
A Novel Statin Compound from Monacolin J Produced Using CYP102A1-Catalyzed Regioselective C-Hydroxylation

 ,
,
Abstract
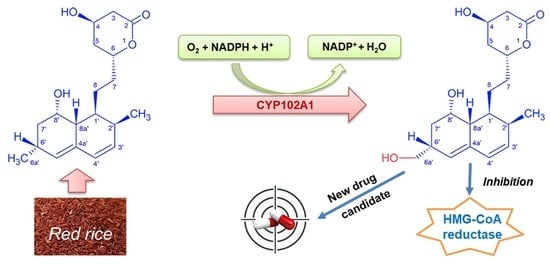
:
1. Introduction
2. Results
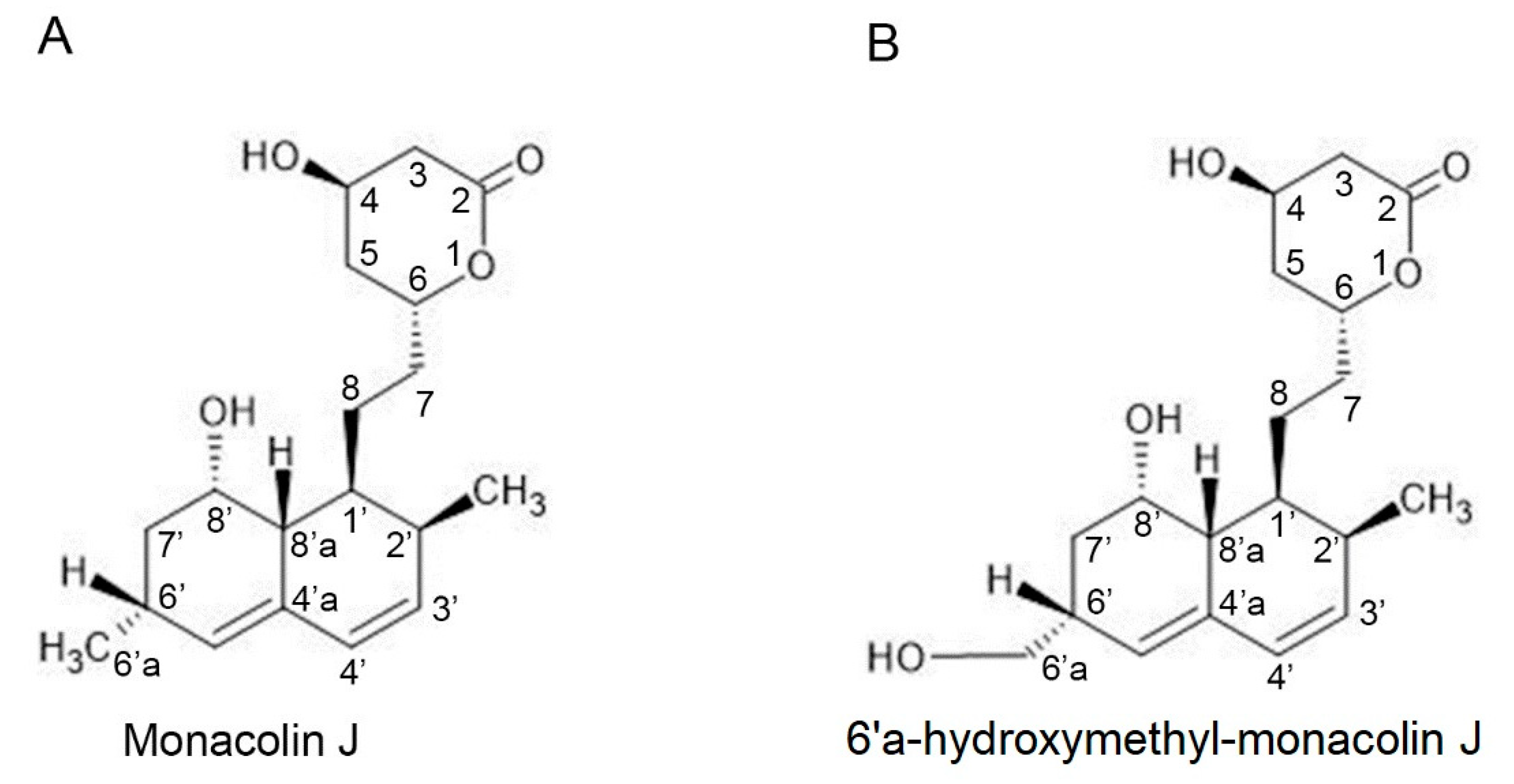
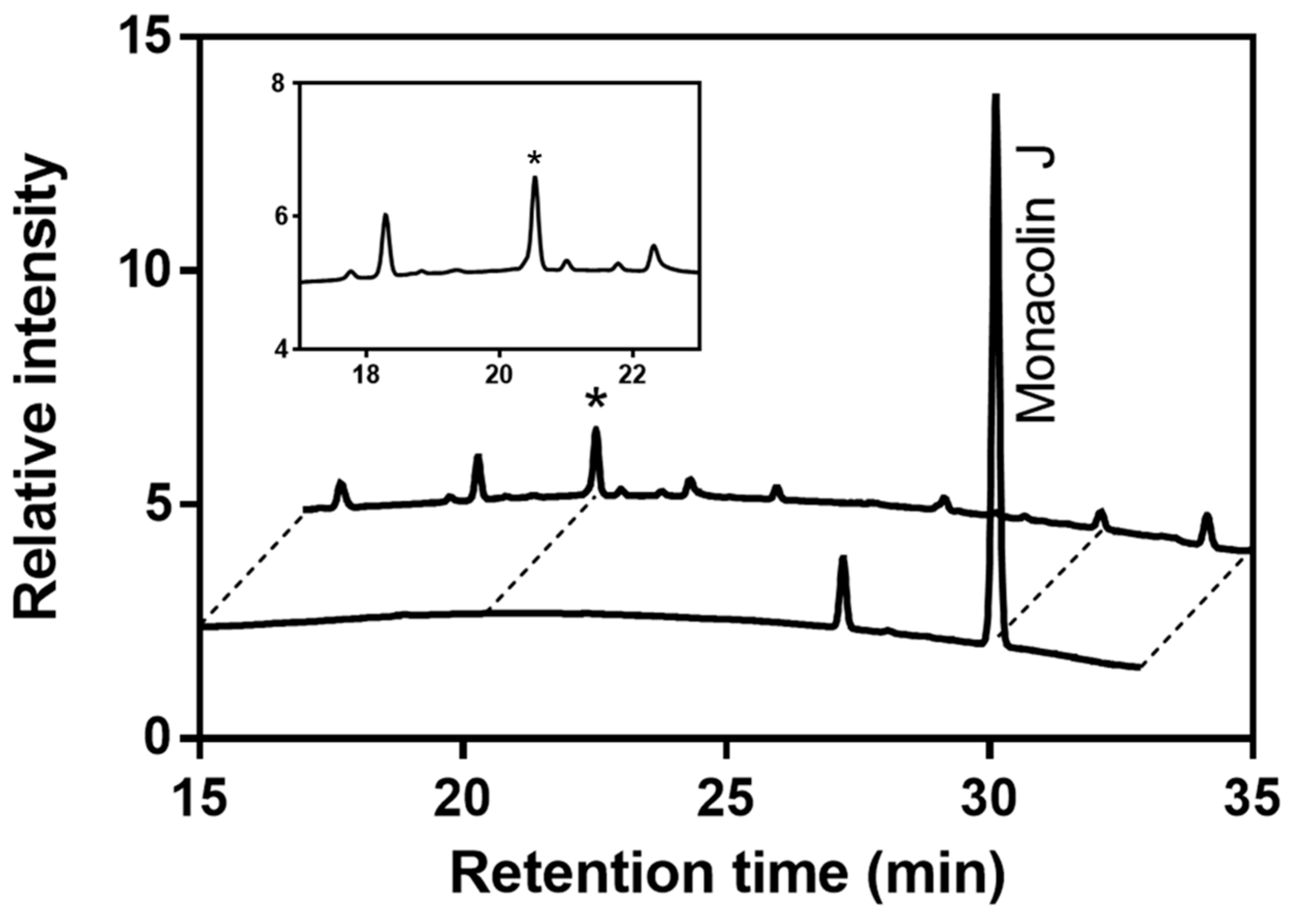
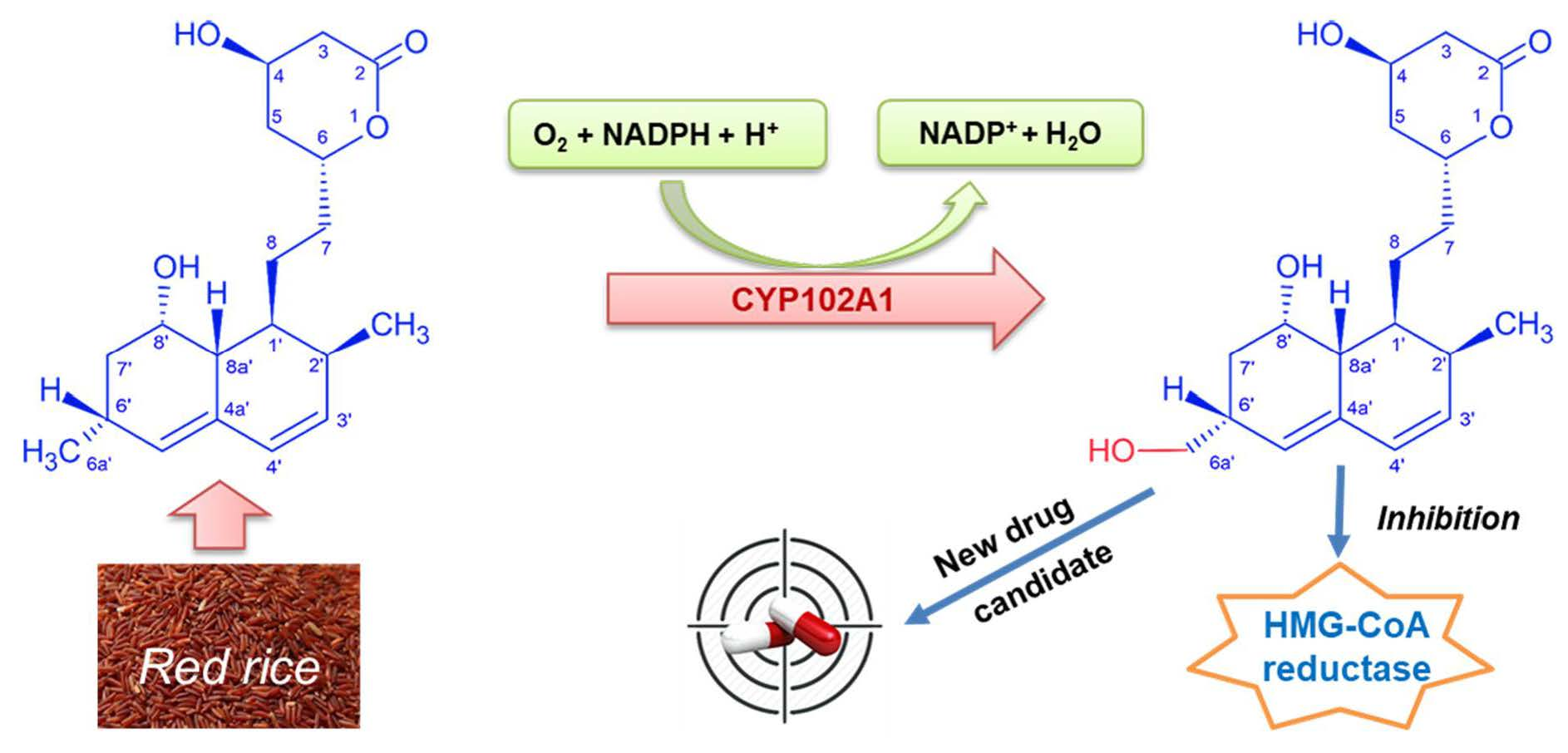
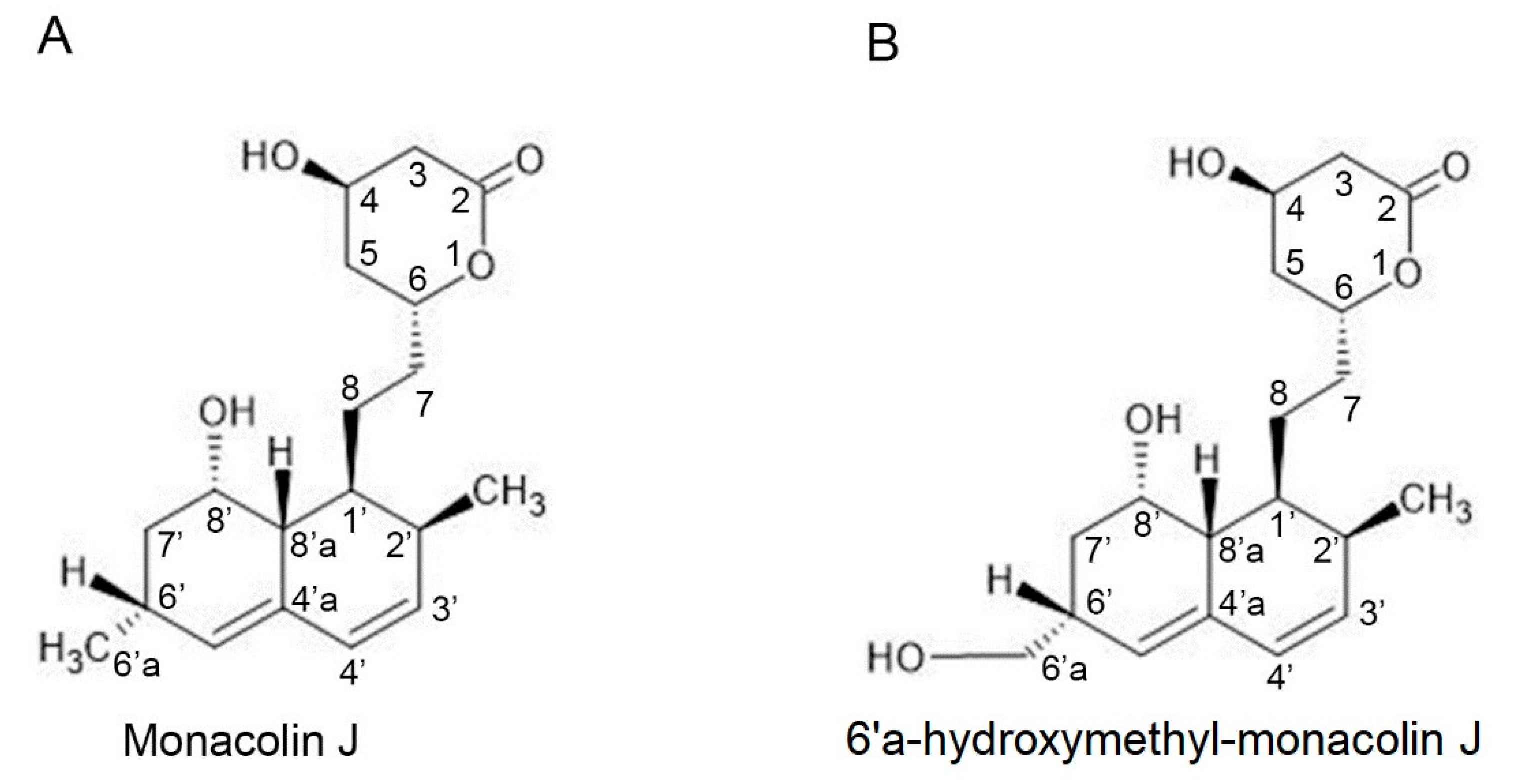
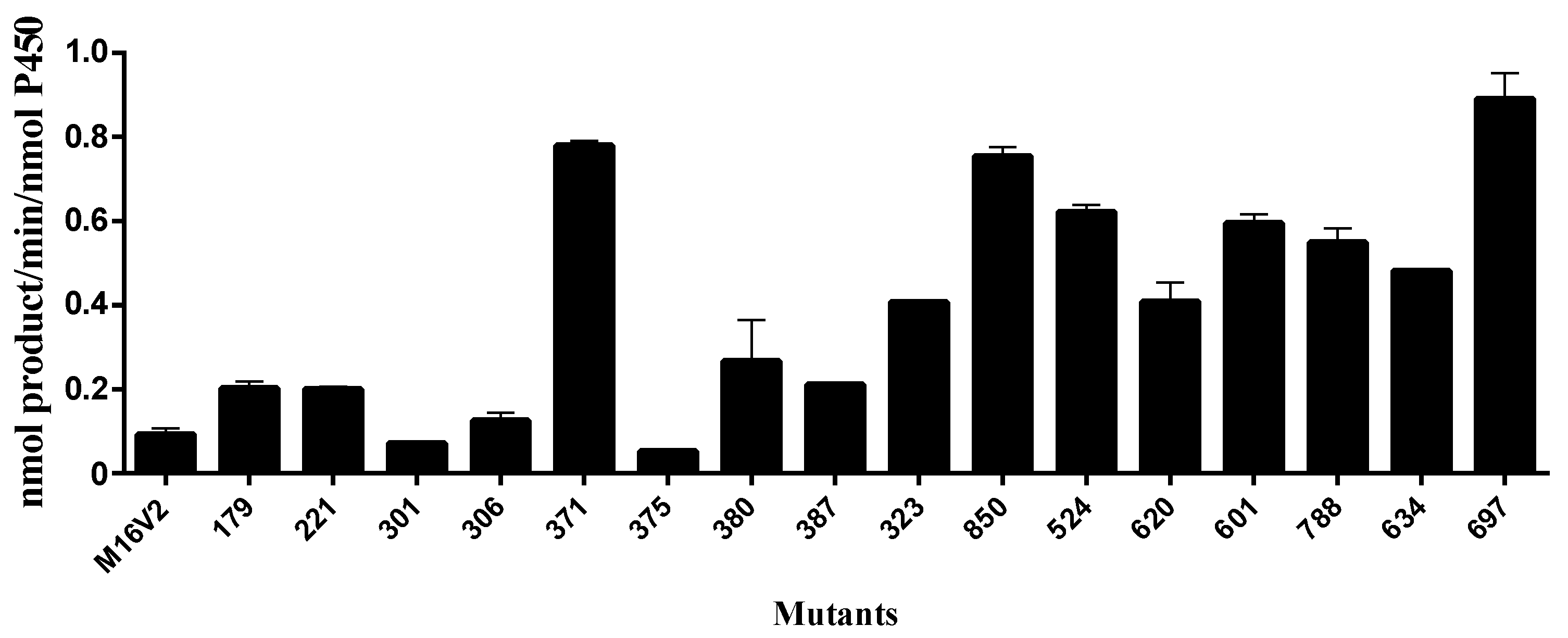
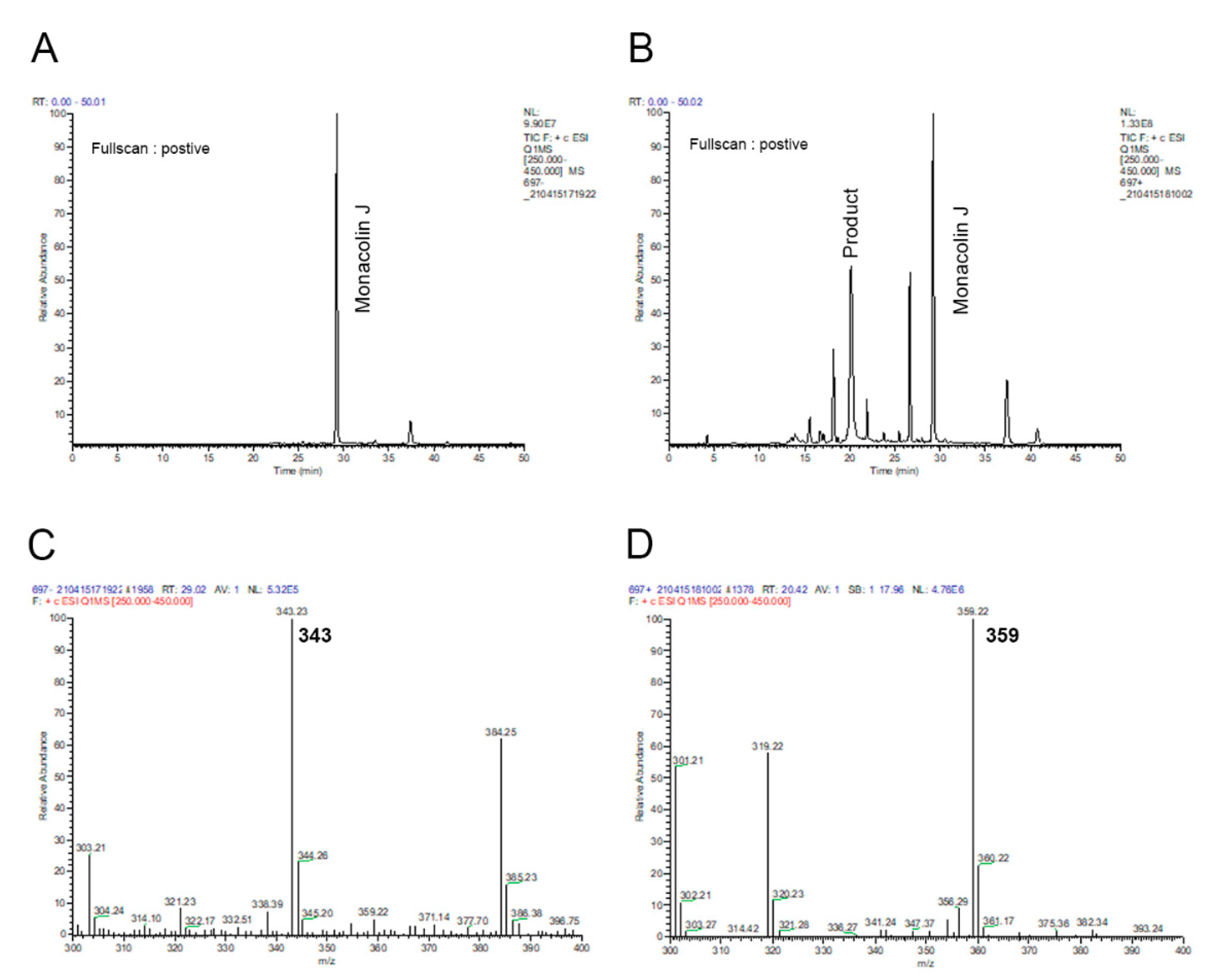
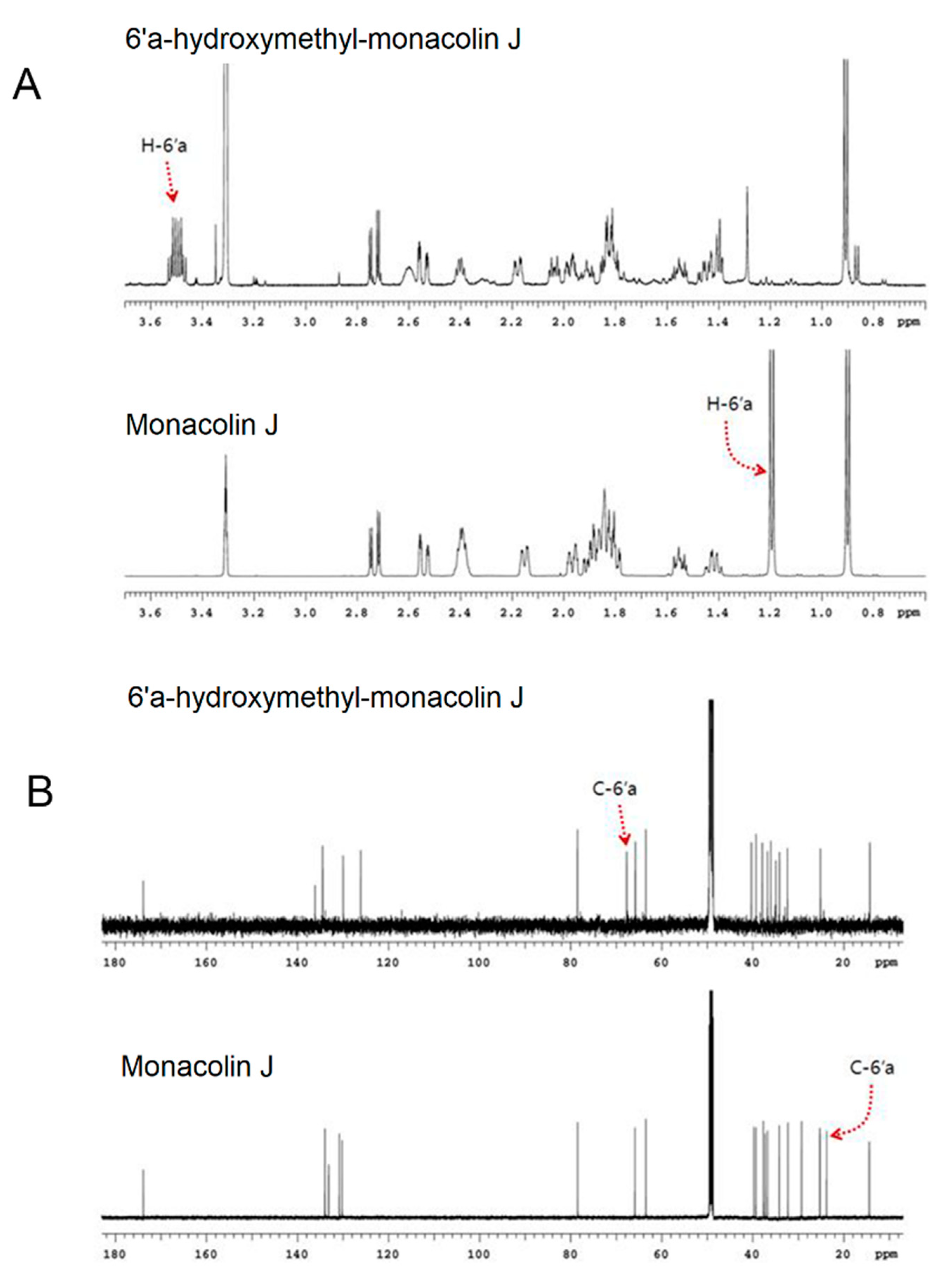
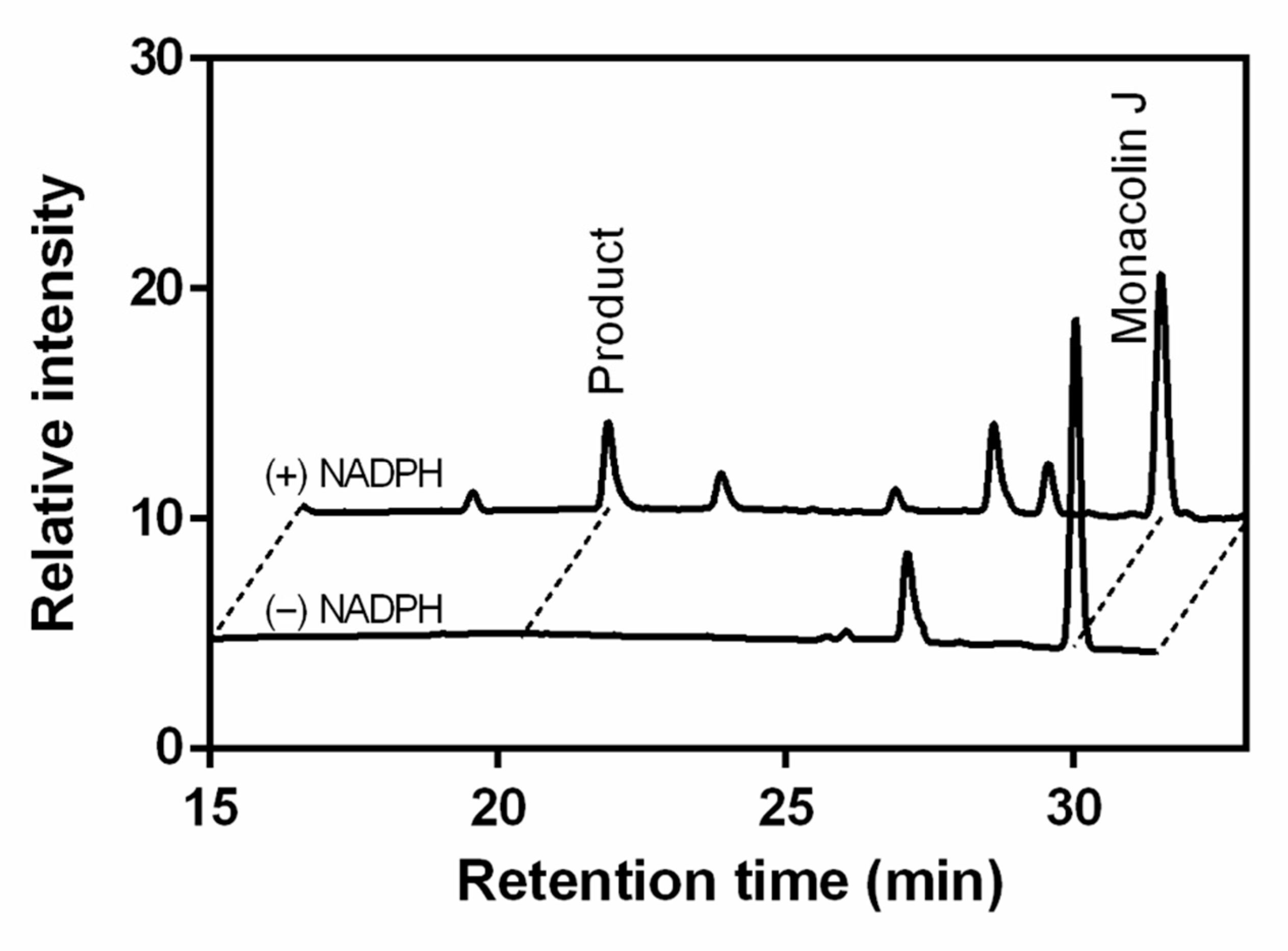
2.1. Monacolin J Oxidation Using CYP102A1 and Identification of the Major Product
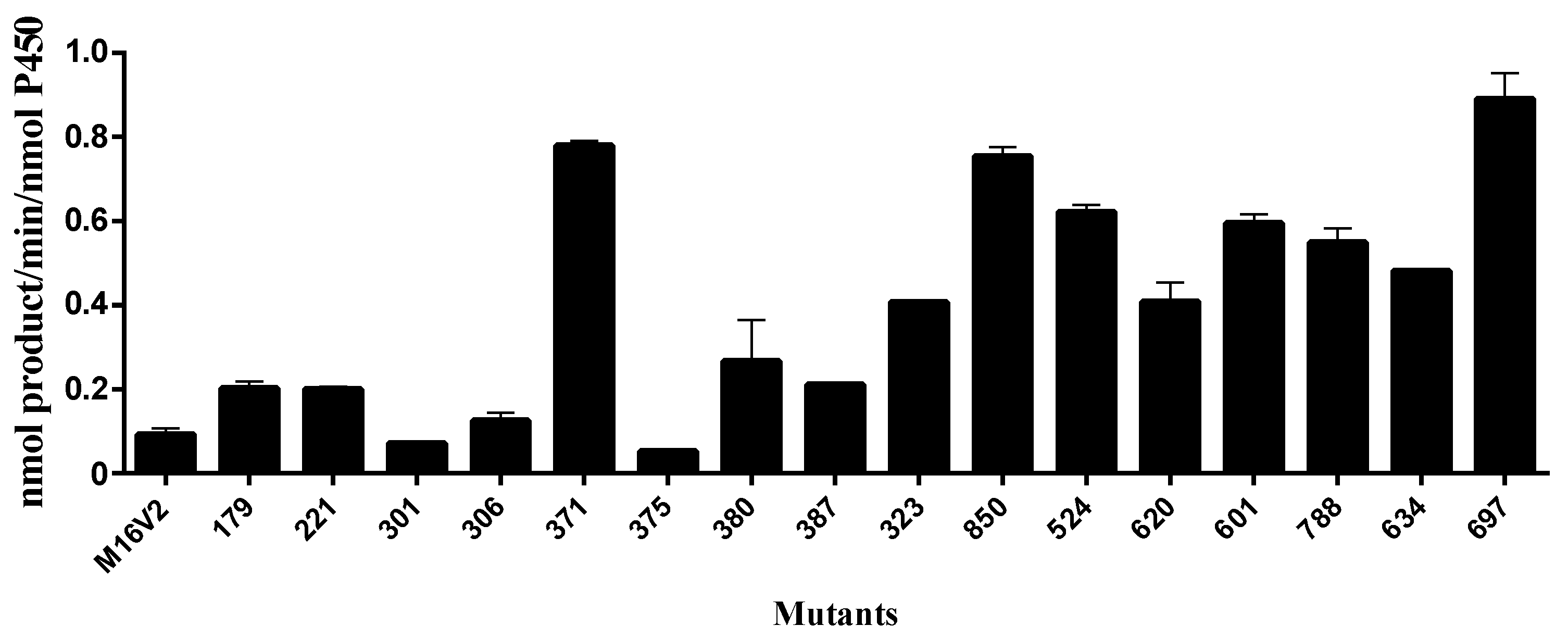
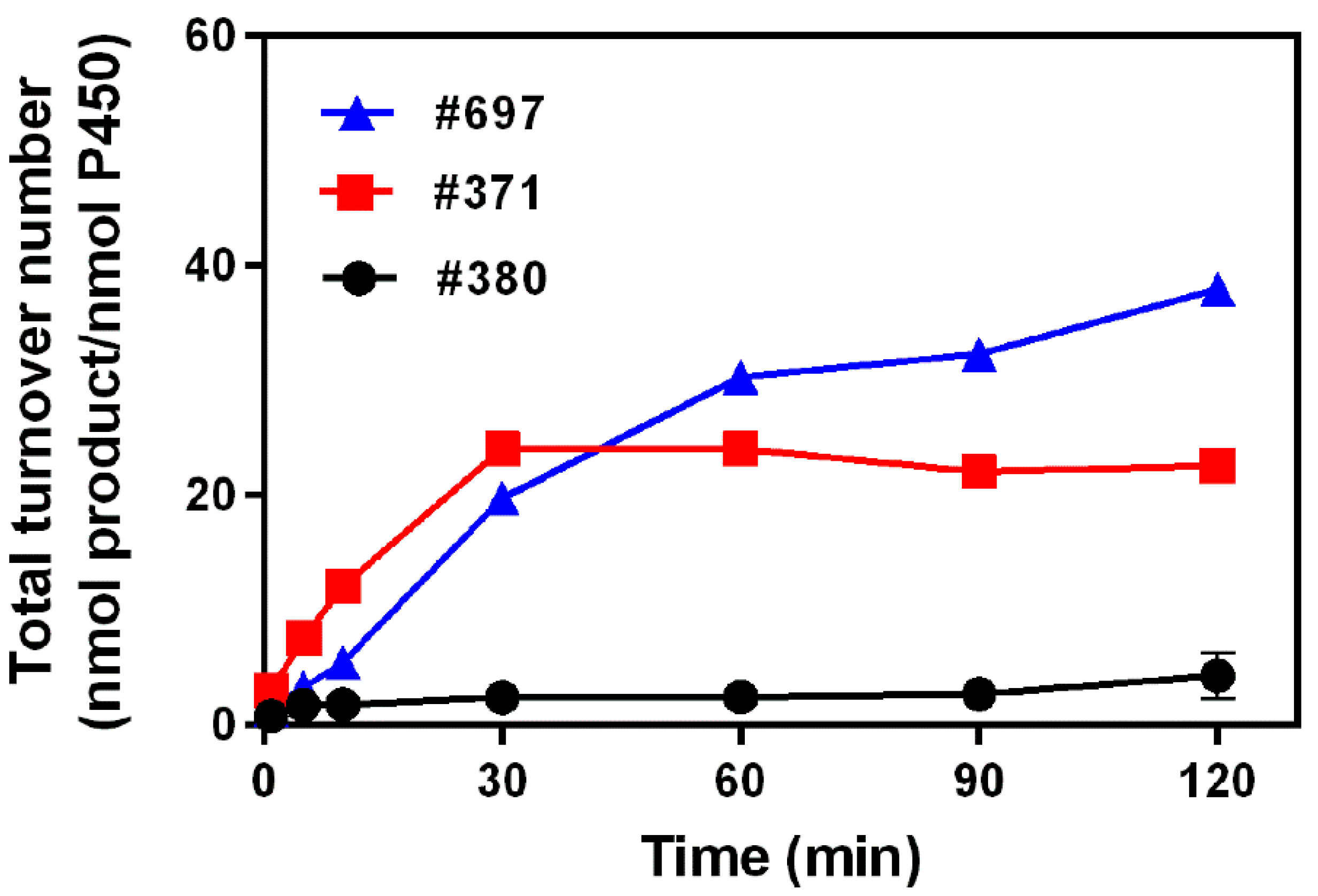
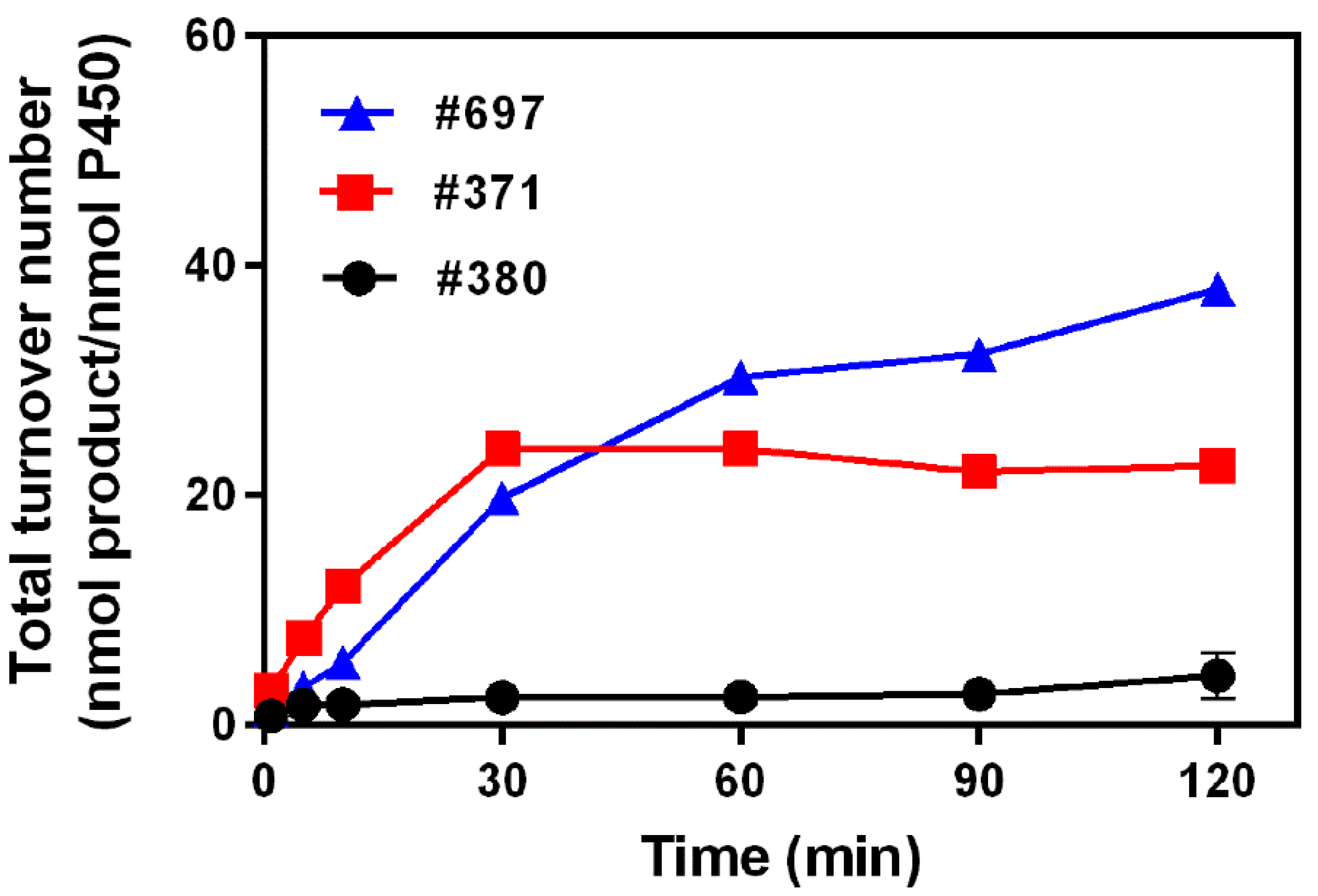
2.2. Kinetics Parameters and Total Turnover Numbers (TTNs) of Monacolin J Hydroxylation Using CYP102A1
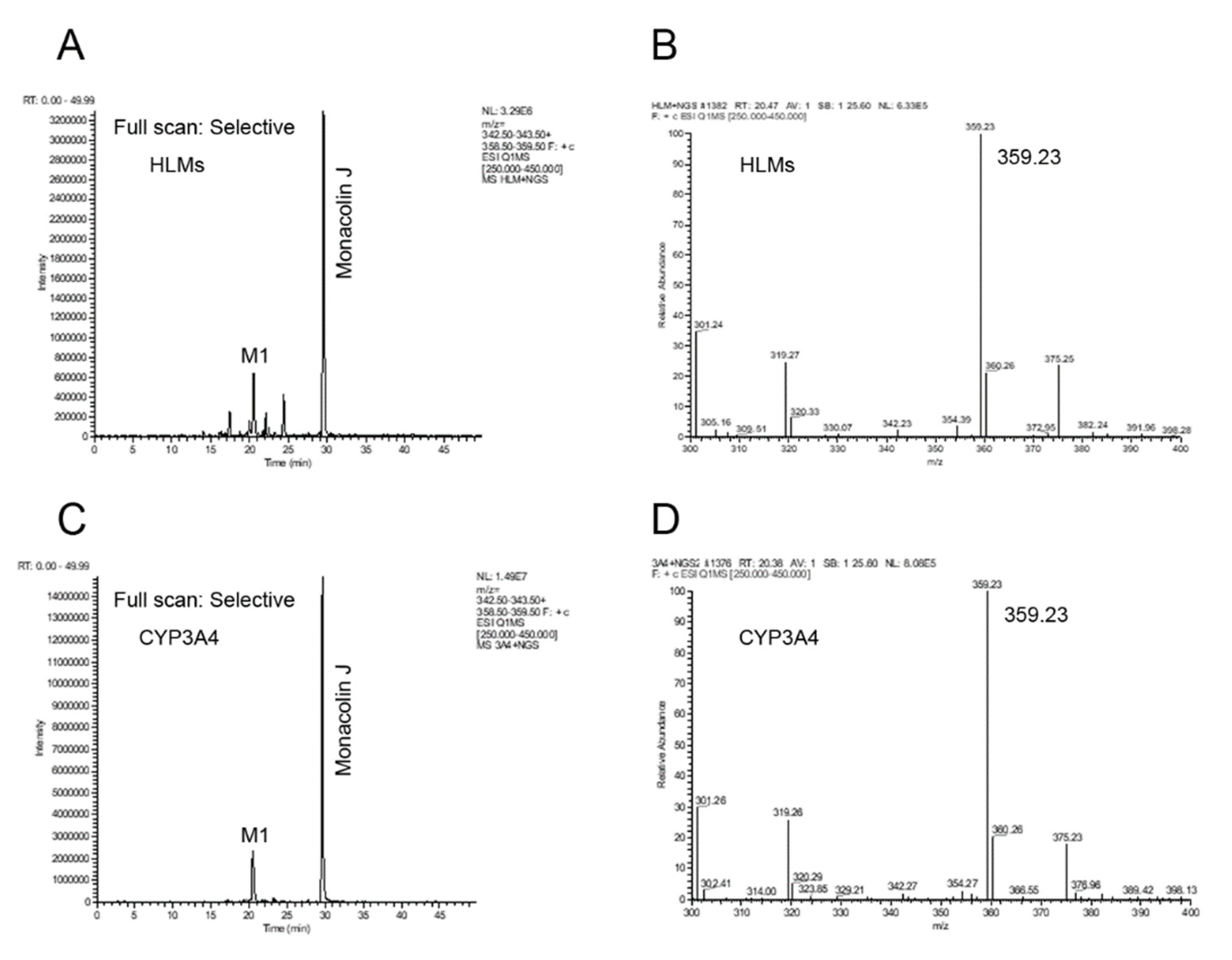
2.3. Hydroxylation of Monacolin J Catalyzed Using Human Liver Microsomes
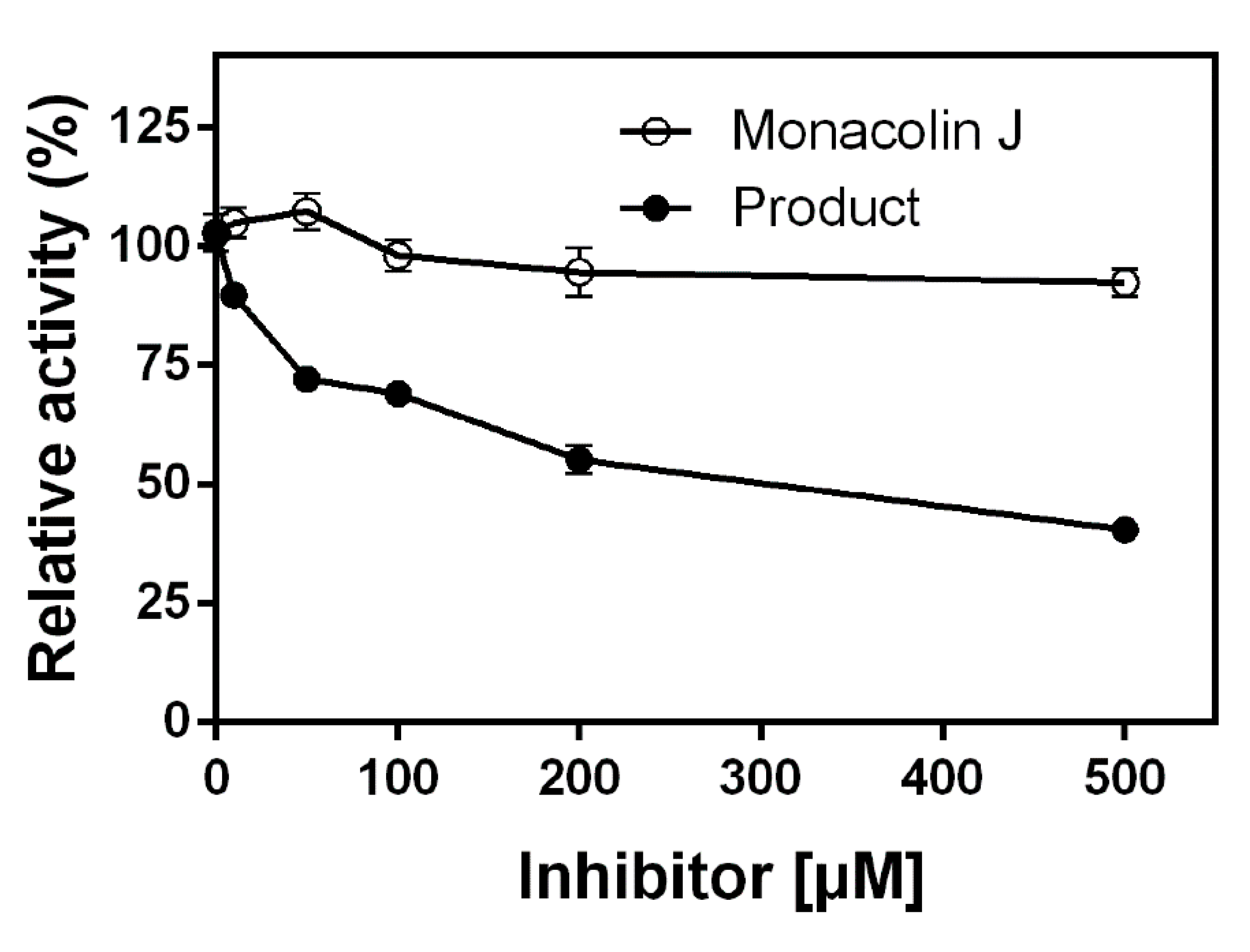
2.4. Inhibitory Effects of Monacolin J and Its Product on HMG-CoA Reductase
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Hydroxylation of Monacolin J Catalyzed Using CYP102A1
4.3. LC–Mass Spectrometric Analysis of a Product of Monacolin J
4.4. NMR Spectroscopy Analysis of Monacolin J Product
4.5. Inhibitory Effects of Monacolin J and Its Metabolite on HMG-CoA Reductase
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Istvan, E.S.; Deisenhofer, J. Structural Mechanism for Statin Inhibition of HMG-CoA Reductase. Science 2001, 292, 1160–1164. [Google Scholar] [CrossRef] [Green Version]
- Istvan, E. Statin Inhibition of HMG-CoA Reductase: A 3-Dimensional View. Atheroscler. Suppl. 2003, 4, 3–8. [Google Scholar] [CrossRef]
- Tsoukas, M.A.; Mantzoros, C.S. Chapter 37—Lipodystrophy syndromes. In Endocrinology: Adult and Pediatric, 7th ed.; Jameson, J.L., De Groot, L.J., de Kretser, D.M., Giudice, L.C., Grossman, A.B., Melmed, S., Potts, J.T., Weir, G.C., Eds.; W.B. Saunders: Philadelphia, PA, USA, 2016; pp. 648–661.e5. ISBN 978-0-323-18907-1. [Google Scholar]
- Murphy, C.; Bennett, K.; Fahey, T.; Shelley, E.; Graham, I.; Kenny, R.A. Statin Use in Adults at High Risk of Cardiovascular Disease Mortality: Cross-Sectional Analysis of Baseline Data from The Irish Longitudinal Study on Ageing (TILDA). BMJ Open 2015, 5, e008017. [Google Scholar] [CrossRef] [Green Version]
- Singh, B.M.; Lamichhane, H.K.; Srivatsa, S.S.; Adhikari, P.; Kshetri, B.J.; Khatiwada, S.; Shrestha, D.B. Role of Statins in the Primary Prevention of Atherosclerotic Cardiovascular Disease and Mortality in the Population with Mean Cholesterol in the Near-Optimal to Borderline High Range: A Systematic Review and Meta-Analysis. Adv. Prev. Med. 2020, 2020, e6617905. [Google Scholar] [CrossRef] [PubMed]
- Almuwaqqat, Z.; Hung, O.; Parashar, S. Chapter 14—Statins in cardio-oncology. In Cardio-Oncology; Gottlieb, R.A., Mehta, P.K., Eds.; Academic Press: Boston, MA, USA, 2017; pp. 209–219. ISBN 978-0-12-803547-4. [Google Scholar]
- Vega, G.L.; Weiner, M.F.; Lipton, A.M.; Von Bergmann, K.; Lutjohann, D.; Moore, C.; Svetlik, D. Reduction in Levels of 24S-Hydroxycholesterol by Statin Treatment in Patients with Alzheimer Disease. Arch. Neurol. 2003, 60, 510–515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shiozawa, A.; Yamaori, S.; Kamijo, S.; Ohmori, S. Effects of Acid and Lactone Forms of Statins on S-Warfarin 7-Hydroxylation Catalyzed by Human Liver Microsomes and Recombinant CYP2C9 Variants (CYP2C9.1 and CYP2C9.3). Drug Metab. Pharmacokinet. 2021, 36, 100364. [Google Scholar] [CrossRef]
- De Angelis, G. The Influence of Statin Characteristics on Their Safety and Tolerability. Int. J. Clin. Pract. 2004, 58, 945–955. [Google Scholar] [CrossRef] [PubMed]
- Barriuso, J.; Nguyen, D.T.; Li, J.W.-H.; Roberts, J.N.; MacNevin, G.; Chaytor, J.L.; Marcus, S.L.; Vederas, J.C.; Ro, D.-K. Double Oxidation of the Cyclic Nonaketide Dihydromonacolin L to Monacolin J by a Single Cytochrome P450 Monooxygenase, LovA. J. Am. Chem. Soc. 2011, 133, 8078–8081. [Google Scholar] [CrossRef]
- Barrios-González, J.; Miranda, R.U. Biotechnological Production and Applications of Statins. Appl. Microbiol. Biotechnol. 2010, 85, 869–883. [Google Scholar] [CrossRef]
- Wen, Q.; Cao, X.; Chen, Z.; Xiong, Z.; Liu, J.; Cheng, Z.; Zheng, Z.; Long, C.; Zheng, B.; Huang, Z. An Overview of Monascus Fermentation Processes for Monacolin K Production. Open Chem. 2020, 18, 10–21. [Google Scholar] [CrossRef]
- Endo, A.; Hasumi, K.; Negishi, S. Monacolins J and L, New Inhibitors of Cholesterol Biosynthesis Produced by Monascus Ruber. J. Antibiot. 1985, 38, 420–422. [Google Scholar] [CrossRef]
- Huang, X.; Liang, Y.; Yang, Y.; Lu, X. Single-Step Production of the Simvastatin Precursor Monacolin J by Engineering of an Industrial Strain of Aspergillus Terreus. Metab. Eng. 2017, 42, 109–114. [Google Scholar] [CrossRef] [PubMed]
- Campoy, S.; Sierra, S.; Suarez, B.; Ramos, M.C.; Velasco, J.; Burgos, J.S.; Adrio, J.L. Semisynthesis of Novel Monacolin J Derivatives: Hypocholesterolemic and Neuroprotective Activities. J. Antibiot. 2010, 63, 499–505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, T.H.H.; Woo, S.-M.; Nguyen, N.A.; Cha, G.-S.; Yeom, S.-J.; Kang, H.-S.; Yun, C.-H. Regioselective Hydroxylation of Naringin Dihydrochalcone to Produce Neoeriocitrin Dihydrochalcone by CYP102A1 (BM3) Mutants. Catalysts 2020, 10, 823. [Google Scholar] [CrossRef]
- Maurer, S.C.; Kühnel, K.; Kaysser, L.A.; Eiben, S.; Schmid, R.D.; Urlacher, V.B. Catalytic Hydroxylation in Biphasic Systems Using CYP102A1 Mutants. Adv. Synth. Catal. 2005, 347, 1090–1098. [Google Scholar] [CrossRef] [Green Version]
- Syntrivanis, L.-D.; Wong, L.L.; Robertson, J. Hydroxylation of Eleuthoside Synthetic Intermediates by P450BM3 (CYP102A1). Eur. J. Org. Chem. 2018, 2018, 6369–6378. [Google Scholar] [CrossRef]
- Kim, K.-H.; Kang, J.-Y.; Kim, D.-H.; Park, S.-H.; Park, S.H.; Kim, D.; Park, K.D.; Lee, Y.J.; Jung, H.-C.; Pan, J.-G.; et al. Generation of Human Chiral Metabolites of Simvastatin and Lovastatin by Bacterial CYP102A1 Mutants. Drug Metab. Dispos. 2011, 39, 140–150. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, T.; Yeom, S.-J.; Yun, C.-H. Production of a Human Metabolite of Atorvastatin by Bacterial CYP102A1 Peroxygenase. Appl. Sci. 2021, 11, 603. [Google Scholar] [CrossRef]
- Nguyen, N.A.; Jang, J.; Le, T.-K.; Nguyen, T.H.H.; Woo, S.-M.; Yoo, S.-K.; Lee, Y.J.; Park, K.D.; Yeom, S.-J.; Kim, G.-J.; et al. Biocatalytic Production of a Potent Inhibitor of Adipocyte Differentiation from Phloretin Using Engineered CYP102A1. J. Agric. Food Chem. 2020, 68, 6683–6691. [Google Scholar] [CrossRef]
- Le, T.-K.; Cha, G.-S.; Jang, H.-H.; Nguyen, T.H.H.; Doan, T.T.M.; Lee, Y.J.; Park, K.D.; Shin, Y.; Kim, D.-H.; Yun, C.-H. Regioselective Hydroxylation Pathway of Tenatoprazole to Produce Human Metabolites by Bacillus Megaterium CYP102A1. Process Biochem. 2019, 87, 95–104. [Google Scholar] [CrossRef]
- Cao, N.T.; Nguyen, N.A.; Le, T.-K.; Cha, G.S.; Park, K.D.; Yun, C.-H. Regioselective Hydroxylation of Oleanolic Acid Catalyzed by Human CYP3A4 to Produce Hederagenenin, a Chiral Metabolite. Catalysts 2021, 11, 267. [Google Scholar] [CrossRef]
- Olesen, O.V.; Linnet, K. Identification of the Human Cytochrome P450 Isoforms Mediating in Vitro N-Dealkylation of Perphenazine. Br. J. Clin. Pharmacol. 2000, 50, 563–571. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; He, M.M.; Niu, W.; Wrighton, S.A.; Li, L.; Liu, Y.; Li, C. Metabolic Capabilities of Cytochrome P450 Enzymes in Chinese Liver Microsomes Compared with Those in Caucasian Liver Microsomes. Br. J. Clin. Pharmacol. 2012, 73, 268–284. [Google Scholar] [CrossRef] [Green Version]
- Available online: https://www.who.int/news-room/fact-sheets/detail/cardiovascular-diseases-(cvds) (accessed on 19 September 2021).
- Kazi, D.S.; Penko, J.M.; Bibbins-Domingo, K. Statins for Primary Prevention of Cardiovascular Disease: Review of Evidence and Recommendations for Clinical Practice. Med. Clin. N. Am. 2017, 101, 689–699. [Google Scholar] [CrossRef] [PubMed]
- Reiner, Ž. Statins in the primary prevention of cardiovascular disease. Nat. Rev. Cardiol. 2013, 10, 453–464. [Google Scholar] [CrossRef] [PubMed]
- Sigglekow, F.; Horsburgh, S.; Parkin, L. Statin adherence is lower in primary than secondary prevention: A national follow-up study of new users. PLoS ONE 2020, 15, e0242424. [Google Scholar] [CrossRef]
- Hope, H.F.; Binkley, G.M.; Fenton, S.; Kitas, G.D.; Verstappen, S.M.M.; Symmons, D.P.M. Systematic review of the predictors of statin adherence for the primary prevention of cardiovascular disease. PLoS ONE 2019, 14, e0201196. [Google Scholar] [CrossRef] [PubMed]
- Brown, F.; Singer, A.; Katz, A.; Konrad, G. Statin-prescribing trends for primary and secondary prevention of cardiovascular disease. Can. Fam. Physician 2017, 63, e495–e503. [Google Scholar]
- de Vries, F.M.; Kolthof, J.; Postma, M.J.; Denig, P.; Hak, E. Efficacy of Standard and Intensive Statin Treatment for the Secondary Prevention of Cardiovascular and Cerebrovascular Events in Diabetes Patients: A Meta-Analysis. PLoS ONE 2014, 9, e111247. [Google Scholar] [CrossRef]
- Virani, S.S. Statins in the primary and secondary prevention of cardiovascular disease in women: Facts and myths. Tex. Heart Inst. J. 2013, 40, 288–289. [Google Scholar]
- Schachter, M. Chemical, Pharmacokinetic and Pharmacodynamic Properties of Statins: An Update. Fundam. Clin. Pharmacol. 2005, 19, 117–125. [Google Scholar] [CrossRef]
- McKenney, J.M. Pharmacologic Characteristics of Statins. Clin Cardiol 2003, 26, 32–38. [Google Scholar] [CrossRef]
- Li, X.; Liu, C.; Duan, Z.; Guo, S. HMG-CoA Reductase Inhibitors from Monascus-Fermented Rice. J. Chem. 2013, 2013, e872056. [Google Scholar] [CrossRef] [Green Version]
- Dansette, P.M.; Jaoen, M.; Pons, C. HMG-CoA Reductase Activity in Human Liver Microsomes: Comparative Inhibition by Statins. Exp. Toxicol. Pathol. 2000, 52, 145–148. [Google Scholar] [CrossRef]
- Kang, J.-Y.; Ryu, S.H.; Park, S.-H.; Cha, G.S.; Kim, D.-H.; Kim, K.-H.; Hong, A.W.; Ahn, T.; Pan, J.-G.; Joung, Y.H.; et al. Chimeric Cytochromes P450 Engineered by Domain Swapping and Random Mutagenesis for Producing Human Metabolites of Drugs. Biotechnol. Bioeng. 2014, 111, 1313–1322. [Google Scholar] [CrossRef]
- Murphy, C.; Deplazes, E.; Cranfield, C.G.; Garcia, A. The Role of Structure and Biophysical Properties in the Pleiotropic Effects of Statins. Int. J. Mol. Sci. 2020, 21, 8745. [Google Scholar] [CrossRef]
- Knights, K.M.; Rowland, A.; Miners, J.O. Renal Drug Metabolism in Humans: The Potential for Drug–Endobiotic Interactions Involving Cytochrome P450 (CYP) and UDP-Glucuronosyltransferase (UGT). Br. J. Clin. Pharmacol. 2013, 76, 587–602. [Google Scholar] [CrossRef] [Green Version]
- Zanger, U.M.; Schwab, M. Cytochrome P450 Enzymes in Drug Metabolism: Regulation of Gene Expression, Enzyme Activities, and Impact of Genetic Variation. Pharmacol. Ther. 2013, 138, 103–141. [Google Scholar] [CrossRef]
- Lins, R.L.; Matthys, K.E.; Verpooten, G.A.; Peeters, P.C.; Dratwa, M.; Stolear, J.; Lameire, N.H. Pharmacokinetics of Atorvastatin and Its Metabolites after Single and Multiple Dosing in Hypercholesterolaemic Haemodialysis Patients. Nephrol. Dial. Transplant. 2003, 18, 967–976. [Google Scholar] [CrossRef] [Green Version]
- Ogu, C.C.; Maxa, J.L. Drug Interactions Due to Cytochrome P450. Bayl. Univ. Med. Cent. Proc. 2000, 13, 421–423. [Google Scholar] [CrossRef]
- Yun, C.-H.; Ahn, T.; Guengerich, F.P.; Yamazaki, H.; Shimada, T. Phospholipase D Activity of Cytochrome P450 in Human Liver Endoplasmic Reticulum. Arch. Biochem. Biophys. 1999, 367, 81–88. [Google Scholar] [CrossRef] [PubMed]
- Yun, C.-H.; Yim, S.-K.; Kim, D.-H.; Ahn, T. Functional Expression of Human Cytochrome P450 Enzymes in Escherichia Coli. Curr. Drug Metab. 2006, 7, 411–429. [Google Scholar] [CrossRef] [PubMed]
- Parikh, A.; Gillam, E.M.J.; Guengerich, F.P. Drug Metabolism by Escherichia Coli Expressing Human Cytochromes P450. Nat. Biotechnol. 1997, 15, 784–788. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.-Y.; Kim, S.-Y.; Kim, D.; Kim, D.-H.; Shin, S.-M.; Park, S.-H.; Kim, K.-H.; Jung, H.-C.; Pan, J.-G.; Joung, Y.H.; et al. Characterization of Diverse Natural Variants of CYP102A1 Found within a Species of Bacillus Megaterium. AMB Express 2011, 1, 1. [Google Scholar] [CrossRef] [Green Version]
- Le, T.-K.; Jang, H.-H.; Nguyen, H.T.H.; Doan, T.T.M.; Lee, G.-Y.; Park, K.D.; Ahn, T.; Joung, Y.H.; Kang, H.-S.; Yun, C.-H. Highly Regioselective Hydroxylation of Polydatin, a Resveratrol Glucoside, for One-Step Synthesis of Astringin, a Piceatannol Glucoside, by P450 BM3. Enzyme Microb. Technol. 2017, 97, 34–42. [Google Scholar] [CrossRef]
- Kim, D.-H.; Ahn, T.; Jung, H.-C.; Pan, J.-G.; Yun, C.-H. Generation of the Human Metabolite Piceatannol from the Anticancer-Preventive Agent Resveratrol by Bacterial Cytochrome P450 BM3. Drug Metab. Dispos. 2009, 37, 932–936. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Enzymes | kcat (min−1) | Km (μM) | kcat/Km (min−1 μM−1) |
|---|---|---|---|
| M16V2 | 0.086 ± 0.025 | 117 ± 97 | 0.00074 ± 0.00065 |
| M380 | 0.24 ± 0.07 | 656 ± 285 | 0.00037 ± 0.00019 |
| M371 | 0.88 ± 0.26 | 527 ± 256 | 0.0017 ± 0.0009 |
| M697 | 1.01 ± 0.14 | 92 ± 42 | 0.011 ± 0.005 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cao, N.T.; Nguyen, N.A.; Park, C.M.; Cha, G.S.; Park, K.D.; Yun, C.-H. A Novel Statin Compound from Monacolin J Produced Using CYP102A1-Catalyzed Regioselective C-Hydroxylation. Pharmaceuticals 2021, 14, 981. https://doi.org/10.3390/ph14100981
Cao NT, Nguyen NA, Park CM, Cha GS, Park KD, Yun C-H. A Novel Statin Compound from Monacolin J Produced Using CYP102A1-Catalyzed Regioselective C-Hydroxylation. Pharmaceuticals. 2021; 14(10):981. https://doi.org/10.3390/ph14100981
Chicago/Turabian StyleCao, Ngoc Tan, Ngoc Anh Nguyen, Chan Mi Park, Gun Su Cha, Ki Deok Park, and Chul-Ho Yun. 2021. "A Novel Statin Compound from Monacolin J Produced Using CYP102A1-Catalyzed Regioselective C-Hydroxylation" Pharmaceuticals 14, no. 10: 981. https://doi.org/10.3390/ph14100981
APA StyleCao, N. T., Nguyen, N. A., Park, C. M., Cha, G. S., Park, K. D., & Yun, C.-H. (2021). A Novel Statin Compound from Monacolin J Produced Using CYP102A1-Catalyzed Regioselective C-Hydroxylation. Pharmaceuticals, 14(10), 981. https://doi.org/10.3390/ph14100981