
Prerequisite Binding Modes Determine the Dynamics of Action of Covalent Agonists of Ion Channel TRPA1



Abstract
:1. Introduction
2. Results and Discussion
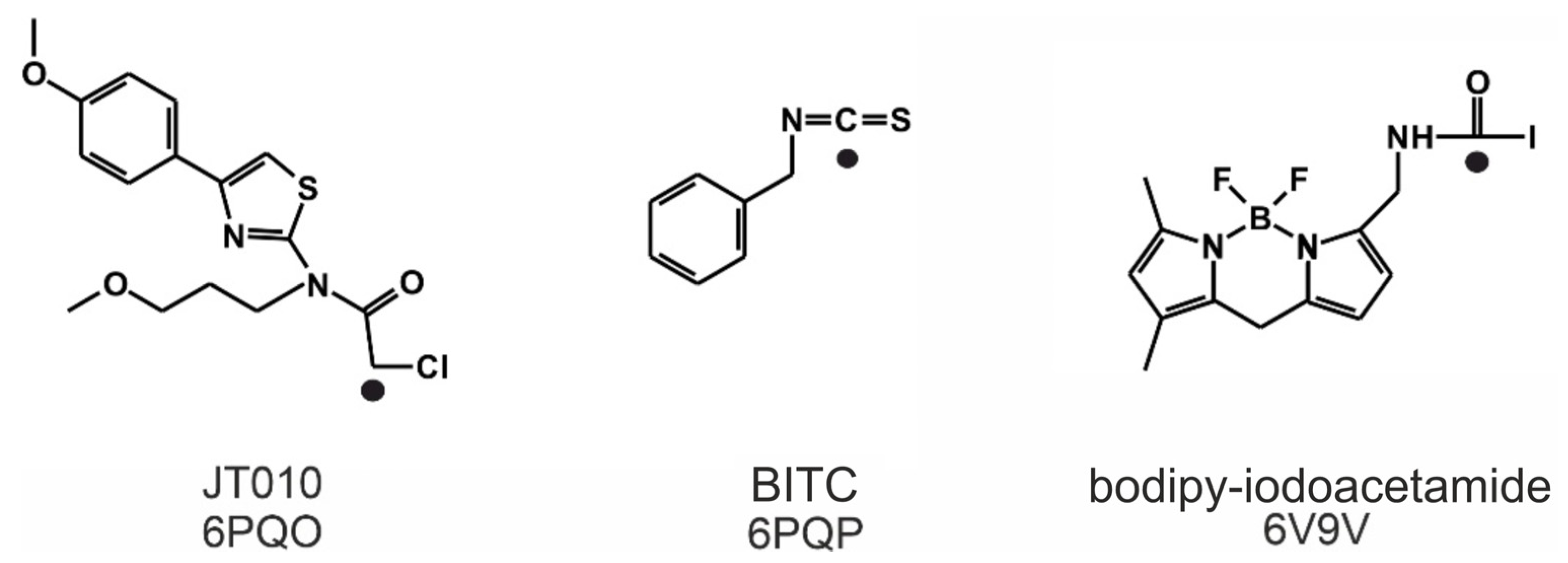
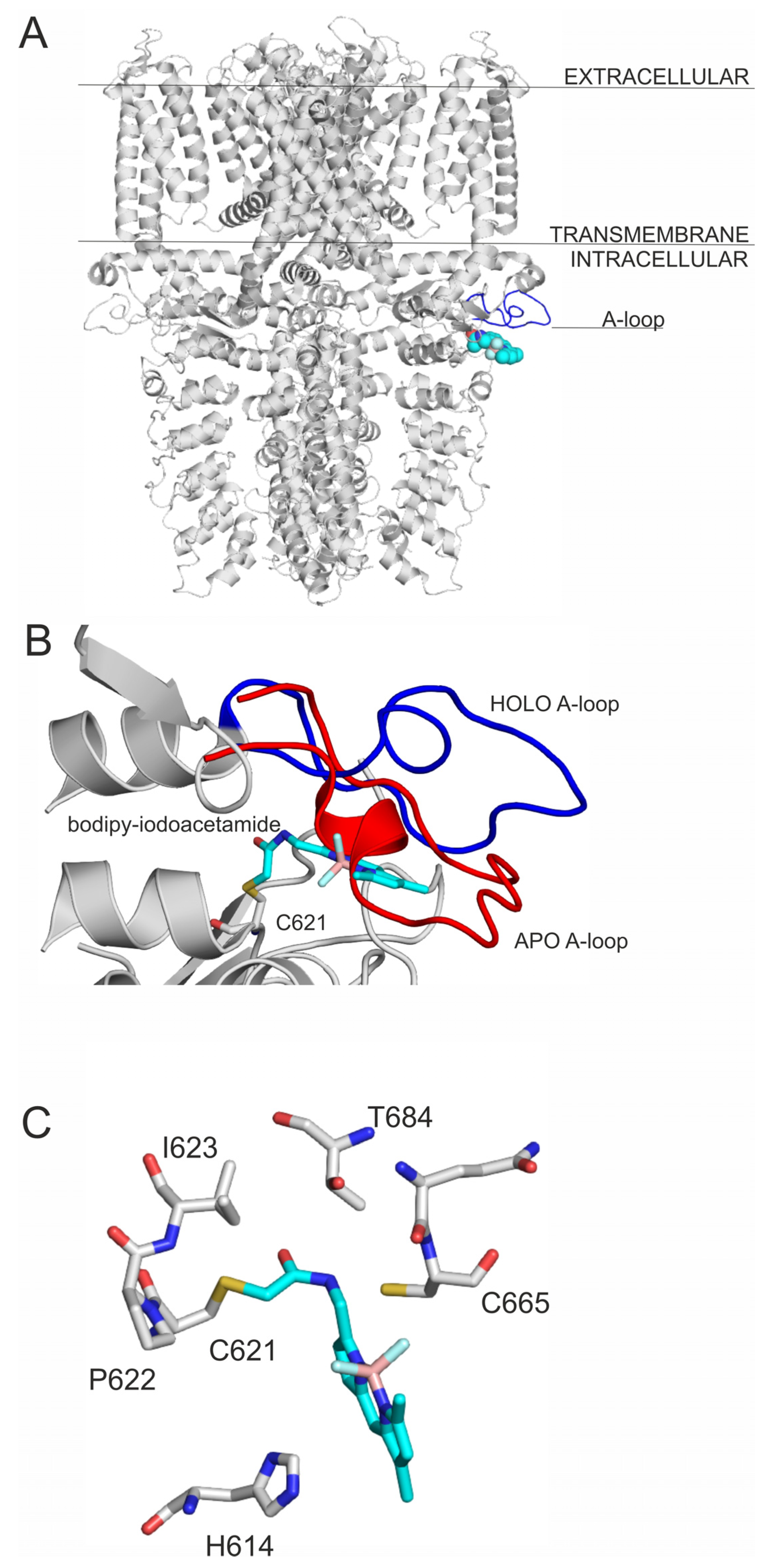
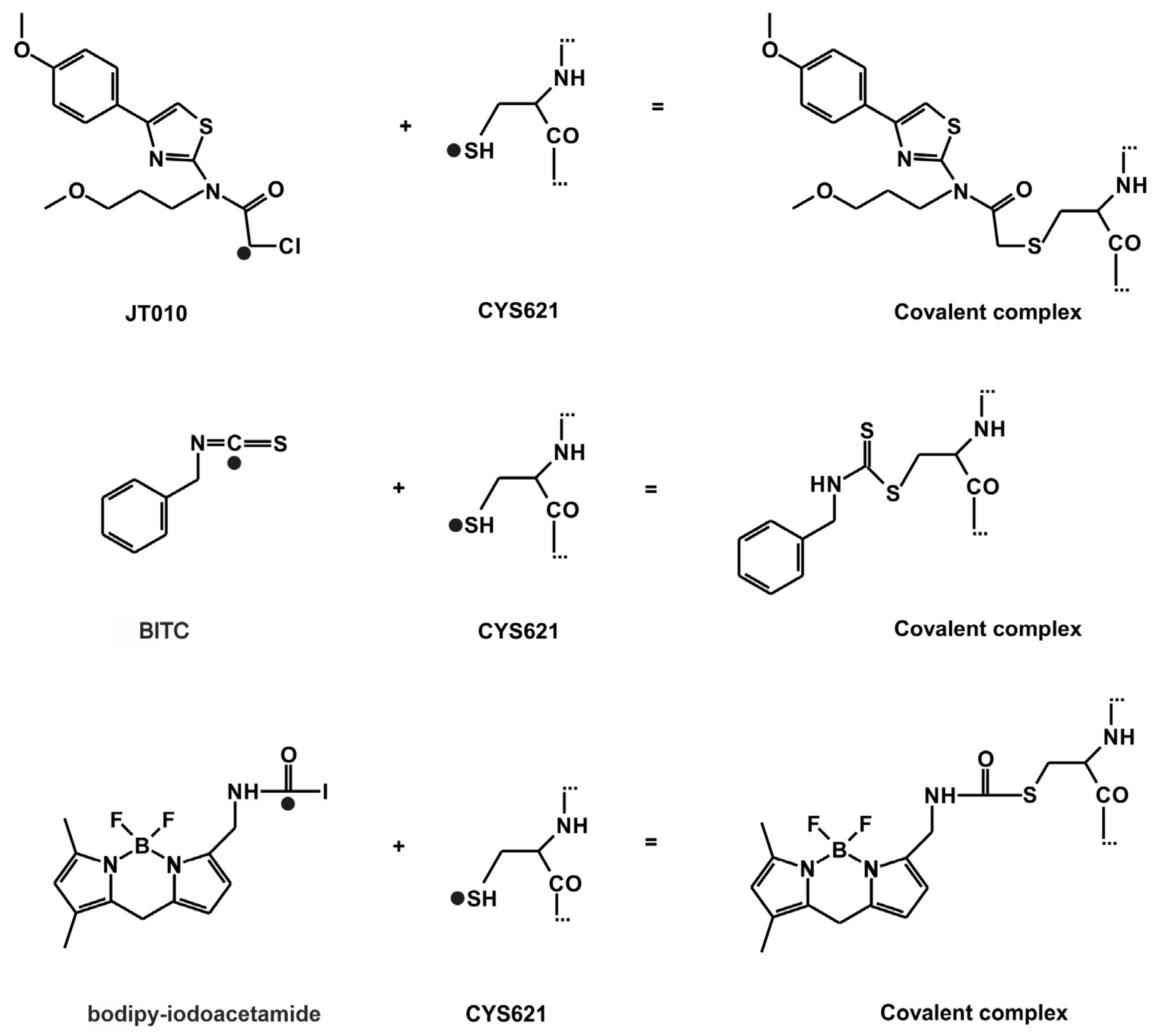
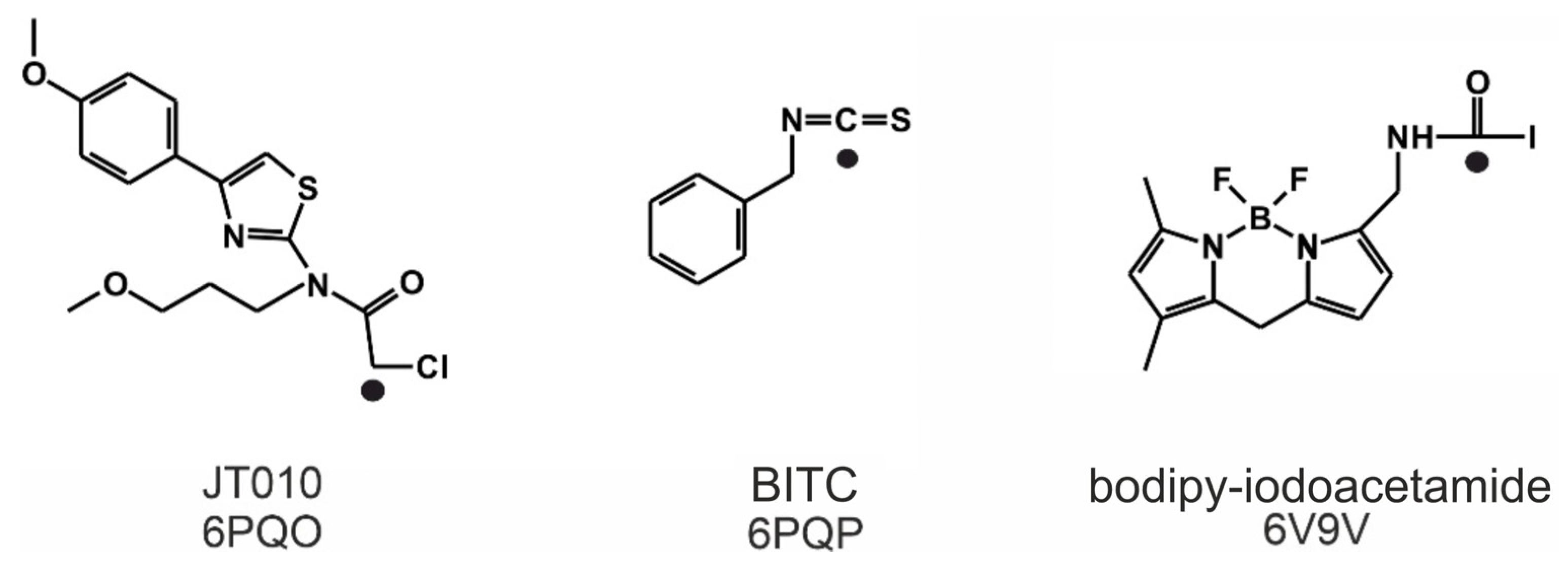
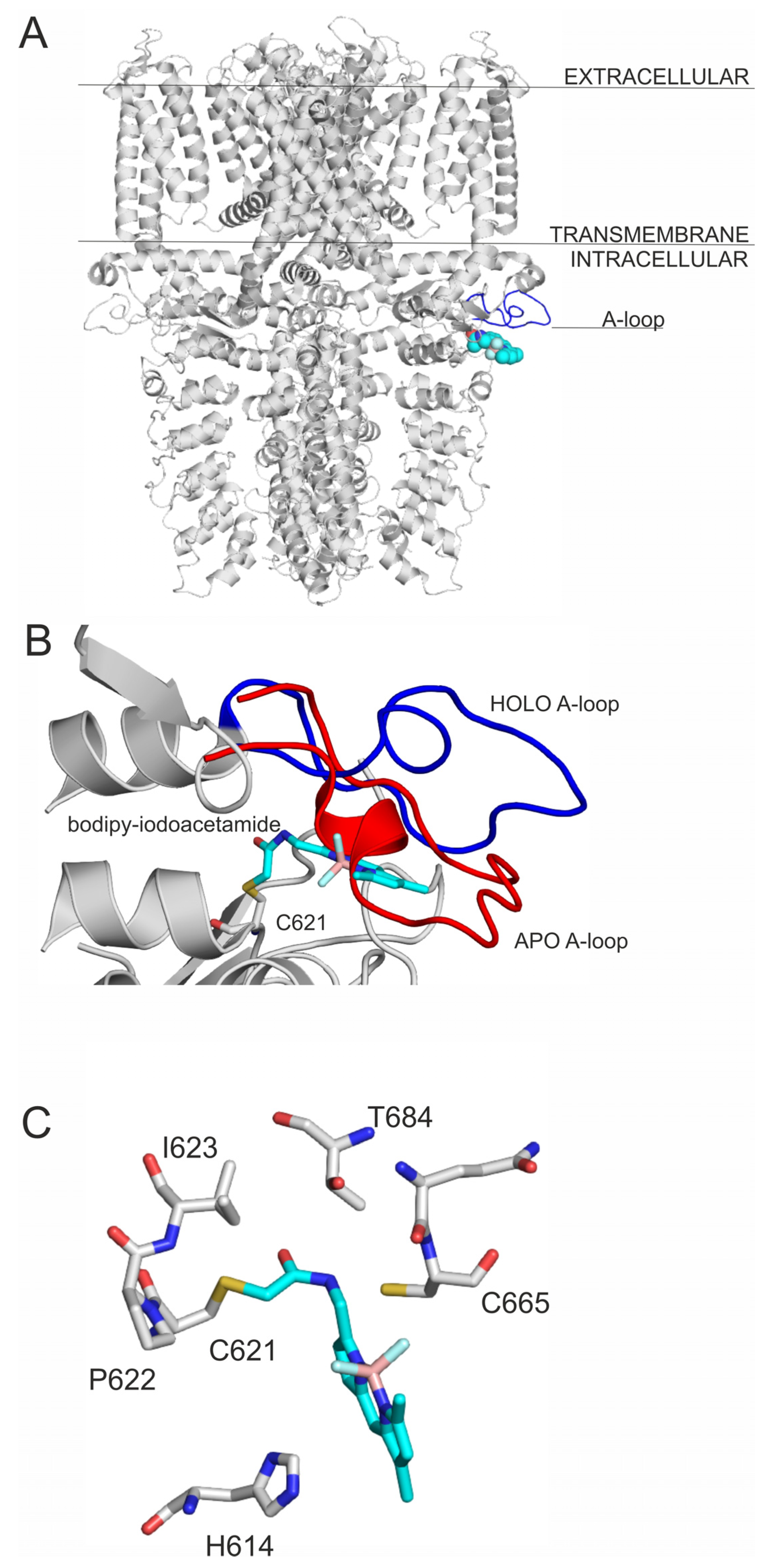
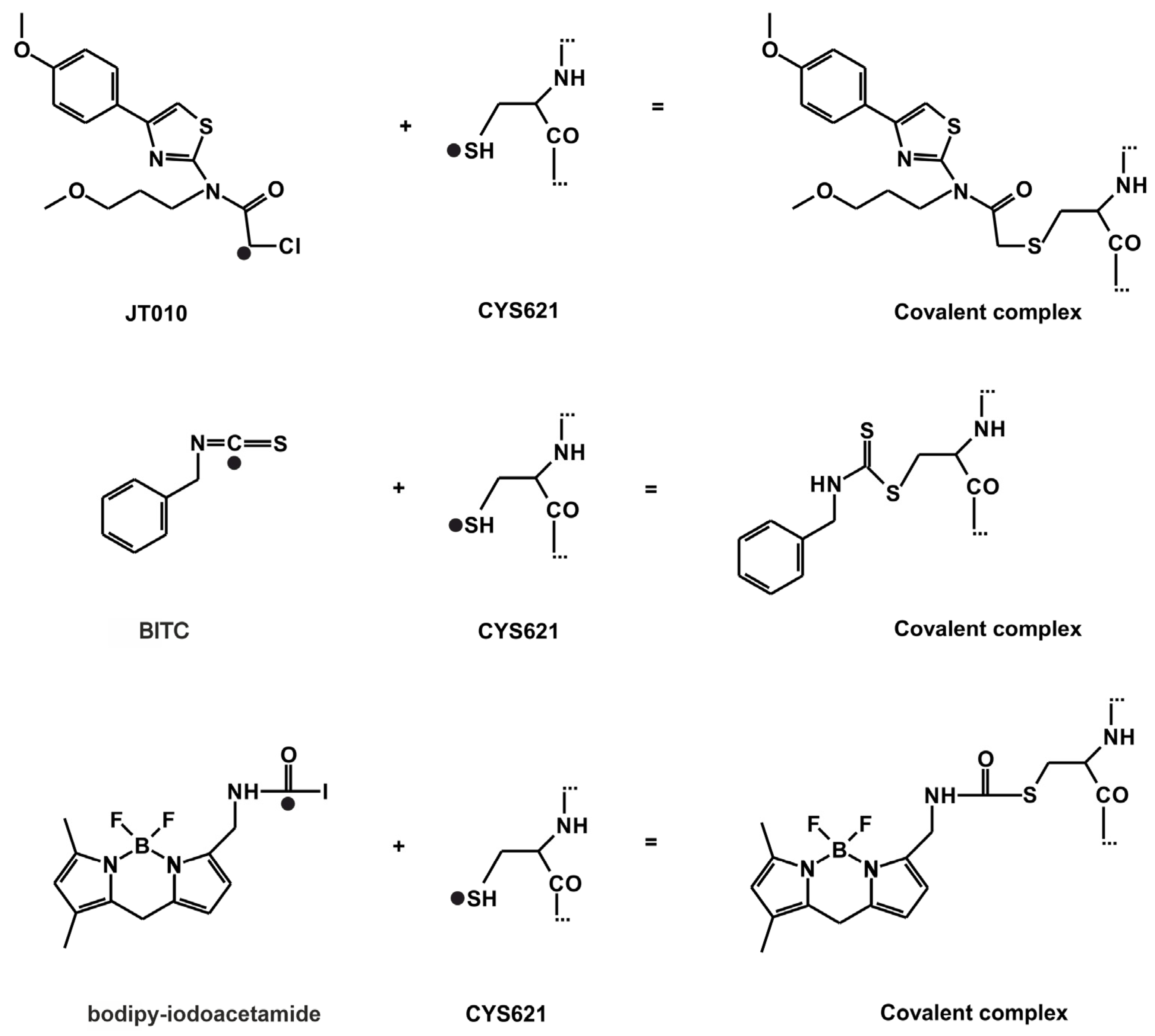
2.1. Final Covalent Binding Modes
2.2. Prerequisite Binding Modes
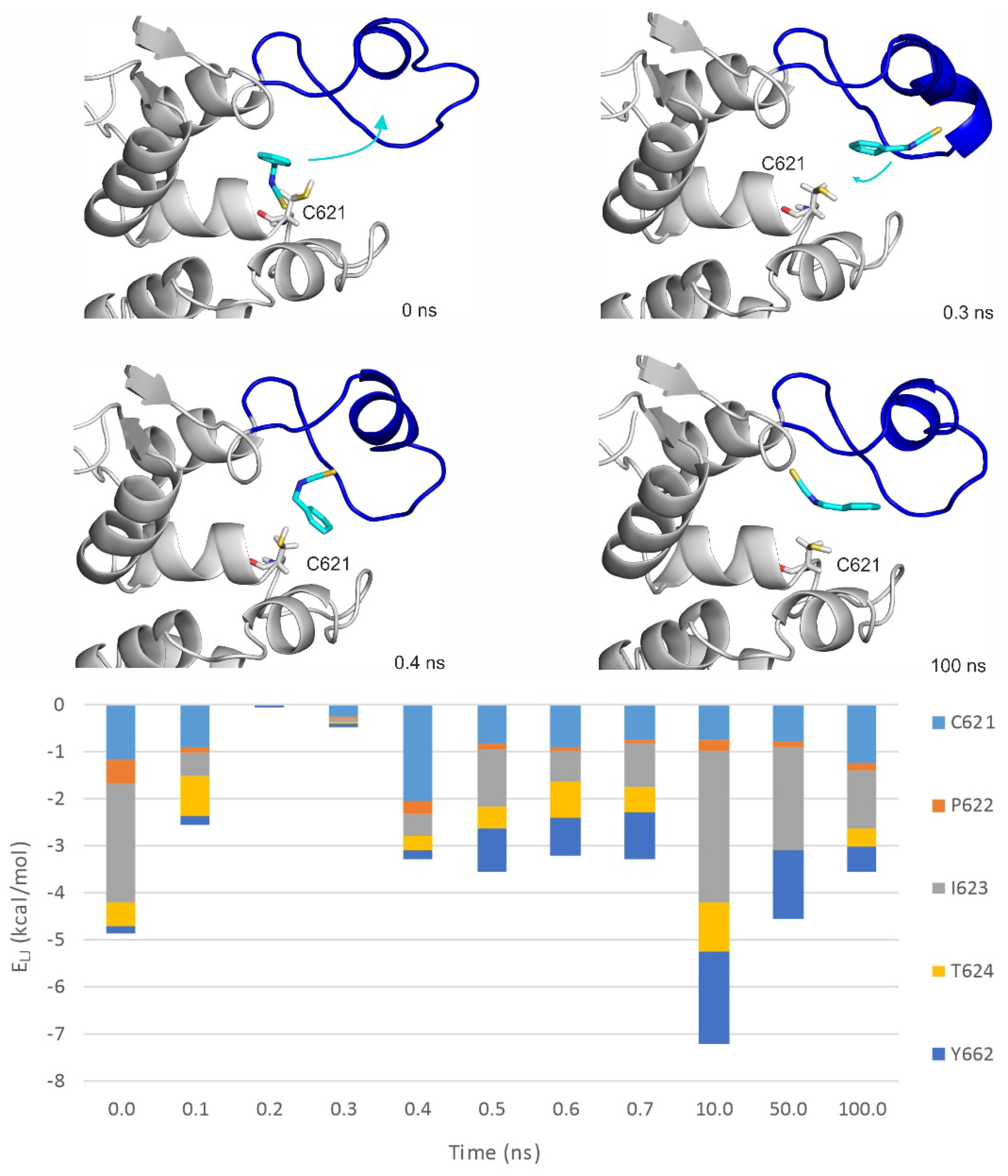
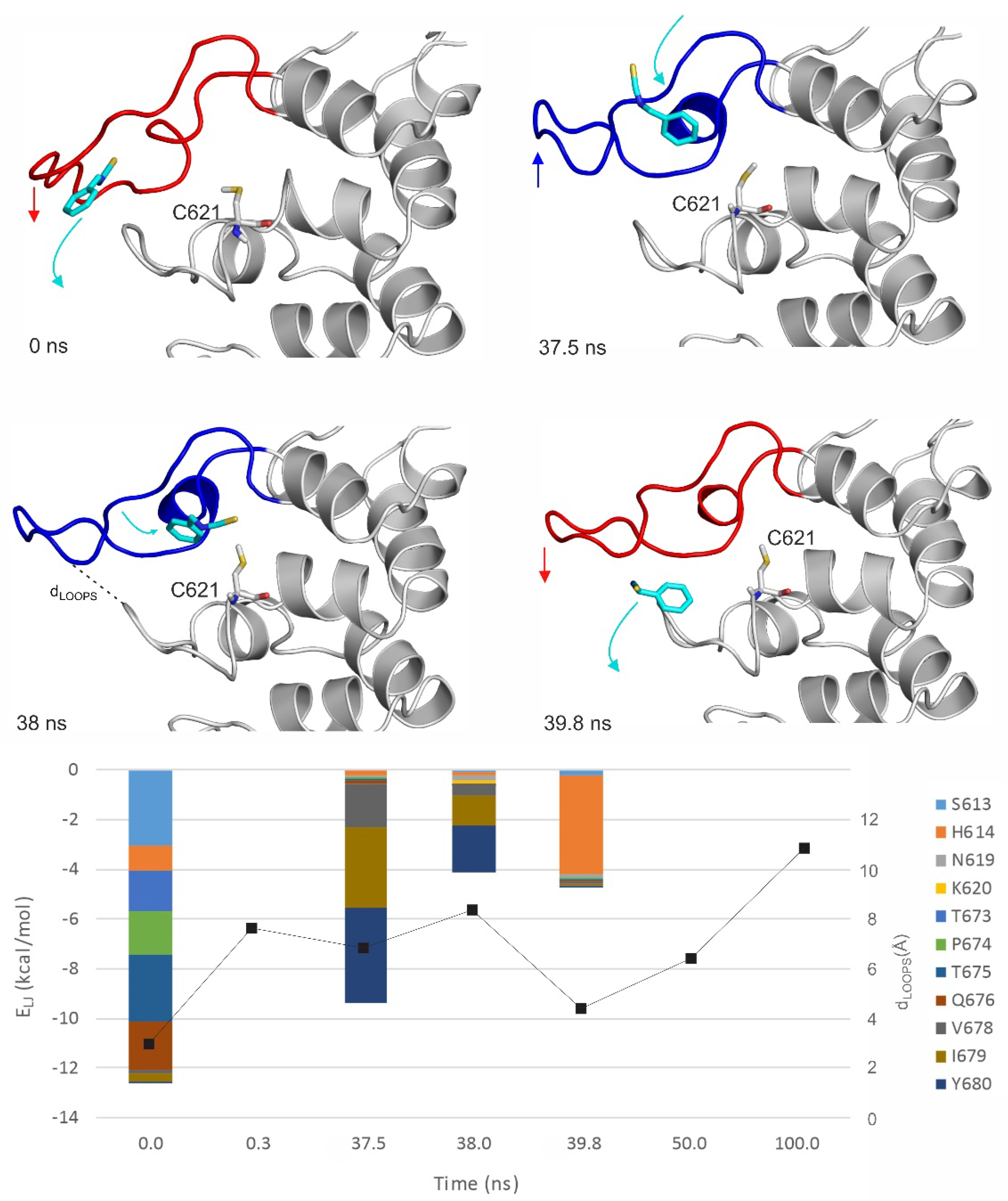
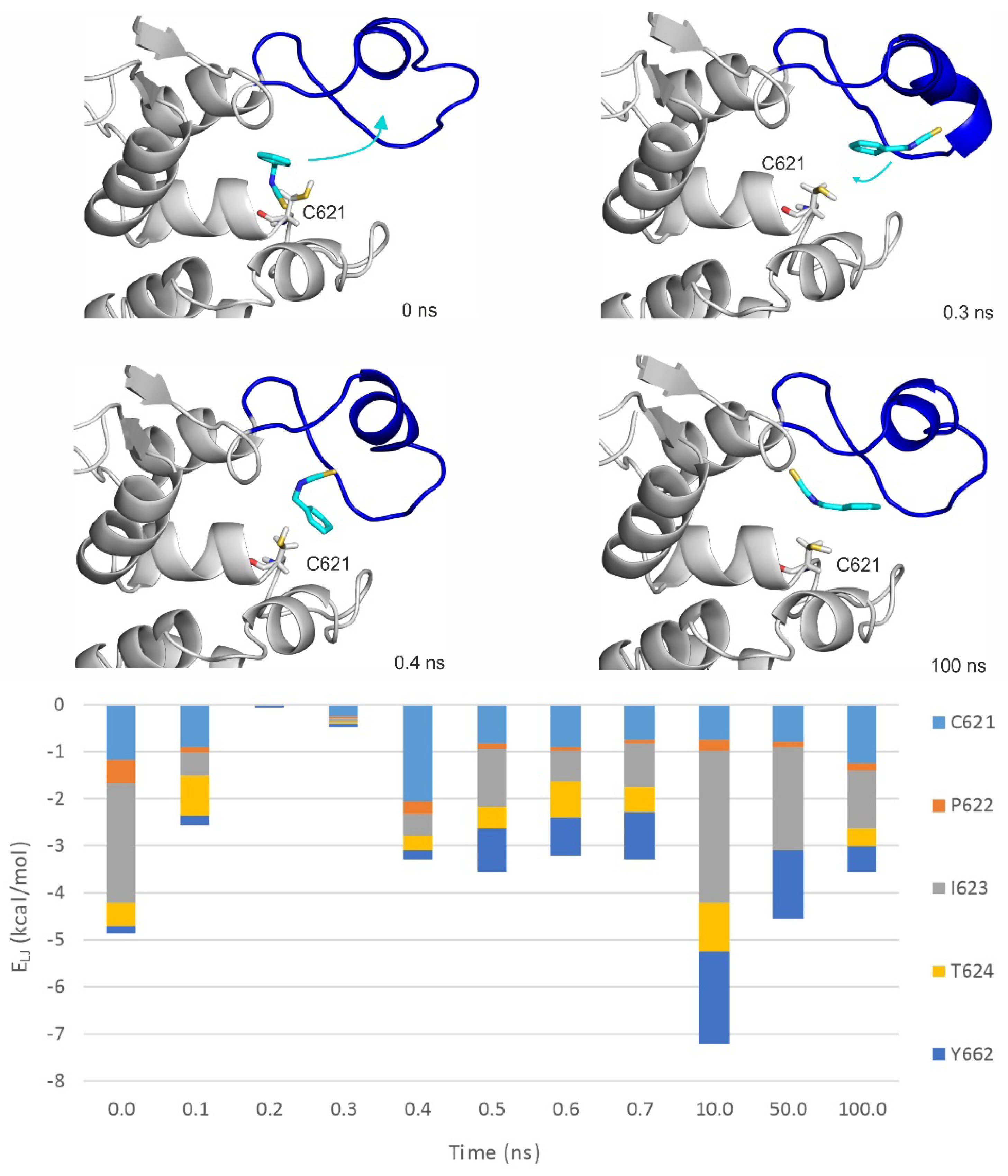
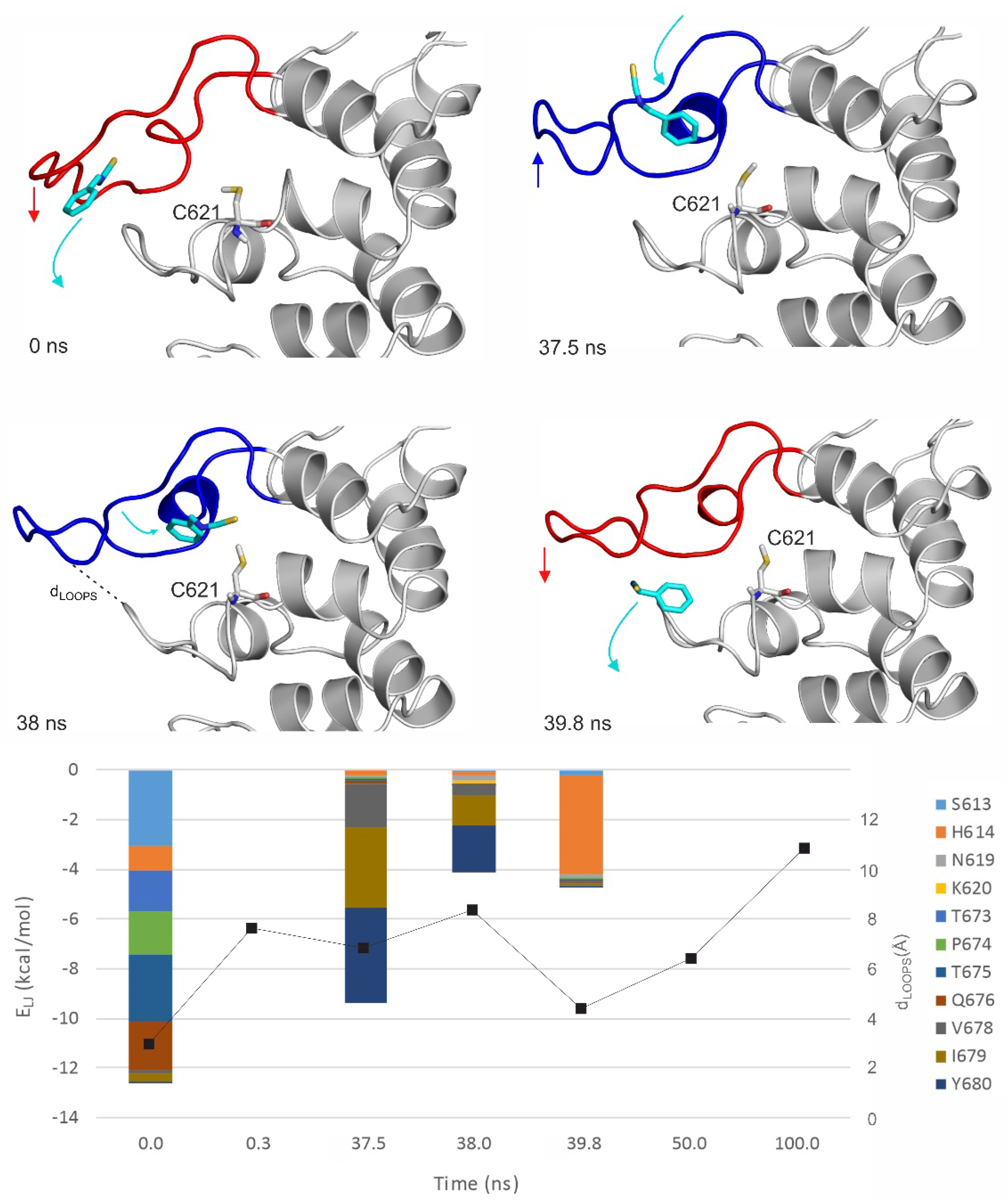
2.3. Ligand Migration Dynamics Connecting Prerequisite and Final Binding Modes
3. Materials and Methods
3.1. Preparation of the Ligand Structures
3.2. Target Preparation
3.3. Covalent Docking with FITTED
3.4. Prerequisite Docking with AutoDock 4.2
3.5. Molecular Dynamics Simulations
3.6. Scoring
3.7. Ranking
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- De Logu, F.; Nassini, R.; Materazzi, S.; Carvalho, G.M.; Nosi, D.; Rossi, D.D.; Marone, I.M.; Ferreira, J.; Li Puma, S.; Benemei, S.; et al. Schwann cell TRPA1 mediates neuroinflammation that sustains macrophage-dependent neuropathic pain in mice. Nat. Commun. 2017, 8, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Paulsen, C.E.; Armache, J.P.; Gao, Y.; Cheng, Y.; Julius, D. Structure of the TRPA1 ion channel suggests regulatory mechanisms. Nature 2015, 520, 511–517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, C.; Reese, R.; Vu, S.; Rougé, L.; Shields, S.D.; Kakiuchi-Kiyota, S.; Chen, H.; Johnson, K.; Shi, Y.P.; Chernov-Rogan, T.; et al. A Non-covalent Ligand Reveals Biased Agonism of the TRPA1 Ion Channel. Neuron 2020, 109, 273–284. [Google Scholar] [CrossRef] [PubMed]
- Takaya, J.; Mio, K.; Shiraishi, T.; Kurokawa, T.; Otsuka, S.; Mori, Y.; Uesugi, M. A Potent and Site-Selective Agonist of TRPA1. J. Am. Chem. Soc. 2015, 137, 15859–15864. [Google Scholar] [CrossRef] [Green Version]
- Pozsgai, G.; Bátai, I.Z.; Pintér, E. Effects of sulfide and polysulfides transmitted by direct or signal transduction-mediated activation of TRPA1 channels. Br. J. Pharmacol. 2019, 176, 628–645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suo, Y.; Wang, Z.; Zubcevic, L.; Hsu, A.L.; He, Q.; Borgnia, M.J.; Ji, R.R.; Lee, S.Y. Structural Insights into Electrophile Irritant Sensing by the Human TRPA1 Channel. Neuron 2020, 105, 882–894. [Google Scholar] [CrossRef]
- Zhao, J.; Lin King, J.V.; Paulsen, C.E.; Cheng, Y.; Julius, D. Irritant-evoked activation and calcium modulation of the TRPA1 receptor. Nature 2020, 585, 141–145. [Google Scholar] [CrossRef]
- Chernov-Rogan, T.; Gianti, E.; Liu, C.; Villemure, E.; Cridland, A.P.; Hu, X.; Ballini, E.; Lange, W.; Deisemann, H.; Li, T.; et al. TRPA1 modulation by piperidine carboxamides suggests an evolutionarily conserved binding site and gating mechanism. Proc. Natl. Acad. Sci. USA 2019, 116, 26008–26019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tseng, W.C.; Pryde, D.C.; Yoger, K.E.; Padilla, K.M.; Antonio, B.M.; Han, S.; Shanmugasundaram, V.; Gerlach, A.C. TRPA1 ankyrin repeat six interacts with a small molecule inhibitor chemotype. Proc. Natl. Acad. Sci. USA 2018, 115, 12301–12306. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.Y.; Chang, R.B.; Waters, H.N.; McKemy, D.D.; Liman, E.R. The nociceptor ion channel TRPA1 is potentiated and inactivated by permeating calcium ions. J. Biol. Chem. 2008, 283, 32691–32703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ábrányi-Balogh, P.; Petri, L.; Imre, T.; Szijj, P.; Scarpino, A.; Hrast, M.; Mitrović, A.; Fonovič, U.P.; Németh, K.; Barreteau, H.; et al. A road map for prioritizing warheads for cysteine targeting covalent inhibitors. Eur. J. Med. Chem. 2018, 160, 94–107. [Google Scholar] [CrossRef] [PubMed]
- Petri, L.; Egyed, A.; Bajusz, D.; Imre, T.; Hetényi, A.; Martinek, T.; Ábrányi-Balogh, P.; Keserű, G.M. An electrophilic warhead library for mapping the reactivity and accessibility of tractable cysteines in protein kinases. Eur. J. Med. Chem. 2020, 207, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Corbeil, C.R.; Englebienne, P.; Moitessier, N. Docking ligands into flexible and solvated macromolecules. 1. Development and validation of FITTED 1.0. J. Chem. Inf. Model. 2007, 47, 435–449. [Google Scholar] [CrossRef]
- Pottel, J.; Therrien, E.; Gleason, J.L.; Moitessier, N. Docking ligands into flexible and solvated macromolecules. 6. Development and application to the docking of HDACs and other zinc metalloenzymes inhibitors. J. Chem. Inf. Model. 2014, 54, 254–265. [Google Scholar] [CrossRef] [PubMed]
- Therrien, E.; Englebienne, P.; Arrowsmith, A.G.; Mendoza-Sanchez, R.; Corbeil, C.R.; Weill, N.; Campagna-Slater, V.; Moitessier, N. Integrating medicinal chemistry, organic/combinatorial chemistry, and computational chemistry for the discovery of selective estrogen receptor modulatorswith FORECASTER, a novel platform for drug discovery. J. Chem. Inf. Model. 2012, 52, 210–224. [Google Scholar] [CrossRef]
- Bálint, M.; Horváth, I.; Mészáros, N.; Hetényi, C. Towards Unraveling the Histone Code by Fragment Blind Docking. Int. J. Mol. Sci. 2019, 20, 422. [Google Scholar] [CrossRef] [Green Version]
- Kevener, H.E.; Zhao, W.; Ball, D.M.; Babaoglu, K.; Qi, J.; White, S.W.; Lee, R.E. Validation of Molecular Docking Programs for Virtual Screening against Dihydropteroate Synthase. J. Chem. Inf. Model. 2009, 49, 444–460. [Google Scholar] [CrossRef] [PubMed]
- Castro-Alvarez, A.; Costa, A.M.; Vilarrasa, J. The Performance of several docking programs at reproducing protein-macrolide-like crystal structures. Molecules 2017, 22, 136. [Google Scholar] [CrossRef] [Green Version]
- Mena-Ulecia, K.; Tiznado, W.; Caballero, J. Study of the differential activity of thrombin inhibitors using docking, QSAR, molecular dynamics, and MM-GBSA. PLoS ONE 2015, 10, 1–21. [Google Scholar]
- Ramírez, D.; Caballero, J. Is It Reliable to Take the Molecular Docking Top Scoring Position as the Best Solution without Considering Available Structural Data? Molecules 2018, 23, 1038. [Google Scholar] [CrossRef] [Green Version]
- Gohlke, H.; Hendlich, M.; Klebe, G. Knowledge-based scoring function to predict protein-ligand interactions. J. Mol. Biol. 2000, 295, 337–356. [Google Scholar] [CrossRef]
- Bálint, M.; Jeszenoi, N.; Horváth, I.; Van Der Spoel, D.; Hetényi, C. Systematic exploration of multiple drug binding sites. J. Cheminform. 2017, 9, 65–79. [Google Scholar] [CrossRef]
- Sotriffer, C. Docking of Covalent Ligands: Challenges and Approaches. Mol. Inform. 2018, 37, 1–12. [Google Scholar] [CrossRef]
- Kumalo, H.M.; Bhakat, S.; Soliman, M.E.S. Theory and applications of covalent docking in drug discovery: Merits and pitfalls. Molecules 2015, 20, 1984–2000. [Google Scholar] [CrossRef] [Green Version]
- Morris, G.M.; Goodsell, D.S.; Halliday, R.S.; Huey, R.; Hart, W.E.; Belew, R.K.; Olson, A.J. Automated docking using a Lamarckian Genetic Algorithm and empirical binding free energy function. J. Comput. Chem. 1998, 19, 1639–1662. [Google Scholar] [CrossRef] [Green Version]
- Shi, S.; Yan, L.; Yang, Y.; Fisher-Shaulsky, J.; Thacher, T. An extensible and systematic force field, ESFF, for molecular modeling of organic, inorganic, and organometallic systems. J. Comput. Chem. 2003, 24, 1059–1076. [Google Scholar] [CrossRef] [PubMed]
- CDiscoVer, 98.0; Accelrys, Inc.: San Diego, CA, USA, 2001.
- Macpherson, L.J.; Dubin, A.E.; Evans, M.J.; Marr, F.; Schultz, P.G.; Cravatt, B.F.; Patapoutian, A. Noxious compounds activate TRPA1 ion channels through covalent modification of cysteines. Nature 2007, 445, 541–545. [Google Scholar] [CrossRef] [PubMed]
- Warren, L.D. The PyMOL Molecular Graphics System; Version 2.0; Schrödinger, LLC.: New York, NY, USA, 2002. [Google Scholar]
- Stewart, J.J.P. Optimization of parameters for semiempirical methods V: Modification of NDDO approximations and application to 70 elements. J. Mol. Model. 2007, 13, 1173–1213. [Google Scholar] [CrossRef] [Green Version]
- Stewart, J.J.P. Optimization of parameters for semiempirical methods VI: More modifications to the NDDO approximations and re-optimization of parameters. J. Mol. Model. 2013, 19, 1–32. [Google Scholar] [CrossRef] [Green Version]
- Gasteiger, J.; Marsili, M. Iterative partial equalization of orbital electronegativity-a rapid access to atomic charges. Tetrahedron 1980, 36, 3219–3228. [Google Scholar] [CrossRef]
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open Babel. J. Cheminform. 2011, 3, 1–14. [Google Scholar]
- Moitessier, N.; Therrien, E.; Hanessian, S. A method for induced-fit docking, scoring, and ranking of flexible ligands. Application to peptidic and pseudopeptidic β-secretase (BACE 1) inhibitors. J. Med. Chem. 2006, 49, 5885–5894. [Google Scholar] [CrossRef]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general Amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef]
- Schrödinger Release 2020-4: Maestro; Schrödinger, LLC.: New York, NY, USA, 2021.
- Dupradeau, F.Y.; Pigache, A.; Zaffran, T.; Savineau, C.; Lelong, R.; Grivel, N.; Lelong, D.; Rosanski, W.; Cieplak, P. The R.E.D. tools: Advances in RESP and ESP charge derivation and force field library building. Phys. Chem. Chem. Phys. 2010, 12, 7821–7839. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmidt, M.W.; Baldridge, K.K.; Boatz, J.A.; Elbert, S.T.; Gordon, M.S.; Jensen, J.H.; Koseki, S.; Matsunaga, N.; Nguyen, K.A.; Su, S.; et al. General atomic and molecular electronic structure system. J. Comput. Chem. 1993, 14, 1347–1363. [Google Scholar] [CrossRef]
- Sousa Da Silva, A.W.; Vranken, W.F. ACPYPE—AnteChamber PYthon Parser interfacE. BMC Res. Notes 2012, 5, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Wang, W.; Kollman, P.A.; Case, D.A. Automatic atom type and bond type perception in molecular mechanical calculations. J. Mol. Graph. Model. 2006, 25, 247–260. [Google Scholar] [CrossRef]
- Zsidó, B.Z.; Börzsei, R.; Szél, V.; Hetényi, C. Determination of Ligand Binding Modes in Hydrated Viral Ion Channels to Foster Drug Design and Repositioning. J. Chem. Inf. Model. 2021, 8, 4011–4022. [Google Scholar] [CrossRef]
- Berman, H.M.; Battistuz, T.; Bhat, T.N.; Bluhm, W.F.; Bourne, P.E.; Burkhardt, K.; Feng, Z.; Gilliland, G.L.; Iype, L.; Jain, S.; et al. The protein data bank. Acta Crystallogr. Sect. D Biol. Crystallogr. 2002, 58, 899–907. [Google Scholar] [CrossRef]
- Zsidó, B.Z.; Hetényi, C. Molecular structure, binding affinity, and biological activity in the epigenome. Int. J. Mol. Sci. 2020, 21, 4143. [Google Scholar] [CrossRef]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; De Beer, T.A.P.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef] [Green Version]
- Van Der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J.C. GROMACS: Fast, flexible, and free. J. Comput. Chem. 2005, 26, 1701–1718. [Google Scholar] [CrossRef]
- Fliszár-Nyúl, E.; Faisal, Z.; Mohos, V.; Derdák, D.; Lemli, B.; Kálai, T.; Sár, C.; Zsidó, B.Z.; Hetényi, C.; Horváth, Á.I.; et al. Interaction of SZV 1287, a novel oxime analgesic drug candidate, and its metabolites with serum albumin. J. Mol. Liq. 2021, 333, 1–10. [Google Scholar] [CrossRef]
- Zsidó, B.Z.; Balog, M.; Erős, N.; Poór, M.; Mohos, V.; Fliszár-Nyúl, E.; Hetényi, C.; Nagane, M.; Hideg, K.; Kálai, T.; et al. Synthesis of spin-labelled bergamottin: A potent CYP3A4 inhibitor with antiproliferative activity. Int. J. Mol. Sci. 2020, 21, 508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohos, V.; Fliszár-Nyúl, E.; Ungvári, O.; Bakos, É.; Kuffa, K.; Bencsik, T.; Zsidó, B.Z.; Hetényi, C.; Telbisz, Á.; Özvegy-Laczka, C.; et al. Effects of chrysin and its major conjugated metabolites chrysin-7-sulfate and chrysin-7-glucuronide on cytochrome P450 enzymes and on OATP, P-gp, BCRP, and MRP2 transporters. Drug Metab. Dispos. 2020, 48, 1064–1073. [Google Scholar] [CrossRef] [PubMed]
- Mohos, V.; Fliszár-Nyúl, E.; Lemli, B.; Zsidó, B.Z.; Hetényi, C.; Mladěnka, P.; Horký, P.; Pour, M.; Poór, M. Testing the pharmacokinetic interactions of 24 colonic flavonoid metabolites with human serum albumin and cytochrome P450 enzymes. Biomolecules 2020, 10, 409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Huey, R.; Morris, G.M.; Olson, A.J.; Goodsell, D.S. A semiempirical free energy force field with charge-based desolvation. J. Comput. Chem. 2007, 28, 1145–1152. [Google Scholar] [CrossRef] [PubMed]
- Hetényi, C.; Van Der Spoel, D. Blind docking of drug-sized compounds to proteins with up to a thousand residues. FEBS Lett. 2006, 580, 1447–1450. [Google Scholar] [CrossRef] [Green Version]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ligand Name | JT010 | BITC | Bodipy-Iodoacetamide |
|---|---|---|---|
| HOLO target | |||
| AAmatch (%) | 100% | 100% | 100% |
| RMSDbest (Å) | 2.28 | 2.05 | 3.87 |
| Rankbest | 1/3 | 1/3 | 1/3 |
| ΔGFD (kcal/mol) | −84.1 | −77.7 | −44.3 |
| NHA c | 23 | 10 | 22 |
| EINHA d (kcal/mol) | 3.66 | 7.77 | 2.01 |
| dcovalent (Å) | 1.8 (1.8) a | 1.8 (1.8) a | 1.8 (1.8) a |
| APO target b | |||
| AAmatch (%) | 100% | 60% | 66.6% |
| RMSDbest (Å) | 6.82 | 4.75 | 6.55 |
| Rankbest | 1/5 | 1/5 | 1/5 |
| ΔGFD (kcal/mol) | −77.4 | −73.8 | −43.1 |
| NHA c | 23 | 10 | 22 |
| EINHA d (kcal/mol) | 3.36 | 7.38 | 1.96 |
| dcovalent (Å) | 1.8 | 1.8 | 1.8 |
| Ligand Name | JT010 | BITC | Bodipy-Iodoacetamide |
|---|---|---|---|
| HOLO target | |||
| ΔGFD (kcal/mol) | −46.1 | −32.4 | −13.7 |
| Rankbest | 10/10 | 1/10 | 8/10 |
| AAmatch (%) | 100% | 100% | 100% |
| dbest (Å) | 3.6 | 4.0 | 8.7 |
| APO target a | |||
| ΔGFD (kcal/mol) | −33.4 | −26.7 | 0.5 |
| Rankbest | 3/5 | 1/5 | 4/5 |
| AAmatch (%) | 100% | 60% | 33.3% |
| dbest (Å) | 3.5 | 3.9 | 3.3 |
| Ligand Name | JT010 | BITC | Bodipy-Iodoacetamide |
|---|---|---|---|
| HOLO target | |||
| ΔGAD (kcal/mol) | −6.8 | −3.8 | −5.9 |
| Rankbest | 1/5 | 1/1 | 4/4 |
| AAmatch (%) | 100% | 80% | 66% |
| dbest (Å) | 3.6 | 6.5 | 4.0 |
| APO target a | |||
| ΔGAD (kcal/mol) | −5.16 | −3.74 | −5.26 |
| Rankbest | 1/3 | 1/2 | 3/5 |
| AAmatch (%) | 50% | 40% | 0% |
| dbest (Å) | 7.5 | 7.2 | 7.3 |
| Simulation Name | TRPA1 | Ligand | Change in A-Loop | Movement of the Agonist |
|---|---|---|---|---|
| MDapo | Apo protein | - | No change in A-loop conformation | - |
| MDholo,PSA | Holo protein | Experimental | No change in A-loop conformation | Unbinding–binding |
| MDrank1 | Apo protein | Rank 1 docked ligand binding mode | A-loop flipping to the active conformation | Dissociation–association |
| MDrank3 | Apo protein | Rank 3 docked ligand binding mode | No change in A-loop conformation | Dissociation |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zsidó, B.Z.; Börzsei, R.; Pintér, E.; Hetényi, C. Prerequisite Binding Modes Determine the Dynamics of Action of Covalent Agonists of Ion Channel TRPA1. Pharmaceuticals 2021, 14, 988. https://doi.org/10.3390/ph14100988
Zsidó BZ, Börzsei R, Pintér E, Hetényi C. Prerequisite Binding Modes Determine the Dynamics of Action of Covalent Agonists of Ion Channel TRPA1. Pharmaceuticals. 2021; 14(10):988. https://doi.org/10.3390/ph14100988
Chicago/Turabian StyleZsidó, Balázs Zoltán, Rita Börzsei, Erika Pintér, and Csaba Hetényi. 2021. "Prerequisite Binding Modes Determine the Dynamics of Action of Covalent Agonists of Ion Channel TRPA1" Pharmaceuticals 14, no. 10: 988. https://doi.org/10.3390/ph14100988
APA StyleZsidó, B. Z., Börzsei, R., Pintér, E., & Hetényi, C. (2021). Prerequisite Binding Modes Determine the Dynamics of Action of Covalent Agonists of Ion Channel TRPA1. Pharmaceuticals, 14(10), 988. https://doi.org/10.3390/ph14100988