Physiol. Metab. 2002, 282, E763–E769. [Google Scholar] [CrossRef][Green Version]
- Tisdale, M.J. Proteolysis-Inducing Factor in Cancer Cachexia. In Cachexia and Wasting: A Modern Approach; Mantovani, G., Anker, S.D., Inui, A., Morley, J.E., Fanelli, F.R., Scevola, D., Schuster, M.W., Yeh, S.-S., Eds.; Springer: Milano, Italy, 2006; pp. 483–488. [Google Scholar]
- Tisdale, M.J. The ‘cancer cachectic factor’. Support. Care Cancer 2003, 11, 73–78. [Google Scholar] [CrossRef]
- Wyke, S.M.; Tisdale, M.J. NF-κB mediates proteolysis-inducing factor induced protein degradation and expression of the ubiquitin–proteasome system in skeletal muscle. Br. J. Cancer 2005, 92, 711–721. [Google Scholar] [CrossRef]
- Tickenbrock, L.; Müller-Tidow, C.; Berdel, W.E.; Serve, H. Emerging Flt3 kinase inhibitors in the treatment of leukaemia. Expert Opin. Emerg. Drugs 2006, 11, 153–165. [Google Scholar] [CrossRef]
- Vazquez-Ortiz, G.; Chisholm, C.; Xu, X.; Lahusen, T.J.; Li, C.; Sakamuru, S.; Huang, R.; Thomas, C.J.; Xia, M.; Deng, C. Drug repurposing screen identifies lestaurtinib amplifies the ability of the poly (ADP-ribose) polymerase 1 inhibitor AG14361 to kill breast cancer associated gene-1 mutant and wild type breast cancer cells. Breast Cancer Res. 2014, 16, R67. [Google Scholar] [CrossRef]
- Miller, S.C.; Huang, R.; Sakamuru, S.; Shukla, S.J.; Attene-Ramos, M.S.; Shinn, P.; Van Leer, D.; Leister, W.; Austin, C.P.; Xia, M. Identification of known drugs that act as inhibitors of NF-κB signaling and their mechanism of action. Biochem. Pharmacol. 2010, 79, 1272–1280. [Google Scholar] [CrossRef] [PubMed]
- Santos, F.P.S.; Kantarjian, H.M.; Jain, N.; Manshouri, T.; Thomas, D.A.; Garcia-Manero, G.; Kennedy, D.; Estrov, Z.; Cortes, J.; Verstovsek, S. Phase 2 study of CEP-701, an orally available JAK2 inhibitor, in patients with primary or post-polycythemia vera/essential thrombocythemia myelofibrosis. Blood 2010, 115, 1131–1136. [Google Scholar] [CrossRef]
- Wylie, B.; Macri, C.; Mintern, J.; Waithman, J. Dendritic Cells and Cancer: From Biology to Therapeutic Intervention. Cancers 2019, 11, 521. [Google Scholar] [CrossRef]
- Koprowska, K.; Czyz, M. Molecular mechanisms of parthenolide’s action: Old drug with a new face. Postep. Hig. Med. Dosw. (Online) 2010, 64, 100–114. [Google Scholar]
- Santos, J.M.O.; Moreira-Pais, A.; Neto, T.; Peixoto da Silva, S.; Oliveira, P.A.; Ferreira, R.; Mendes, J.; Bastos, M.; Lopes, C.; Casaca, F.; et al. Dimethylaminoparthenolide reduces the incidence of dysplasia and ameliorates a wasting syndrome in HPV16-transgenic mice. Drug Dev. Res. 2019, 80, 824–830. [Google Scholar] [CrossRef]
- Wang, R.; Bhat-Nakshatri, P.; Padua, M.B.; Prasad, M.S.; Anjanappa, M.; Jacobson, M.; Finnearty, C.; Sefcsik, V.; McElyea, K.; Redmond, R.; et al. Pharmacological Dual Inhibition of Tumor and Tumor-Induced Functional Limitations in a Transgenic Model of Breast Cancer. Mol. Cancer Ther. 2017, 16, 2747–2758. [Google Scholar] [CrossRef] [PubMed]
- Alfonso, L.; Ai, G.; Spitale, R.C.; Bhat, G.J. Molecular targets of aspirin and cancer prevention. Br. J. Cancer 2014, 111, 61–67. [Google Scholar] [CrossRef] [PubMed]
- D’Acquisto, F. Inhibition of Nuclear Factor Kappa B (NF-B): An Emerging Theme in Anti-Inflammatory Therapies. Mol. Interv. 2002, 2, 22–35. [Google Scholar] [CrossRef] [PubMed]
- Das, K.M. Sulfasalazine Therapy in Inflammatory Bowel Disease. Gastroenterol. Clin. N. Am. 1989, 18, 1–20. [Google Scholar] [CrossRef]
- Wahl, C.; Liptay, S.; Adler, G.; Schmid, R.M. Sulfasalazine: A potent and specific inhibitor of nuclear factor kappa B. J. Clin. Investig. 1998, 101, 1163–1174. [Google Scholar] [CrossRef]
- Plosker, G.L.; Croom, K.F. Sulfasalazine. Drugs 2005, 65, 1825–1849. [Google Scholar] [CrossRef]
- Smith, H.J.; Tisdale, M.J. Signal transduction pathways involved in proteolysis-inducing factor induced proteasome expression in murine myotubes. Br. J. Cancer 2003, 89, 1783–1788. [Google Scholar] [CrossRef]
- Gomes-Marcondes, M.C.C.; Smith, H.J.; Cooper, J.C.; Tisdale, M.J. Development of an in-vitro model system to investigate the mechanism of muscle protein catabolism induced by proteolysis-inducing factor. Br. J. Cancer 2002, 86, 1628–1633. [Google Scholar] [CrossRef]
- Shojima, K.; Sato, A.; Hanaki, H.; Tsujimoto, I.; Nakamura, M.; Hattori, K.; Sato, Y.; Dohi, K.; Hirata, M.; Yamamoto, H.; et al. Wnt5a promotes cancer cell invasion and proliferation by receptor-mediated endocytosis-dependent and -independent mechanisms, respectively. Sci. Rep. 2015, 5, 8042. [Google Scholar] [CrossRef] [PubMed]
- Johnson, M.; Gernsheimer, T.; Johansen, K. Essential thrombocytosis: Underemphasized cause of large-vessel thrombosis. J. Vasc. Surg. 1995, 22, 443–449. [Google Scholar] [CrossRef]
- Kajiguchi, T.; Kamoshita, S.; Ito, T.; Yagi, M.; Kimura, T. Drug-induced interstitial pneumonitis in essential thrombocythemia treated with anagrelide. Rinsho Ketsueki 2017, 58, 119–125. [Google Scholar]
- Goppelt-Struebe, M.; Wolter, D.; Resch, K. Glucocorticoids inhibit prostaglandin synthesis not only at the level of phospholipase A2 but also at the level of cyclo-oxygenase/PGE isomerase. Br. J. Pharmacol. 1989, 98, 1287–1295. [Google Scholar] [CrossRef]
- Briegel, J.; Kellermann, W.; Forst, H.; Haller, M.; Bittl, M.; Hoffmann, G.E.; Büchler, M.; Uhl, W.; Peter, K. Low-dose hydrocortisone infusion attenuates the systemic inflammatory response syndrome. J. Mol. Med. 1994, 72, 782–787. [Google Scholar] [CrossRef]
- Hengge, U.R.; Ruzicka, T.; Schwartz, R.A.; Cork, M. Adverse effects of topical glucocorticosteroids. J. Am. Acad. Dermatol. 2006, 54, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Wyke, S.M.; Khal, J.; Tisdale, M.J. Signalling pathways in the induction of proteasome expression by proteolysis-inducing factor in murine myotubes. Cell. Signal. 2005, 17, 67–75. [Google Scholar] [CrossRef] [PubMed]
- Tisdale, M.J. Mechanisms of Cancer Cachexia. Physiol. Rev. 2009, 89, 381–410. [Google Scholar] [CrossRef] [PubMed]
- Kortmansky, J.; Schwartz, G.K. Bryostatin-1: A Novel PKC Inhibitor in Clinical Development. Cancer Investig. 2003, 21, 924–936. [Google Scholar] [CrossRef] [PubMed]
- Madhusudan, S.; Protheroe, A.; Propper, D.; Han, C.; Corrie, P.; Earl, H.; Hancock, B.; Vasey, P.; Turner, A.; Balkwill, F.; et al. A multicentre phase II trial of bryostatin-1 in patients with advanced renal cancer. Br. J. Cancer 2003, 89, 1418–1422. [Google Scholar] [CrossRef]
- Lumachi, F.; Brunello, A.; Maruzzo, M.; Basso, U.; Basso, S.M. Treatment of Estrogen Receptor-Positive Breast Cancer. Curr. Med. Chem. 2013, 20, 596–604. [Google Scholar] [CrossRef]
- O’Brian, C.A.; Liskamp, R.; Solomon, D.H.; Weinstein, I.B. Inhibition of protein kinase C by tamoxifen. Cancer Res. 1985, 45, 2462–2465. [Google Scholar]
- Mourits, M.J.; de Vries, E.G.E.; Willemse, P.H.; Hoor, K.A.T.; Hollema, H.; van der Zee, A.G. Tamoxifen treatment and gynecologic side effects: A review. Obstet. Gynecol. 2001, 97, 855–866. [Google Scholar] [CrossRef]
- Tsoli, M.; Robertson, G. Cancer cachexia: Malignant inflammation, tumorkines, and metabolic mayhem. Trends Endocrinol. Metab. 2013, 24, 174–183. [Google Scholar] [CrossRef]
- Vaitkus, J.A.; Celi, F.S. The role of adipose tissue in cancer-associated cachexia. Exp. Biol. Med. 2016, 242, 473–481. [Google Scholar] [CrossRef]
- Deng, H.; Li, W. Monoacylglycerol lipase inhibitors: Modulators for lipid metabolism in cancer malignancy, neurological and metabolic disorders. Acta Pharm. Sin. B 2019, 10, 582–602. [Google Scholar] [CrossRef]
- Cisar, J.S.; Weber, O.D.; Clapper, J.R.; Blankman, J.L.; Henry, C.L.; Simon, G.M.; Alexander, J.P.; Jones, T.K.; Ezekowitz, R.A.B.; O’Neill, G.P.; et al. Identification of ABX-1431, a Selective Inhibitor of Monoacylglycerol Lipase and Clinical Candidate for Treatment of Neurological Disorders. J. Med. Chem. 2018, 61, 9062–9084. [Google Scholar] [CrossRef] [PubMed]
- Jiang, M.; van der Stelt, M. Activity-Based Protein Profiling Delivers Selective Drug Candidate ABX-1431, a Monoacylglycerol Lipase Inhibitor, To Control Lipid Metabolism in Neurological Disorders. J. Med. Chem. 2018, 61, 9059–9061. [Google Scholar] [CrossRef] [PubMed]
- Mannelli, M.; Gamberi, T.; Magherini, F.; Fiaschi, T. The Adipokines in Cancer Cachexia. Int. J. Mol. Sci. 2020, 21, 4860. [Google Scholar] [CrossRef]
- Das, S.K.; Hoefler, G. The role of triglyceride lipases in cancer associated cachexia. Trends Mol. Med. 2013, 19, 292–301. [Google Scholar] [CrossRef] [PubMed]
- Petrovic, N.; Walden, T.B.; Shabalina, I.G.; Timmons, J.A.; Cannon, B.; Nedergaard, J. Chronic Peroxisome Proliferator-activated Receptor γ (PPARγ) Activation of Epididymally Derived White Adipocyte Cultures Reveals a Population of Thermogenically Competent, UCP1-containing Adipocytes Molecularly Distinct from Classic Brown Adipocytes. J. Biol. Chem. 2010, 285, 7153–7164. [Google Scholar] [CrossRef]
- Hasan, A.U.; Ohmori, K.; Hashimoto, T.; Kamitori, K.; Yamaguchi, F.; Rahman, A.; Tokuda, M.; Kobori, H. PPARγ activation mitigates glucocorticoid receptor-induced excessive lipolysis in adipocytes via homeostatic crosstalk. J. Cell. Biochem. 2017, 119, 4627–4635. [Google Scholar] [CrossRef] [PubMed]
- You, W.; Xu, Z.; Sun, Y.; Valencak, T.G.; Wang, Y.; Shan, T. GADD45α drives brown adipose tissue formation through upregulating PPARγ in mice. Cell Death Dis. 2020, 11, 585. [Google Scholar] [CrossRef]
- Colson, C.; Batrow, P.-L.; Gautier, N.; Rochet, N.; Ailhaud, G.; Peiretti, F.; Amri, E.-Z. The Rosmarinus Bioactive Compound Carnosic Acid Is a Novel PPAR Antagonist That Inhibits the Browning of White Adipocytes. Cells 2020, 9, 2433. [Google Scholar] [CrossRef]
- Jin, J.; Miao, C.; Wang, Z.; Zhang, W.; Zhang, X.; Xie, X.; Lu, W. Design and synthesis of aryloxypropanolamine as β3-adrenergic receptor antagonist in cancer and lipolysis. Eur. J. Med. Chem. 2018, 150, 757–770. [Google Scholar] [CrossRef]
- Tabuchi, C.; Sul, H.S. Signaling Pathways Regulating Thermogenesis. Front. Endocrinol. 2021, 12. [Google Scholar] [CrossRef]
- Frishman, W.H. Beta-adrenergic receptor blockers. Adverse effects and drug interactions. Hypertension 1988, 11, II21-9. [Google Scholar] [CrossRef] [PubMed]
- Canová, N.; Lincová, D.; Farghali, H. Inconsistent role of nitric oxide on lipolysis in isolated rat adipocytes. Physiol. Res. 2004, 54. [Google Scholar]
- Canová, N.K.; Lincová, D.; Kmoníčková, E.; Kameníková, L.; Farghali, H. Nitric oxide production from rat adipocytes is modulated by β3-adrenergic receptor agonists and is involved in a cyclic AMP-dependent lipolysis in adipocytes. Nitric Oxide 2006, 14, 200–211. [Google Scholar] [CrossRef] [PubMed]
- Jeck, T.; Edmonds, D.; Mengden, T.; Schubert, M.; Renz, I.; Weisser, B.; Vetter, W. Betablocking drugs in essential hypertension: Transdermal bupranolol compared with oral metoprolol. Int. J. Clin. Pharmacol. Res. 1992, 12, 139–148. [Google Scholar]
- Dong, Y.; Ishikawa, H.; Wu, Y.; Yoshitomi, T. Vasodilatory mechanism of levobunolol on vascular smooth muscle cells. Exp. Eye Res. 2007, 84, 1039–1046. [Google Scholar] [CrossRef]
- Lin, L.; Wang, Y.; Chen, Y.; Liu, M. Bradyarrhythmias secondary to topical levobunolol hydrochloride solution. Clin. Interv. Aging 2014, 9, 1741–1745. [Google Scholar] [CrossRef]
- Donckier, J.E.; Massart, P.-E.; Van Mechelen, H.; Heyndrickx, G.R.; Gauthier, C.; Balligand, J.-L. Cardiovascular effects of beta 3-adrenoceptor stimulation in perinephritic hypertension. Eur. J. Clin. Investig. 2001, 31, 681–689. [Google Scholar] [CrossRef]
- Yang, W.; Wolter, N.E.; Cushing, S.L.; Pope, E.; Wolter, J.K.; Propst, E.J. Propranolol versus nadolol for treatment of pediatric subglottic hemangioma. Int. J. Pediatr. Otorhinolaryngol. 2021, 144, 110688. [Google Scholar] [CrossRef]
- Jia, Y.; Leung, S.-W. Drug efficacy in treating stable angina pectoris: A protocol for network meta-analysis of randomised controlled trials. BMJ Open 2014, 4, e005453. [Google Scholar] [CrossRef]
- McGillis, E.; Baumann, T.; Leroy, J. Death Associated with Nadolol for Infantile Hemangioma: A Case for Improving Safety. Pediatrics 2019, 145, e20191035. [Google Scholar] [CrossRef]
- Lantz, M.; Calissendorff, J.; Träisk, F.; Tallstedt, L.; Planck, T.; Törring, O.; Hallengren, B.; Åsman, P. Adjuvant Treatment of Graves’ Disease with Diclofenac: Safety, Effects on Ophthalmopathy and Antibody Concentrations. Eur. Thyroid. J. 2016, 5, 50–56. [Google Scholar] [CrossRef]
- Suffredini, A.F.; Fantuzzi, G.; Badolato, R.; Oppenheim, J.J.; O’Grady, N.P. New Insights into the Biology of the Acute Phase Response. J. Clin. Immunol. 1999, 19, 203–214. [Google Scholar] [CrossRef] [PubMed]
- Gruys, E.; Toussaint, M.; Niewold, T.; Koopmans, S. Acute phase reaction and acute phase proteins. J. Zhejiang Univ. A 2005, 6, 1045–1056. [Google Scholar] [CrossRef]
- Stephens, N.A.; Skipworth, R.J.; Fearon, K.C.H. Cachexia, survival and the acute phase response. Curr. Opin. Support. Palliat. Care 2008, 2, 267–274. [Google Scholar] [CrossRef] [PubMed]
- Barber, M.D.; Fearon, K.C.H.; McMillan, D.; Slater, C.; Ross, J.A.; Preston, T. Liver export protein synthetic rates are increased by oral meal feeding in weight-losing cancer patients. Am. J. Physiol. Metab. 2000, 279, E707–E714. [Google Scholar] [CrossRef] [PubMed]
- Moshage, H. Cytokines and the hepatic acute phase response. J. Pathol. 1997, 181, 257–266. [Google Scholar] [CrossRef]
- Sanford, D.; Luong, L.; Gabalski, A.; Oh, S.; Vu, J.P.; Pisegna, J.R.; Germano, P. An Intraperitoneal Treatment with Calcitonin Gene-Related Peptide (CGRP) Regulates Appetite, Energy Intake/Expenditure, and Metabolism. J. Mol. Neurosci. 2018, 67, 28–37. [Google Scholar] [CrossRef]
- Haruta, I.; Fuku, Y.; Kinoshita, K.; Yoneda, K.; Morinaga, A.; Amitani, M.; Amitani, H.; Asakawa, A.; Sugawara, H.; Takeda, Y.; et al. One-year intranasal application of growth hormone releasing peptide-2 improves body weight and hypoglycemia in a severely emaciated anorexia nervosa patient. J. Cachex Sarcopenia Muscle 2015, 6, 237–241. [Google Scholar] [CrossRef]
- Granado, M.; Priego, T.; Martín, A.I.; Villanúa, M.; López-Calderón, A. Anti-inflammatory effect of the ghrelin agonist growth hormone-releasing peptide-2 (GHRP-2) in arthritic rats. Am. J. Physiol. Metab. 2005, 288, E486–E492. [Google Scholar] [CrossRef]
- Xu, X.-B.; Pang, J.-J.; Cao, J.-M.; Ni, C.; Xu, R.-K.; Peng, X.-Z.; Yu, X.-X.; Guo, S.; Chen, M.-C.; Chen, C. GH-releasing peptides improve cardiac dysfunction and cachexia and suppress stress-related hormones and cardiomyocyte apoptosis in rats with heart failure. Am. J. Physiol. Circ. Physiol. 2005, 289, H1643–H1651. [Google Scholar] [CrossRef] [PubMed]
- Doi, N.; Hirotani, C.; Ukai, K.; Shimada, O.; Okuno, T.; Kurasaki, S.; Kiyofuji, T.; Ikegami, R.; Futamata, M.; Nakagawa, T.; et al. Pharmacological Characteristics of KP-102 (GHRP-2), a Potent Growth Hormone-Releasing Peptide. Arzneimittelforschung 2004, 54, 857–867. [Google Scholar] [CrossRef]
- Klaus, B.; Sachse, R.; Ammer, N.; Kelepouris, N.; Ostrow, V. Safety, tolerability, pharmacokinetics, and pharmacodynamics of macimorelin in healthy adults: Results of a single-dose, randomized controlled study. Growth Horm. IGF Res. 2020, 52, 101321. [Google Scholar] [CrossRef]
- Campbell, G.A.; Patrie, J.T.; Gaylinn, B.D.; Thorner, M.O.; Bolton, W.K. Oral ghrelin receptor agonist MK-0677 increases serum insulin-like growth factor 1 in hemodialysis patients: A randomized blinded study. Nephrol. Dial. Transplant. 2017, 33, 523–530. [Google Scholar] [CrossRef]
- Holubová, M.; Špolcová, A.; Demianova, Z.; Sykora, D.; Fehrentz, J.A.; Martinez, J.; Štofková, A.; Jurčovičová, J.; Drápalová, J.; Lacinová, Z.; et al. Ghrelin Agonist JMV 1843 Increases Food Intake, Body Weight and Expression of Orexigenic Neuropeptides in Mice. Physiol. Res. 2013, 62, 435–444. [Google Scholar] [CrossRef]
- Nass, R.; Pezzoli, S.S.; Oliveri, M.C.; Patrie, J.T.; Harrell, F.E.; Clasey, J.L.; Heymsfield, S.B.; Bach, M.A.; Vance, M.L.; Thorner, M.O. Effects of an Oral Ghrelin Mimetic on Body Composition and Clinical Outcomes in Healthy Older Adults. Ann. Intern. Med. 2008, 149, 601–611. [Google Scholar] [CrossRef]
- Lasseter, K.C.; Shaughnessy, L.; Cummings, D.; Pezzullo, J.C.; Wargin, W.; Gagnon, R.; Oliva, J.; Kosutic, G. Ghrelin Agonist (TZP-101): Safety, Pharmacokinetics and Pharmacodynamic Evaluation in Healthy Volunteers: A Phase I, First-in-Human Study. J. Clin. Pharmacol. 2008, 48, 193–202. [Google Scholar] [CrossRef] [PubMed]
- Nass, R.; Gaylinn, B.D.; Thorner, M.O. The ghrelin axis in disease: Potential therapeutic indications. Mol. Cell. Endocrinol. 2011, 340, 106–110. [Google Scholar] [CrossRef]
- Curtis, N.J.; Mooney, L.; Hopcroft, L.; Michopoulos, F.; Whalley, N.; Zhong, H.; Murray, C.; Logie, A.; Revill, M.; Byth, K.F.; et al. Pre-clinical pharmacology of AZD3965, a selective inhibitor of MCT1: DLBCL, NHL and Burkitt’s lymphoma anti-tumor activity. Oncotarget 2017, 8, 69219–69236. [Google Scholar] [CrossRef] [PubMed]
- Braga, M.; Kaliszczak, M.; Carroll, L.; Schug, Z.T.; Heinzmann, K.; Baxan, N.; Benito, A.; Valbuena, G.N.; Stribbling, S.; Beckley, A.; et al. Tracing Nutrient Flux Following Monocarboxylate Transporter-1 Inhibition with AZD3965. Cancers 2020, 12, 1703. [Google Scholar] [CrossRef]
- Noble, R.A.; Bell, N.; Blair, H.; Sikka, A.; Thomas, H.; Phillips, N.; Nakjang, S.; Miwa, S.; Crossland, R.; Rand, V.; et al. Inhibition of monocarboxyate transporter 1 by AZD3965 as a novel therapeutic approach for diffuse large B-cell lymphoma and Burkitt lymphoma. Haematologica 2017, 102, 1247–1257. [Google Scholar] [CrossRef] [PubMed]
- Halford, S.E.R.; Jones, P.; Wedge, S.; Hirschberg, S.; Katugampola, S.; Veal, G.; Payne, G.; Bacon, C.; Potter, S.; Griffin, M.; et al. A first-in-human first-in-class (FIC) trial of the monocarboxylate transporter 1 (MCT1) inhibitor AZD3965 in patients with advanced solid tumours. J. Clin. Oncol. 2017, 35, 2516. [Google Scholar] [CrossRef]
- Campos, C.A.; Bowen, A.; Han, S.; Wisse, B.E.; Palmiter, R.D.; Schwartz, M.W. Cancer-induced anorexia and malaise are mediated by CGRP neurons in the parabrachial nucleus. Nat. Neurosci. 2017, 20, 934–942. [Google Scholar] [CrossRef] [PubMed]
- Gao, B.; Yang, Y.; Wang, Z.; Sun, Y.; Chen, Z.; Zhu, Y.; Wang, Z. Efficacy and Safety of Rimegepant for the Acute Treatment of Migraine: Evidence from Randomized Controlled Trials. Front. Pharmacol. 2020, 10, 1577. [Google Scholar] [CrossRef]
- Zhang, Z.; Shu, Y.; Diao, Y.; Du, Y.; Chen, L.; Liu, Y.; Du, B. Calcitonin gene-related peptide receptor antagonist ubrogepant for the treatment of acute migraine. Medicine 2021, 100, e24741. [Google Scholar] [CrossRef]
- Boinpally, R.; Jakate, A.; Butler, M.; Borbridge, L.; Periclou, A. Single-Dose Pharmacokinetics and Safety of Atogepant in Adults with Hepatic Impairment: Results from an Open-Label, Phase 1 Trial. Clin. Pharmacol. Drug Dev. 2021, 10, 726–733. [Google Scholar] [CrossRef] [PubMed]
- Ajona, D.M.; Pérez-Rodríguez, A.; Goadsby, P.J. Gepants, calcitonin-gene-related peptide receptor antagonists: What could be their role in migraine treatment? Curr. Opin. Neurol. 2020, 33, 309–315. [Google Scholar] [CrossRef] [PubMed]
- Grossberg, A.J.; Scarlett, J.M.; Marks, D.L. Hypothalamic mechanisms in cachexia. Physiol. Behav. 2010, 100, 478–489. [Google Scholar] [CrossRef]
- DeBoer, M.D. Update on melanocortin interventions for cachexia: Progress toward clinical application. Nutrition 2010, 26, 146–151. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, M.W.; Woods, S.C.; Porte, D.; Seeley, R.; Baskin, D.G. Central nervous system control of food intake. Nature 2000, 404, 661–671. [Google Scholar] [CrossRef] [PubMed]
- Laviano, A.; Seelaender, M.; Rianda, S.; Silvério, R.; Fanelli, F.R. Neuroinflammation: A contributing factor to the pathogenesis of cancer cachexia. Crit. Rev. Oncog. 2012, 17, 247–252. [Google Scholar] [CrossRef]
- Marks, D.L.; Ling, N.; Cone, R.D. Role of the central melanocortin system in cachexia. Cancer Res. 2001, 61, 1432–1438. [Google Scholar]
- Laviano, A.; Inui, A.; Marks, D.L.; Meguid, M.M.; Pichard, C.; Fanelli, F.R.; Seelaender, M. Neural control of the anorexia-cachexia syndrome. Am. J. Physiol. Metab. 2008, 295, E1000–E1008. [Google Scholar] [CrossRef]
- Tecott, L.H. Serotonin and the Orchestration of Energy Balance. Cell Metab. 2007, 6, 352–361. [Google Scholar] [CrossRef] [PubMed]
- Cangiano, C.; Cascino, A.; Ceci, F.; Laviano, A.; Mulieri, M.; Muscaritoli, M.; Rossi-Fanelli, F. Plasma and CSF tryptophan in cancer anorexia. J. Neural Transm. 1990, 81, 225–233. [Google Scholar] [CrossRef]
- Heisler, L.K.; Jobst, E.E.; Sutton, G.M.; Zhou, L.; Borok, E.; Thornton-Jones, Z.; Liu, H.Y.; Zigman, J.M.; Balthasar, N.; Kishi, T.; et al. Serotonin Reciprocally Regulates Melanocortin Neurons to Modulate Food Intake. Neuron 2006, 51, 239–249. [Google Scholar] [CrossRef] [PubMed]
- Mylecharane, E.J. 5-HT2 receptor antagonists and migraine therapy. J. Neurol. 1991, 238, S45–S52. [Google Scholar] [CrossRef] [PubMed]
- Aronson, J.K. (Ed.) Pizotifen. In Meyler’s Side Effects of Drugs, 16th ed.; Elsevier: Oxford, UK, 2016; p. 801. [Google Scholar]
- Marcoli, M.; Maura, G.; Tortarolo, M.; Raiteri, M. Trazodone is a potent agonist at 5-HT2C receptors mediating inhibition of the N-methyl-D-aspartate/nitric oxide/cyclic GMP pathway in rat cerebellum. J. Pharmacol. Exp. Ther. 1998, 285, 983–986. [Google Scholar]
- Bossini, L.; Coluccia, A.; Casolaro, I.; Benbow, J.; Amodeo, G.; de Giorgi, R.; Fagiolini, A. Off-Label Trazodone Prescription: Evidence, Benefits and Risks. Curr. Pharm. Des. 2015, 21, 3343–3351. [Google Scholar] [CrossRef]
- Bossini, L.; Casolaro, I.; Koukouna, D.; Cecchini, F.; Fagiolini, A. Off-label uses of trazodone: A review. Expert Opin. Pharmacother. 2012, 13, 1707–1717. [Google Scholar] [CrossRef] [PubMed]
- Stahl, S.M.; Shayegan, D.K. The psychopharmacology of ziprasidone: Receptor-binding properties and real-world psychiatric practice. J. Clin. Psychiatry 2003, 64, 6–12. [Google Scholar] [PubMed]
- Herrick-Davis, K.; Grinde, E.; Teitler, M. Inverse agonist activity of atypical antipsychotic drugs at human 5-hydroxytryptamine2C receptors. J. Pharmacol. Exp. Ther. 2000, 295, 226–232. [Google Scholar]
- Khokhar, J.Y.; Henricks, A.M.; Sullivan, E.D.; Green, A.I. Unique Effects of Clozapine: A Pharmacological Perspective. Adv. Pharmacol. 2018, 82, 137–162. [Google Scholar] [CrossRef] [PubMed]
- Kirk, S.L.; Glazebrook, J.; Grayson, B.; Neill, J.C.; Reynolds, G.P. Olanzapine-induced weight gain in the rat: Role of 5-HT2C and histamine H1 receptors. Psychopharmacology 2009, 207, 119–125. [Google Scholar] [CrossRef]
- Bindels, L.B.; Neyrinck, A.; Loumaye, A.; Catry, E.; Walgrave, H.; Cherbuy, C.; Leclercq, S.; van Hul, M.; Plovier, H.; Pachikian, B.; et al. Increased gut permeability in cancer cachexia: Mechanisms and clinical relevance. Oncotarget 2018, 9, 18224–18238. [Google Scholar] [CrossRef]
- Klein, G.L.; Petschow, B.W.; Shaw, A.L.; Weaver, E. Gut barrier dysfunction and microbial translocation in cancer cachexia. Curr. Opin. Support. Palliat. Care 2013, 7, 361–367. [Google Scholar] [CrossRef]
- Peng, S.-N. Effects of rhein on intestinal epithelial tight junction in IgA nephropathy. World J. Gastroenterol. 2013, 19, 4137–4145. [Google Scholar] [CrossRef] [PubMed]
- König, J.; Wells, J.; Cani, P.; Garcia-Rodenas, C.L.; Macdonald, T.; Mercenier, A.; Whyte, J.; Troost, F.; Brummer, R.-J. Human Intestinal Barrier Function in Health and Disease. Clin. Transl. Gastroenterol. 2016, 7, e196. [Google Scholar] [CrossRef]
- Fasano, A. Zonulin, regulation of tight junctions, and autoimmune diseases. Ann. N. Y. Acad. Sci. 2012, 1258, 25–33. [Google Scholar] [CrossRef]
- Enomoto, H.; Yeatts, J.; Carbajal, L.; Krishnan, B.R.; Madan, J.P.; Laumas, S.; Blikslager, A.T.; Messenger, K.M. In vivo assessment of a delayed release formulation of larazotide acetate indicated for celiac disease using a porcine model. PLoS ONE 2021, 16, e0249179. [Google Scholar] [CrossRef]
- Troisi, J.; Venutolo, G.; Terracciano, C.; Carri, M.D.; Di Micco, S.; Landolfi, A.; Fasano, A. The Therapeutic use of the Zonulin Inhibitor AT-1001 (Larazotide) for a Variety of Acute and Chronic Inflammatory Diseases. Curr. Med. Chem. 2021, 28, 5788–5807. [Google Scholar] [CrossRef] [PubMed]
- Leffler, D.A.; Kelly, C.P.; Abdallah, H.Z.; Colatrella, A.M.; Harris, L.A.; Leon, F.; Arterburn, L.A.; Paterson, B.M.; Lan, Z.H.; Murray, J. A Randomized, Double-Blind Study of Larazotide Acetate to Prevent the Activation of Celiac Disease During Gluten Challenge. Am. J. Gastroenterol. 2012, 107, 1554–1562. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Liang, R.; Zhang, W.; Tian, K.; Li, J.; Chen, X.; Yu, T.; Chen, Q. Faecalibacterium prausnitzii—derived microbial anti-inflammatory molecule regulates intestinal integrity in diabetes mellitus mice via modulating tight junction protein expression. J. Diabetes 2019, 12, 224–236. [Google Scholar] [CrossRef] [PubMed]
- Fischer, A.; Gluth, M.; Pape, U.-F.; Wiedenmann, B.; Theuring, F.; Baumgart, D.C. Adalimumab prevents barrier dysfunction and antagonizes distinct effects of TNF-α on tight junction proteins and signaling pathways in intestinal epithelial cells. Am. J. Physiol. Liver Physiol. 2013, 304, G970–G979. [Google Scholar] [CrossRef] [PubMed]
- Pavelka, K.; Bruyère, O.; Cooper, C.; Kanis, J.A.; Leeb, B.F.; Maheu, E.; Martel-Pelletier, J.; Monfort, J.; Pelletier, J.-P.; Rizzoli, R.; et al. Diacerein: Benefits, Risks and Place in the Management of Osteoarthritis. An Opinion-Based Report from the ESCEO. Drugs Aging 2016, 33, 75–85. [Google Scholar] [CrossRef]
- Louthrenoo, W.; Nilganuwong, S.; Nanagara, R.; Siripaitoon, B.; Basset, S.C. Diacerein for the treatment of rheumatoid arthritis in patients with inadequate response to methotrexate: A pilot randomized, double-blind, placebo-controlled add-on trial. Clin. Rheumatol. 2019, 38, 2461–2471. [Google Scholar] [CrossRef]
- Nowrouzi-Sohrabi, P.; Tabrizi, R.; Jalali, M.; Jamali, N.; Rezaei, S.; Hessami, K.; Keshavarz, P.; Shahabi, S.; Kolahi, A.-A.; Carson-Chahhoud, K.; et al. Effects of Diacerein Intake on Cardiometabolic Profiles in Type 2 Diabetics: A Systematic Review and Meta-Analysis of Clinical Trials. Curr. Med. Chem. 2021, 28, 840–852. [Google Scholar] [CrossRef]
- Zhuang, S.; Zhong, J.; Zhou, Q.; Zhong, Y.; Liu, P.; Liu, Z. Rhein protects against barrier disruption and inhibits inflammation in intestinal epithelial cells. Int. Immunopharmacol. 2019, 71, 321–327. [Google Scholar] [CrossRef]
- Nishii, N.; Oshima, T.; Li, M.; Eda, H.; Nakamura, K.; Tamura, A.; Ogawa, T.; Yamasaki, T.; Kondo, T.; Kono, T.; et al. Lubiprostone Induces Claudin-1 and Protects Intestinal Barrier Function. Pharmacology 2019, 105, 102–108. [Google Scholar] [CrossRef]
- Eguchi, T.; Yoshizaki, T.; Takagi, M.; Ikeoka, S.; Hashimura, H.; Okamoto, N.; Matsumoto, M.; Matsuda, T.; Miura, T.; Momose, K.; et al. Risk Factors for Adverse Events in Patients with Chronic Constipation Following Lubiprostone Administration. Dig. Dis. 2020, 39, 10–15. [Google Scholar] [CrossRef]
- Masoudi-Sobhanzadeh, Y.; Omidi, Y.; Amanlou, M.; Masoudi-Nejad, A. Drug databases and their contributions to drug repurposing. Genomics 2019, 112, 1087–1095. [Google Scholar] [CrossRef]
- Pawson, A.J.; Sharman, J.; Benson, H.; Faccenda, E.; Alexander, S.; Buneman, O.P.; Davenport, A.P.; McGrath, J.C.; Peters, J.A.; Southan, C.; et al. The IUPHAR/BPS Guide to pharmacology: An expert-driven knowledgebase of drug targets and their ligands. Nucleic Acids Res. 2013, 42, D1098–D1106. [Google Scholar] [CrossRef]
- Wishart, D.S. DrugBank: A comprehensive resource for in silico drug discovery and exploration. Nucleic Acids Res. 2006, 34, D668–D672. [Google Scholar] [CrossRef]
- Tanoli, Z.; Seemab, U.; Scherer, A.; Wennerberg, K.; Tang, J.; Vähä-Koskela, M. Exploration of databases and methods supporting drug repurposing: A comprehensive survey. Brief. Bioinform. 2020, 22, 1656–1678. [Google Scholar] [CrossRef] [PubMed]
- Schirrmacher, V. Mitochondria at Work: New Insights into Regulation and Dysregulation of Cellular Energy Supply and Metabolism. Biomedicines 2020, 8, 526. [Google Scholar] [CrossRef] [PubMed]
- Kitaoka, Y.; Miyazaki, M.; Kikuchi, S. Voluntary exercise prevents abnormal muscle mitochondrial morphology in cancer cachexia mice. Physiol. Rep. 2021, 9, e15016. [Google Scholar] [CrossRef]
- Antunes, D.; Padrão, A.I.; Maciel, E.; Santinha, D.; Oliveira, P.; Vitorino, R.; Gonçalves, D.; Colaço, B.J.A.; Pires, M.J.; Nunes, C.; et al. Molecular insights into mitochondrial dysfunction in cancer-related muscle wasting. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2014, 1841, 896–905. [Google Scholar] [CrossRef] [PubMed]
- Julienne, C.M.; Dumas, J.-F.; Goupille, C.; Pinault, M.; Berri, C.; Collin, A.; Tesseraud, S.; Couet, C.; Servais, S. Cancer cachexia is associated with a decrease in skeletal muscle mitochondrial oxidative capacities without alteration of ATP production efficiency. J. Cachex Sarcopenia Muscle 2012, 3, 265–275. [Google Scholar] [CrossRef] [PubMed]
- Argiles, J.M.; Fontes-Oliveira, C.C.; Toledo, M.; López-Soriano, F.J.; Busquets, S. Cachexia: A problem of energetic inefficiency. J. Cachex Sarcopenia Muscle 2014, 5, 279–286. [Google Scholar] [CrossRef]
- Vanderveen, B.N.; Fix, D.K.; Carson, J.A. Disrupted Skeletal Muscle Mitochondrial Dynamics, Mitophagy, and Biogenesis during Cancer Cachexia: A Role for Inflammation. Oxidative Med. Cell. Longev. 2017, 2017, 3292087. [Google Scholar] [CrossRef]
- Pulley, J.M.; Rhoads, J.P.; Jerome, R.N.; Challa, A.; Erreger, K.B.; Joly, M.M.; Lavieri, R.R.; Perry, K.E.; Zaleski, N.M.; Shirey-Rice, J.K.; et al. Using What We Already Have: Uncovering New Drug Repurposing Strategies in Existing Omics Data. Annu. Rev. Pharmacol. Toxicol. 2020, 60, 333–352. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| Cachexia Phenotypes | Molecular Pathways and Components |
|---|---|
| Inflammation | Increased levels of tumor necrosis factor alpha (TNF-α), interleukin (IL)-6, IL-1, interferon gamma (IFN-γ), and IL-8 |
| Skeletal and cardiac muscle wasting | Up-regulation of the ubiquitin-mediated proteasome degradation system (UPS) |
| Autophagy | |
| Calcium-activated protease calpains | |
| Low circulating levels of insulin-like growth factor 1 (IGF-1) | |
| Insulin resistance | |
| Myostatin | |
| Proteolysis-inducing factor (PIF) | |
| Impaired mitochondrial metabolism | |
| Adipose tissue depletion | Lipolysis |
| Inhibition of lipogenesis | |
| Browning | |
| Hepatic metabolic changes | Acute-phase response |
| Altered energy balance | Tumor metabolism and inflammation might increase resting energy expenditure and simultaneously decrease energy intake (anorexia), shifting the scale towards negative energy balance |
| Central neuroinflammation | Inflammatory cytokines bind to receptors on hypothalamic neuronal populations, triggering an acute illness response, leading to anorexia, weight loss, skeletal muscle-protein catabolism, and lipolysis. Neuropeptide Y (NPY), melanocortins, and serotonin involved. |
| Gastrointestinal tract malfunction | Impaired barrier function and malabsorption |
| Phenotype | Molecular Pathways and Components | Drugs |
|---|---|---|
| Inflammation | TNF-α | Adalimumab |
| Ozoralizumab | ||
| Golimumab | ||
| Certolizumab pegol | ||
| Remtolumab | ||
| Chloroquine | ||
| Amrinone | ||
| Pomalidomide | ||
| Glycyrrhizic acid | ||
| IL-6 | Sirukumab | |
| Olamkicept | ||
| Vobarilizumab | ||
| Satralizumab | ||
| IL-1 | Lutikizumab | |
| Gevokizumab | ||
| Canakinumab | ||
| Rilonacept | ||
| Isunakinra | ||
| Anakinra | ||
| IL-8 | AZD5069 | |
| Reparixin | ||
| Elubirixin | ||
| IFN-γ | Fontolizumab | |
| Glucosamine |
| Phenotype | Molecular Pathways and Components | Drugs |
|---|---|---|
| Skeletal and cardiac muscle wasting | UPS | Pomalidomide |
| Iberdomide | ||
| Bortezomib | ||
| Carfilzomib | ||
| Ixazomib | ||
| Oprozomib | ||
| VLX1570 | ||
| KZR-616 | ||
| Autophagy | Fedratinib | |
| Critzotinib | ||
| Fostamatinib | ||
| Calcium-activated protease calpains | Aloxistatin | |
| Alicapistat | ||
| Insulin resistance | Trodusquemine | |
| PIF | Lestaurtinib | |
| Parthelonide | ||
| Acetylsalicylic acid | ||
| Sulfasalazine | ||
| Anagrelide | ||
| Varespladib methyl | ||
| Darapladib | ||
| AK 106-001616 | ||
| Budesonide | ||
| Hydrocortisone | ||
| Bryostatin 1 | ||
| Tamoxifen |
| Phenotype | Molecular Pathways and Components | Drugs |
|---|---|---|
| Adipose tissue depletion | Lipolysis | ABX-1431 |
| Inhibition of lipogenesis | Glycyrrhizic acid | |
| WAT browning | Brupanolol | |
| Levobunolol | ||
| Nadolol | ||
| Diclofenac |
| Phenotype | Molecular Pathways and Components | Drugs |
|---|---|---|
| Altered energy balance | Ghrelin | Pralmorelin |
| Macimorelin | ||
| Ibutamoren | ||
| Ulimorelin | ||
| MCT1 | AZD3965 | |
| CGRP receptor | Rimegepant | |
| Ubrogepant | ||
| Telcagepant | ||
| Atogepant |
| Phenotype | Molecular Pathways and Components | Drugs |
|---|---|---|
| Neuroinflammation | Pizotifen | |
| Trazodone | ||
| Serotonin | Ziprazidone | |
| Clozapine | ||
| Olanzapine | ||
| Sertindole |
| Phenotype | Molecular Pathways and Components | Drugs |
|---|---|---|
| Gastrointestinal tract: Impaired barrier function | Zonulin | Larazotide |
| ZO-1 and claudins | Diacerein | |
| Lubiprostone |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Santos, J.M.O.; Costa, A.C.; Dias, T.R.; Satari, S.; Costa e Silva, M.P.; da Costa, R.M.G.; Medeiros, R. Towards Drug Repurposing in Cancer Cachexia: Potential Targets and Candidates. Pharmaceuticals 2021, 14, 1084. https://doi.org/10.3390/ph14111084
Santos JMO, Costa AC, Dias TR, Satari S, Costa e Silva MP, da Costa RMG, Medeiros R. Towards Drug Repurposing in Cancer Cachexia: Potential Targets and Candidates. Pharmaceuticals. 2021; 14(11):1084. https://doi.org/10.3390/ph14111084
Chicago/Turabian StyleSantos, Joana M. O., Alexandra C. Costa, Tânia R. Dias, Setareh Satari, Maria Paula Costa e Silva, Rui M. Gil da Costa, and Rui Medeiros. 2021. "Towards Drug Repurposing in Cancer Cachexia: Potential Targets and Candidates" Pharmaceuticals 14, no. 11: 1084. https://doi.org/10.3390/ph14111084
APA StyleSantos, J. M. O., Costa, A. C., Dias, T. R., Satari, S., Costa e Silva, M. P., da Costa, R. M. G., & Medeiros, R. (2021). Towards Drug Repurposing in Cancer Cachexia: Potential Targets and Candidates. Pharmaceuticals, 14(11), 1084. https://doi.org/10.3390/ph14111084