Substance Use Disorder in the COVID-19 Pandemic: A Systematic Review of Vulnerabilities and Complications


Abstract
1. Introduction
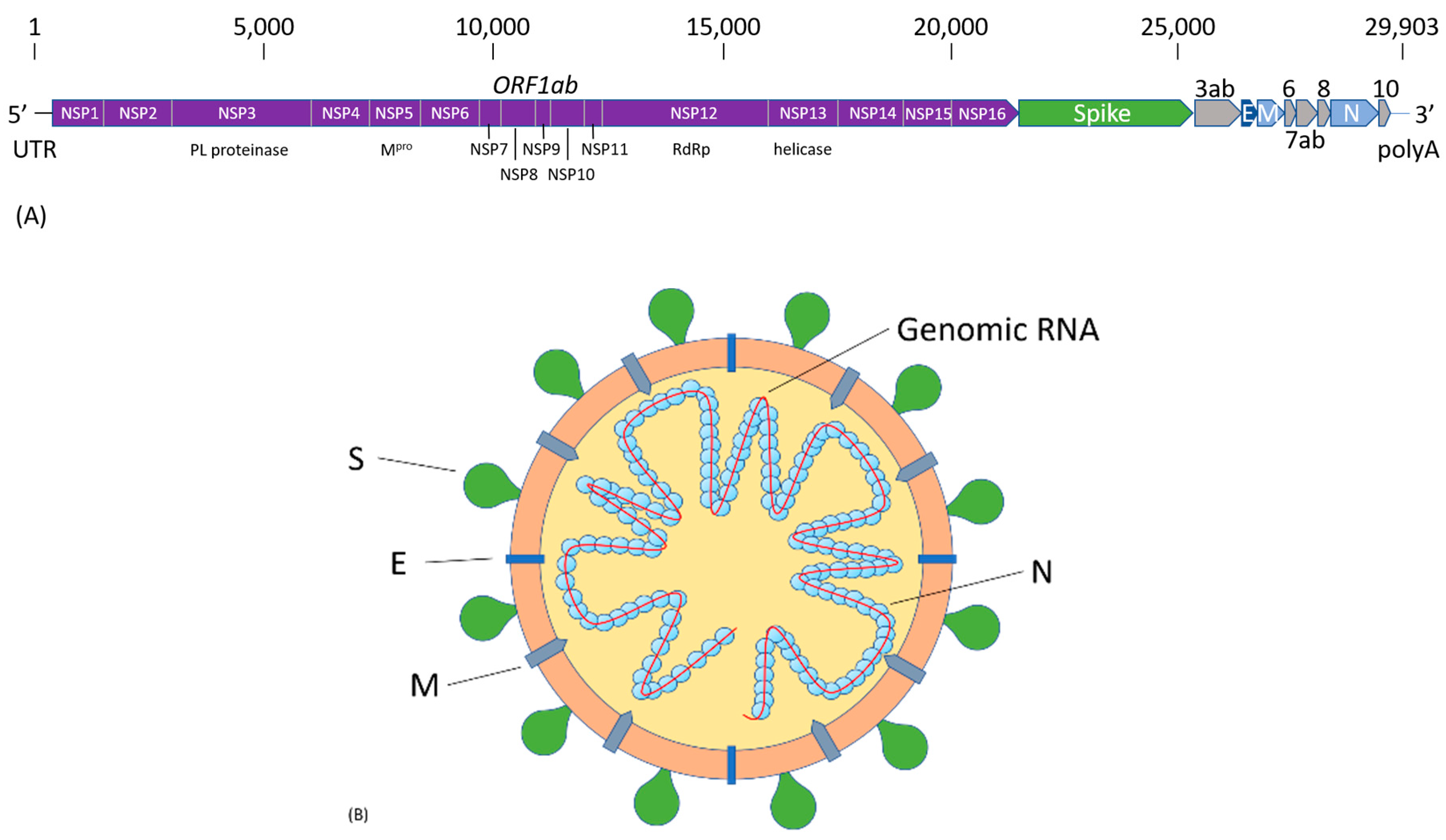
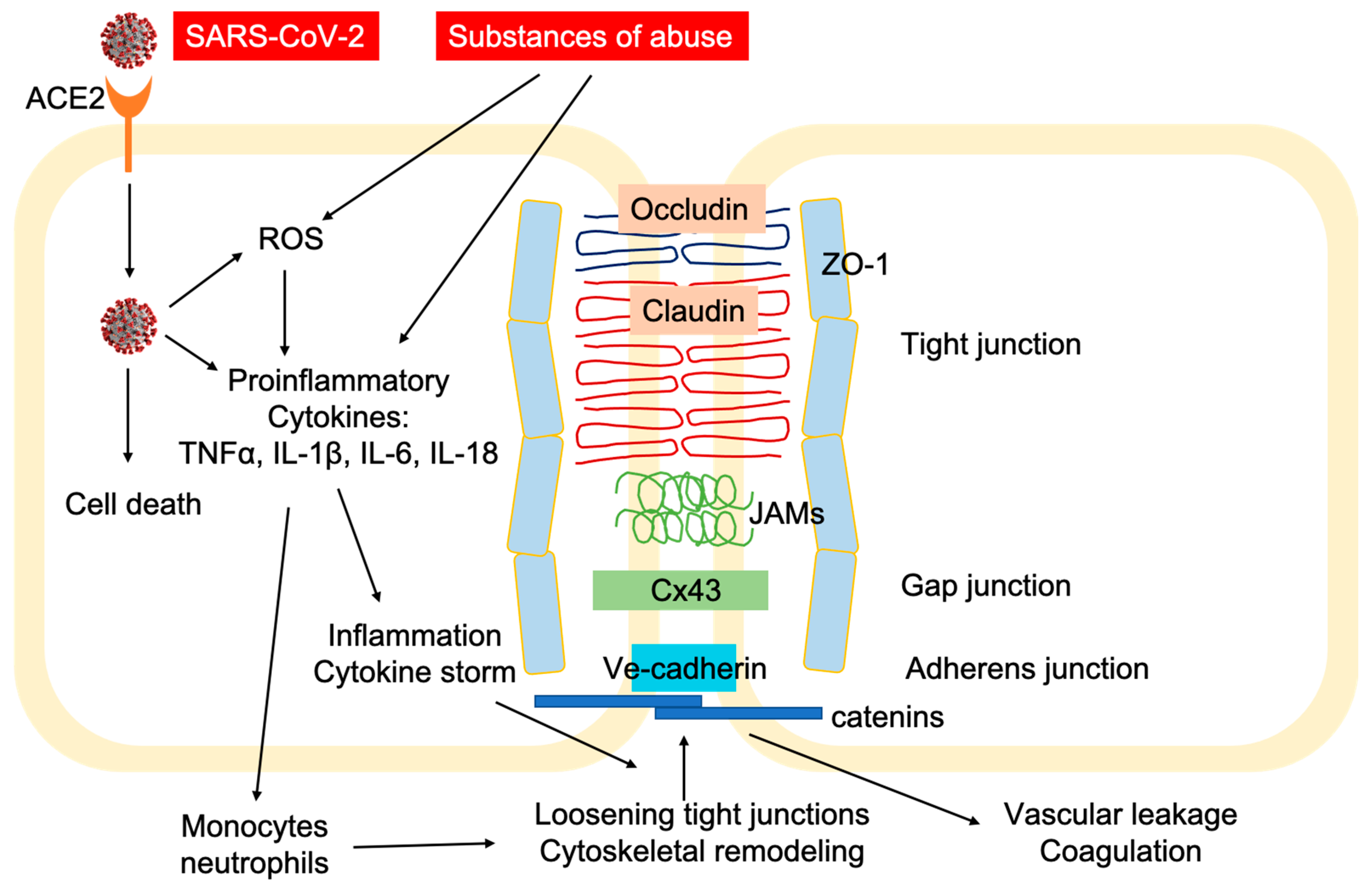
2. SARS-CoV-2 Infection and COVID-19 Pathogenesis
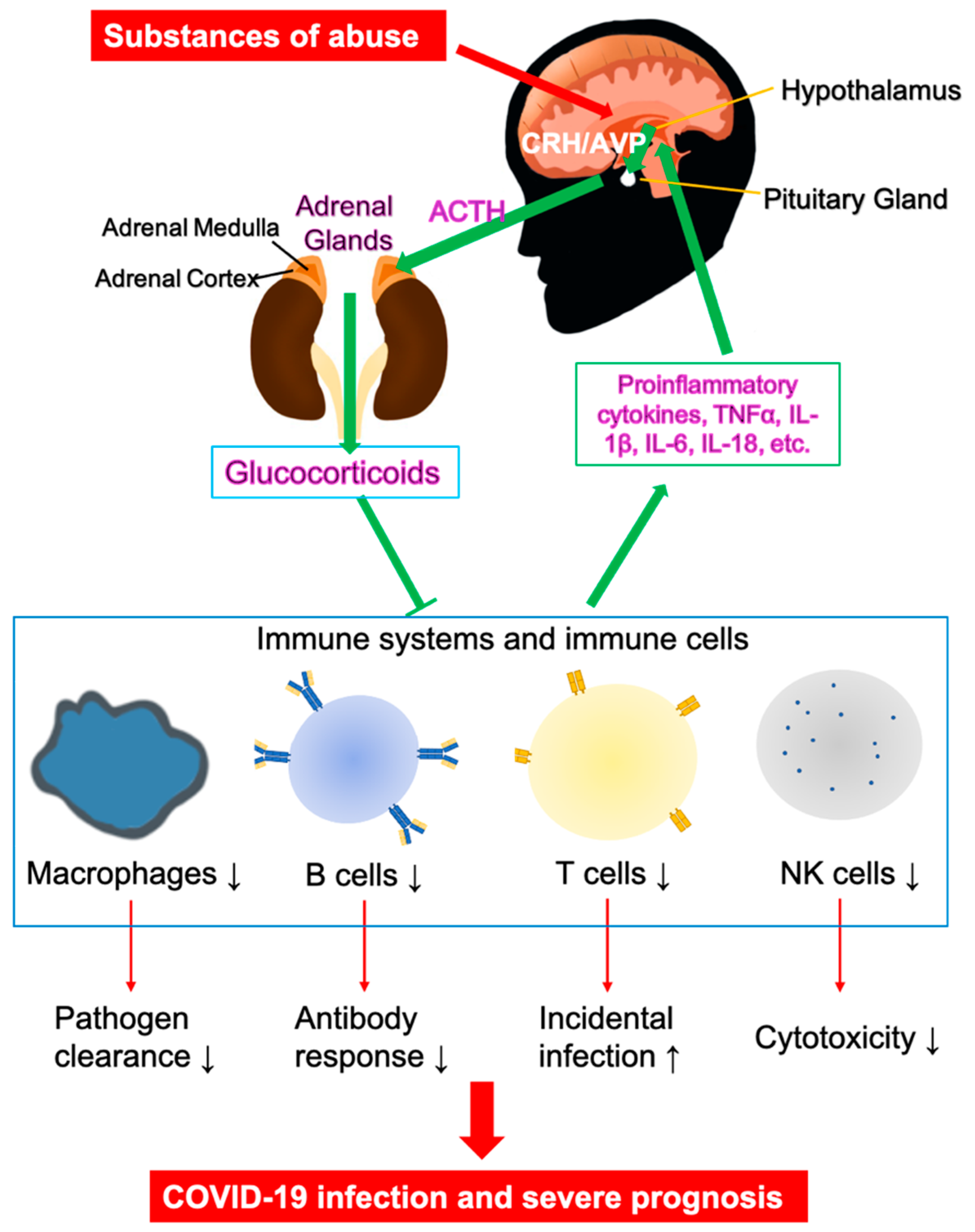
3. Vulnerability of Substance Use Disorders (SUDs) in COVID-19
4. Effects of Commonly Abused Substances on COVID-19
4.1. Nicotine
4.2. Alcohol
4.3. Marijuana
4.4. Opioids
4.5. Cocaine
4.6. Amphetamine, Methamphetamine (METH), and 3,4-Methylenedioxymethamphetamine (MDMA, Ecstasy)
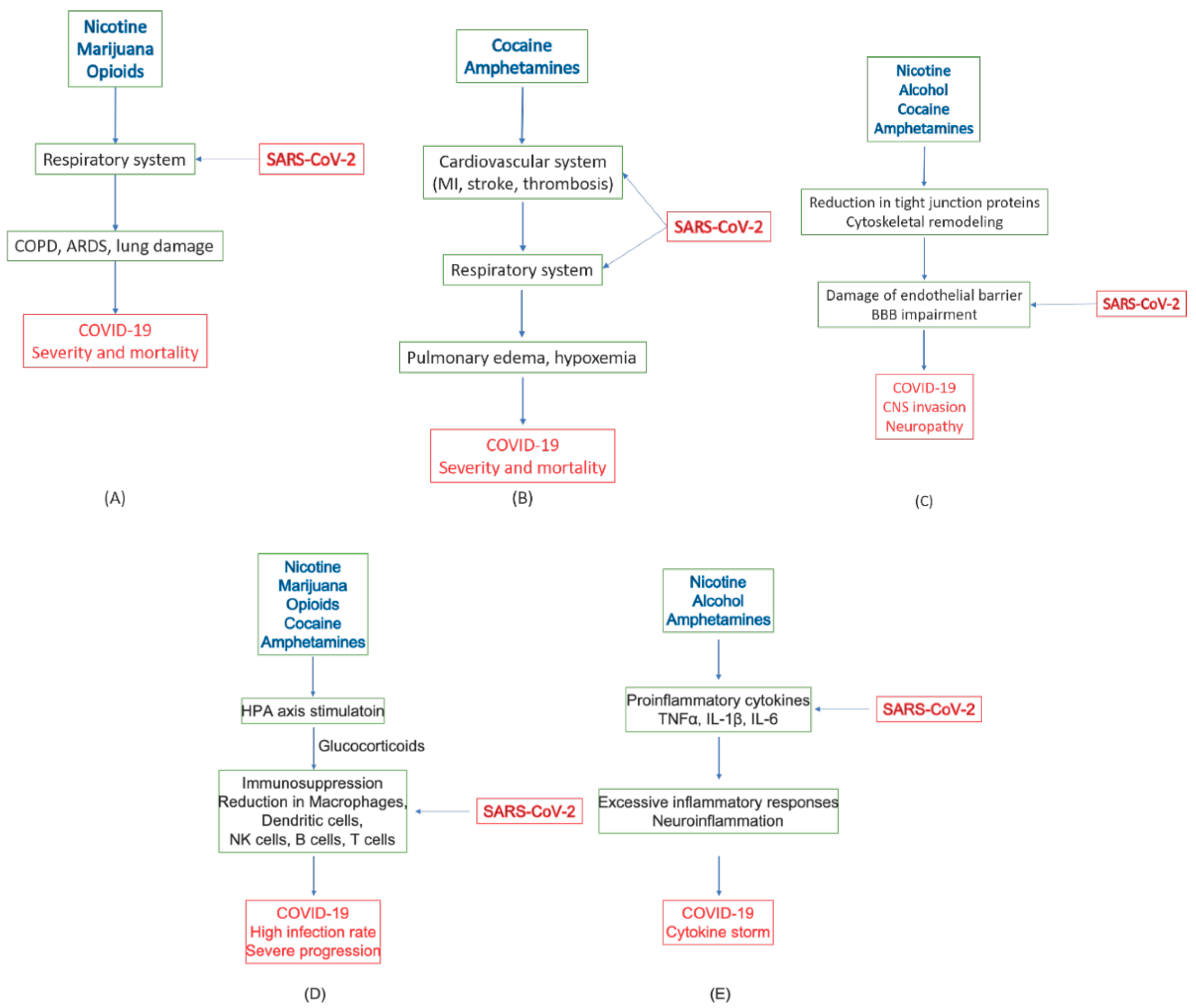
4.7. Summary of Roles of Substances of Abuse in COVID-19
5. Strategies of Treatment and Prevention for Individuals with SUDs
5.1. General Approaches for Treatments and Vaccines
5.2. Challenges for Individuals with SUDs
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Coronaviridae Study Group of the International Committee on Taxonomy of Viruses. The species Severe acute respiratory syndrome-related coronavirus: Classifying 2019-nCoV and naming it SARS-CoV-2. Nat. Microbiol. 2020, 5, 536–544. [Google Scholar] [CrossRef]
- Zhu, N.; Zhang, D.; Wang, W.; Li, X.; Yang, B.; Song, J.; Zhao, X.; Huang, B.; Shi, W.; Lu, R.; et al. A Novel Coronavirus from Patients with Pneumonia in China, 2019. N. Engl. J. Med. 2020, 382, 727–733. [Google Scholar] [CrossRef]
- World Health Organization. WHO Coronavirus Disease (COVID-19) Dashboard. Available online: https://covid19.who.int/?gclid=CjwKCAjw_-D3BRBIEiwAjVMy7IzWSCeKBbqAB9-TFuuZjmGDDMEdt6ZvGZjjKeqBgN3kIA6kRl9XEhoCG9kQAvD_BwE (accessed on 28 June 2020).
- Johns Hopkins University & Medicine. Coronavirus Resource Center. Available online: https://coronavirus.jhu.edu/ (accessed on 28 June 2020).
- World Health Organization. WHO Director-General’s Opening Remarks at the Media Briefing on COVID-19–11 March 2020. Available online: https://www.who.int/dg/speeches/detail/who-director-general-s-opening-remarks-at-the-media-briefing-on-covid-19---11-march-2020 (accessed on 17 March 2020).
- United Nations Office on Drugs and Crime. World Drug Report 2019. Available online: https://wdr.unodc.org/wdr2019/ (accessed on 25 June 2020).
- Substance Abuse and Mental Health Services Administration. Key Substance Use and Mental Health Indicators in the United States: Results from the 2018 National Survey on Drug Use and Health; Center for Behavioral Health Statistics and Quality: Rockville, MD, USA, 2019.
- Volkow, N.D. Collision of the COVID-19 and Addiction Epidemics. Ann. Intern. Med. 2020, M20-1212. [Google Scholar] [CrossRef]
- Madison, M.C.; Landers, C.T.; Gu, B.-H.; Chang, C.-Y.; Tung, H.-Y.; You, R.; Hong, M.J.; Baghaei, N.; Song, L.-Z.; Porter, P.; et al. Electronic cigarettes disrupt lung lipid homeostasis and innate immunity independent of nicotine. J. Clin. Investig. 2020, 129, 4290–4304. [Google Scholar] [CrossRef]
- Sundar, I.K.; Nevid, M.Z.; Friedman, A.E.; Rahman, I. Cigarette smoke induces distinct histone modifications in lung cells: Implications for the pathogenesis of COPD and lung cancer. J. Proteome Res. 2014, 13, 982–996. [Google Scholar] [CrossRef] [PubMed]
- Aldington, S.; Williams, M.; Nowitz, M.; Weatherall, M.; Pritchard, A.; McNaughton, A.; Robinson, G.; Beasley, R. Effects of cannabis on pulmonary structure, function and symptoms. Thorax 2007, 62, 1058–1063. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; McGoogan, J.M. Characteristics of and Important Lessons from the Coronavirus Disease 2019 (COVID-19) Outbreak in China: Summary of a Report of 72 314 Cases From the Chinese Center for Disease Control and Prevention. JAMA 2020. [Google Scholar] [CrossRef]
- Alqahtani, J.S.; Oyelade, T.; Aldhahir, A.M.; Alghamdi, S.M.; Almehmadi, M.; Alqahtani, A.S.; Quaderi, S.; Mandal, S.; Hurst, J.R. Prevalence, Severity and Mortality associated with COPD and Smoking in patients with COVID-19: A Rapid Systematic Review and Meta-Analysis. PLoS ONE 2020, 15, e0233147. [Google Scholar] [CrossRef]
- Alexander, G.C.; Stoller, K.B.; Haffajee, R.L.; Saloner, B. An Epidemic in the Midst of a Pandemic: Opioid Use Disorder and COVID-19. Ann. Intern. Med. 2020, M20-1141. [Google Scholar] [CrossRef]
- Becker, W.C.; Fiellin, D.A. When Epidemics Collide: Coronavirus Disease 2019 (COVID-19) and the Opioid Crisis. Ann. Intern. Med. 2020, M20-1210. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.X.; Kwong, C.; Swaminathan, A.; Gohil, A.; Crawford, M.H. Clinical Characteristics and Outcome of Methamphetamine-Associated Pulmonary Arterial Hypertension and Dilated Cardiomyopathy. JACC Heart Fail. 2018, 6, 209–218. [Google Scholar] [CrossRef] [PubMed]
- Lynch, F.L.; Peterson, E.L.; Lu, C.Y.; Hu, Y.; Rossom, R.C.; Waitzfelder, B.E.; Owen-Smith, A.A.; Hubley, S.; Prabhakar, D.; Keoki Williams, L.; et al. Substance use disorders and risk of suicide in a general US population: A case control study. Addict. Sci. Clin. Pract. 2020, 15, 14. [Google Scholar] [CrossRef]
- Farhoudian, A.; Baldacchino, A.; Clark, N.; Gerra, G.; Ekhtiari, H.; Dom, G.; Mokri, A.; Sadeghi, M.; Nematollahi, P.; Demasi, M.; et al. COVID-19 and Substance Use Disorders: Recommendations to a Comprehensive Healthcare Response. An International Society of Addiction Medicine (ISAM) Practice and Policy Interest Group Position Paper. Basic Clin. Neurosci. J. 2020, 11, 133–146. [Google Scholar] [CrossRef]
- Cui, J.; Li, F.; Shi, Z.-L. Origin and evolution of pathogenic coronaviruses. Nat. Rev. Microbiol. 2019, 17, 181–192. [Google Scholar] [CrossRef] [PubMed]
- Zhou, P.; Yang, X.-L.; Wang, X.-G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.-R.; Zhu, Y.; Li, B.; Huang, C.-L.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef]
- Wu, F.; Zhao, S.; Yu, B.; Chen, Y.M.; Wang, W.; Song, Z.G.; Hu, Y.; Tao, Z.W.; Tian, J.H.; Pei, Y.Y.; et al. A new coronavirus associated with human respiratory disease in China. Nature 2020, 579, 265–269. [Google Scholar] [CrossRef]
- Letko, M.; Marzi, A.; Munster, V. Functional assessment of cell entry and receptor usage for SARS-CoV-2 and other lineage B betacoronaviruses. Nat. Microbiol. 2020, 5, 562–569. [Google Scholar] [CrossRef]
- Wrapp, D.; Wang, N.; Corbett, K.S.; Goldsmith, J.A.; Hsieh, C.L.; Abiona, O.; Graham, B.S.; McLellan, J.S. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science 2020, 367, 1260–1263. [Google Scholar] [CrossRef]
- Walls, A.C.; Park, Y.J.; Tortorici, M.A.; Wall, A.; McGuire, A.T.; Veesler, D. Structure, Function, and Antigenicity of the SARS-CoV-2 Spike Glycoprotein. Cell 2020, 181, 281–292.e286. [Google Scholar] [CrossRef]
- Tai, W.; He, L.; Zhang, X.; Pu, J.; Voronin, D.; Jiang, S.; Zhou, Y.; Du, L. Characterization of the receptor-binding domain (RBD) of 2019 novel coronavirus: Implication for development of RBD protein as a viral attachment inhibitor and vaccine. Cell. Mol. Immunol. 2020. [Google Scholar] [CrossRef]
- Vankadari, N.; Wilce, J.A. Emerging WuHan (COVID-19) coronavirus: Glycan shield and structure prediction of spike glycoprotein and its interaction with human CD26. Emerg. Microbes Infect. 2020, 9, 601–604. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, Y.; Allen, J.D.; Wrapp, D.; McLellan, J.S.; Crispin, M. Site-specific glycan analysis of the SARS-CoV-2 spike. Science 2020. [Google Scholar] [CrossRef]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Krüger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280.e278. [Google Scholar] [CrossRef] [PubMed]
- Sungnak, W.; Huang, N.; Bécavin, C.; Berg, M.; Queen, R.; Litvinukova, M.; Talavera-López, C.; Maatz, H.; Reichart, D.; Sampaziotis, F.; et al. SARS-CoV-2 entry factors are highly expressed in nasal epithelial cells together with innate immune genes. Nat. Med. 2020, 26, 681–687. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Geng, M.; Peng, Y.; Meng, L.; Lu, S. Molecular immune pathogenesis and diagnosis of COVID-19. J. Pharm. Anal. 2020, 10, 102–108. [Google Scholar] [CrossRef]
- Thoms, M.; Buschauer, R.; Ameismeier, M.; Koepke, L.; Denk, T.; Hirschenberger, M.; Kratzat, H.; Hayn, M.; Mackens-Kiani, T.; Cheng, J.; et al. Structural basis for translational shutdown and immune evasion by the Nsp1 protein of SARS-CoV-2. bioRxiv 2020. [Google Scholar] [CrossRef]
- Srinivasan, S.; Cui, H.; Gao, Z.; Liu, M.; Lu, S.; Mkandawire, W.; Narykov, O.; Sun, M.; Korkin, D. Structural Genomics of SARS-CoV-2 Indicates Evolutionary Conserved Functional Regions of Viral Proteins. Viruses 2020, 12, 360. [Google Scholar] [CrossRef]
- Van Dorp, L.; Acman, M.; Richard, D.; Shaw, L.P.; Ford, C.E.; Ormond, L.; Owen, C.J.; Pang, J.; Tan, C.C.S.; Boshier, F.A.T.; et al. Emergence of genomic diversity and recurrent mutations in SARS-CoV-2. Infect. Genet. Evol. 2020. [Google Scholar] [CrossRef]
- Hu, J.; He, C.L.; Gao, Q.; Zhang, G.J.; Cao, X.X.; Long, Q.X.; Deng, H.J.; Huang, L.Y.; Chen, J.; Wang, K.; et al. The D614G mutation of SARS-CoV-2 spike protein enhances viral infectivity and decreases neutralization sensitivity to individual convalescent sera. bioRxiv 2020. [Google Scholar] [CrossRef]
- Zhang, L.; Jackson, C.B.; Mou, H.; Ojha, A.; Rangarajan, E.S.; Izard, T.; Farzan, M.; Choe, H. The D614G mutation in the SARS-CoV-2 spike protein reduces S1 shedding and increases infectivity. bioRxiv 2020. [Google Scholar] [CrossRef]
- Korber, B.; Fischer, W.; Gnanakaran, S.; Yoon, H.; Theiler, J.; Abfalterer, W.; Foley, B.; Giorgi, E.; Bhattacharya, T.; Parker, M.; et al. Spike mutation pipeline reveals the emergence of a more transmissible form of SARS-CoV-2. bioRxiv 2020. [Google Scholar] [CrossRef]
- Petrilli, C.M.; Jones, S.A.; Yang, J.; Rajagopalan, H.; O’Donnell, L.F.; Chernyak, Y.; Tobin, K.; Cerfolio, R.J.; Francois, F.; Horwitz, L.I. Factors associated with hospitalization and critical illness among 4103 patients with COVID-19 disease in New York City. medRxiv 2020. [Google Scholar] [CrossRef]
- Ji, W.; Huh, K.; Kang, M.; Hong, J.; Bae, G.H.; Lee, R.; Na, Y.; Choi, H.; Gong, S.Y.; Choi, Y.H.; et al. Effect of Underlying Comorbidities on the Infection and Severity of COVID-19 in Korea: A Nationwide Case-Control Study. J. Korean Med. Sci. 2020, 35, e237. [Google Scholar] [CrossRef] [PubMed]
- Slaunwhite, A.K.; Gan, W.Q.; Xavier, C.; Zhao, B.; Buxton, J.A.; Desai, R. Overdose and risk factors for coronavirus disease 2019. Drug Alcohol Depend. 2020, 212, 108047. [Google Scholar] [CrossRef]
- Clay, J.M.; Parker, M.O. Alcohol use and misuse during the COVID-19 pandemic: A potential public health crisis? Lancet Public Health 2020, 5. [Google Scholar] [CrossRef]
- Sun, Y.; Li, Y.; Bao, Y.; Meng, S.; Sun, Y.; Schumann, G.; Kosten, T.; Strang, J.; Lu, L.; Shi, J. Brief Report: Increased Addictive Internet and Substance Use Behavior During the COVID-19 Pandemic in China. Am. J. Addict. 2020, 29, 268–270. [Google Scholar] [CrossRef]
- Cao, W.; Li, T. COVID-19: Towards understanding of pathogenesis. Cell Res. 2020, 30, 367–369. [Google Scholar] [CrossRef]
- Ziegler, C.G.K.; Allon, S.J.; Nyquist, S.K.; Mbano, I.M.; Miao, V.N.; Tzouanas, C.N.; Cao, Y.; Yousif, A.S.; Bals, J.; Hauser, B.M.; et al. SARS-CoV-2 Receptor ACE2 Is an Interferon-Stimulated Gene in Human Airway Epithelial Cells and Is Detected in Specific Cell Subsets across Tissues. Cell 2020, 181, 1016–1035.e1019. [Google Scholar] [CrossRef]
- Ravindra, N.G.; Alfajaro, M.M.; Gasque, V.; Wei, J.; Filler, R.B.; Huston, N.C.; Wan, H.; Szigeti-Buck, K.; Wang, B.; Montgomery, R.R.; et al. Single-cell longitudinal analysis of SARS-CoV-2 infection in human bronchial epithelial cells. bioRxiv 2020. [Google Scholar] [CrossRef]
- Hamming, I.; Timens, W.; Bulthuis, M.L.; Lely, A.T.; Navis, G.; van Goor, H. Tissue distribution of ACE2 protein, the functional receptor for SARS coronavirus. A first step in understanding SARS pathogenesis. J. Pathol. 2004, 203, 631–637. [Google Scholar] [CrossRef]
- Varga, Z.; Flammer, A.J.; Steiger, P.; Haberecker, M.; Andermatt, R.; Zinkernagel, A.S.; Mehra, M.R.; Schuepbach, R.A.; Ruschitzka, F.; Moch, H. Endothelial cell infection and endotheliitis in COVID-19. Lancet 2020, 395, 1417–1418. [Google Scholar] [CrossRef]
- Ackermann, M.; Verleden, S.E.; Kuehnel, M.; Haverich, A.; Welte, T.; Laenger, F.; Vanstapel, A.; Werlein, C.; Stark, H.; Tzankov, A.; et al. Pulmonary Vascular Endothelialitis, Thrombosis, and Angiogenesis in Covid-19. N. Engl. J. Med. 2020. [Google Scholar] [CrossRef] [PubMed]
- Bauer, H.C.; Krizbai, I.A.; Bauer, H.; Traweger, A. “You Shall Not Pass”-tight junctions of the blood brain barrier. Front. Neurosci. 2014, 8, 392. [Google Scholar] [CrossRef] [PubMed]
- Luissint, A.C.; Artus, C.; Glacial, F.; Ganeshamoorthy, K.; Couraud, P.O. Tight junctions at the blood brain barrier: Physiological architecture and disease-associated dysregulation. Fluids Barriers CNS 2012, 9, 23. [Google Scholar] [CrossRef] [PubMed]
- Löscher, W.; Potschka, H. Drug resistance in brain diseases and the role of drug efflux transporters. Nat. Rev. Neurosci. 2005, 6, 591–602. [Google Scholar] [CrossRef]
- Miller, D.S.; Cannon, R.E. Signaling pathways that regulate basal ABC transporter activity at the blood-brain barrier. Curr. Pharm. Des. 2014, 20, 1463–1471. [Google Scholar] [CrossRef]
- Stamatovic, S.M.; Johnson, A.M.; Keep, R.F.; Andjelkovic, A.V. Junctional proteins of the blood-brain barrier: New insights into function and dysfunction. Tissue Barriers 2016, 4, e1154641. [Google Scholar] [CrossRef]
- Baig, A.M.; Khaleeq, A.; Ali, U.; Syeda, H. Evidence of the COVID-19 Virus Targeting the CNS: Tissue Distribution, Host-Virus Interaction, and Proposed Neurotropic Mechanisms. ACS Chem. Neurosci. 2020, 11, 995–998. [Google Scholar] [CrossRef]
- Li, Y.C.; Bai, W.Z.; Hashikawa, T. The neuroinvasive potential of SARS-CoV2 may play a role in the respiratory failure of COVID-19 patients. J. Med. Virol. 2020, 92, 552–555. [Google Scholar] [CrossRef]
- Moriguchi, T.; Harii, N.; Goto, J.; Harada, D.; Sugawara, H.; Takamino, J.; Ueno, M.; Sakata, H.; Kondo, K.; Myose, N.; et al. A first case of meningitis/encephalitis associated with SARS-Coronavirus-2. Int. J. Infect. Dis. 2020, 94, 55–58. [Google Scholar] [CrossRef]
- Li, H.; Xue, Q.; Xu, X. Involvement of the Nervous System in SARS-CoV-2 Infection. Neurotox. Res. 2020, 38, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Mao, L.; Jin, H.; Wang, M.; Hu, Y.; Chen, S.; He, Q.; Chang, J.; Hong, C.; Zhou, Y.; Wang, D.; et al. Neurologic Manifestations of Hospitalized Patients with Coronavirus Disease 2019 in Wuhan, China. JAMA Neurol. 2020, 77, 683–690. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Liu, L.; Zhang, D.; Xu, J.; Dai, H.; Tang, N.; Su, X.; Cao, B. SARS-CoV-2 and viral sepsis: Observations and hypotheses. Lancet 2020, 395, 1517–1520. [Google Scholar] [CrossRef]
- Coperchini, F.; Chiovato, L.; Croce, L.; Magri, F.; Rotondi, M. The cytokine storm in COVID-19: An overview of the involvement of the chemokine/chemokine-receptor system. Cytokine Growth Factor Rev. 2020, 53, 25–32. [Google Scholar] [CrossRef]
- Bouhaddou, M.; Memon, D.; Meyer, B.; White, K.M.; Rezelj, V.V.; Correa Marrero, M.; Polacco, B.J.; Melnyk, J.E.; Ulferts, S.; Kaake, R.M.; et al. The Global Phosphorylation Landscape of SARS-CoV-2 Infection. Cell 2020. [Google Scholar] [CrossRef]
- Huang, C.; Wang, Y.; Li, X.; Ren, L.; Zhao, J.; Hu, Y.; Zhang, L.; Fan, G.; Xu, J.; Gu, X.; et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 2020, 395, 497–506. [Google Scholar] [CrossRef]
- Liu, J.; Li, S.; Liu, J.; Liang, B.; Wang, X.; Wang, H.; Li, W.; Tong, Q.; Yi, J.; Zhao, L.; et al. Longitudinal characteristics of lymphocyte responses and cytokine profiles in the peripheral blood of SARS-CoV-2 infected patients. EBioMedicine 2020, 55, 102763. [Google Scholar] [CrossRef]
- Gong, J.; Dong, H.; Xia, S.Q.; Huang, Y.Z.; Wang, D.; Zhao, Y.; Liu, W.; Tu, S.; Zhang, M.; Wang, Q.; et al. Correlation Analysis Between Disease Severity and Inflammation-related Parameters in Patients with COVID-19 Pneumonia. medRxiv 2020. [Google Scholar] [CrossRef]
- Iwasaki, A.; Pillai, P.S. Innate immunity to influenza virus infection. Nat. Rev. Immunol. 2014, 14, 315–328. [Google Scholar] [CrossRef]
- Liu, T.; Zhang, J.; Yang, Y.; Ma, H.; Li, Z.; Zhang, J.; Cheng, J.; Zhang, X.; Zhao, Y.; Xia, Z.; et al. The potential role of IL-6 in monitoring severe case of coronavirus disease 2019. medRxiv 2020. [Google Scholar] [CrossRef]
- Broggi, A.; Ghosh, S.; Sposito, B.; Spreafico, R.; Balzarini, F.; Lo Cascio, A.; Clementi, N.; De Santis, M.; Mancini, N.; Granucci, F.; et al. Type III interferons disrupt the lung epithelial barrier upon viral recognition. Science 2020, eabc3545. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Ye, L.; Ye, L.; Li, B.; Gao, B.; Zeng, Y.; Kong, L.; Fang, X.; Zheng, H.; Wu, Z.; et al. Up-regulation of IL-6 and TNF-alpha induced by SARS-coronavirus spike protein in murine macrophages via NF-kappaB pathway. Virus Res. 2007, 128, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Zhou, Y.-H.; Yang, Z.-Q. The cytokine storm of severe influenza and development of immunomodulatory therapy. Cell. Mol. Immunol. 2016, 13, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Aller, M.A.; Arias, J.L.; Lorente, L.; Nava, M.P.; Duran, H.J.; Arias, J. Neuro-immune-endocrine functional system and vascular pathology. Med. Hypotheses 2001, 57, 561–569. [Google Scholar] [CrossRef]
- Meazza, C.; Pagani, S.; Travaglino, P.; Bozzola, M. Effect of growth hormone (GH) on the immune system. Pediatr. Endocrinol. Rev. 2004, 1 (Suppl. 3), 490–495. [Google Scholar]
- Hosoi, T.; Nomura, Y. Functional role of acetylcholine in the immune system. Front. Biosci. 2004, 9, 2414–2419. [Google Scholar] [CrossRef][Green Version]
- Fuchs, F.; Damm, J.; Gerstberger, R.; Roth, J.; Rummel, C. Activation of the inflammatory transcription factor nuclear factor interleukin-6 during inflammatory and psychological stress in the brain. J. Neuroinflammation 2013, 10, 905. [Google Scholar] [CrossRef]
- Pivneva, T.A. Microglia in normal condition and pathology. Fiziol. Zh. 2008, 54, 81–89. [Google Scholar]
- Sternberg, E.M. Neural regulation of innate immunity: A coordinated nonspecific host response to pathogens. Nat. Rev. Immunol. 2006, 6, 318–328. [Google Scholar] [CrossRef]
- Armario, A. Activation of the hypothalamic-pituitary-adrenal axis by addictive drugs: Different pathways, common outcome. Trends Pharmacol. Sci. 2010, 31, 318–325. [Google Scholar] [CrossRef]
- Tisoncik, J.R.; Korth, M.J.; Simmons, C.P.; Farrar, J.; Martin, T.R.; Katze, M.G. Into the eye of the cytokine storm. Microbiol. Mol. Biol. Rev. 2012, 76, 16–32. [Google Scholar] [CrossRef] [PubMed]
- Huang, K.J.; Su, I.J.; Theron, M.; Wu, Y.C.; Lai, S.K.; Liu, C.C.; Lei, H.Y. An interferon-gamma-related cytokine storm in SARS patients. J. Med. Virol. 2005, 75, 185–194. [Google Scholar] [CrossRef] [PubMed]
- Sajja, R.K.; Rahman, S.; Cucullo, L. Drugs of abuse and blood-brain barrier endothelial dysfunction: A focus on the role of oxidative stress. J. Cereb. Blood Flow Metab. 2016, 36, 539–554. [Google Scholar] [CrossRef] [PubMed]
- Howard, G.; Wagenknecht, L.E.; Burke, G.L.; Diez-Roux, A.; Evans, G.W.; McGovern, P.; Nieto, F.J.; Tell, G.S. Cigarette smoking and progression of atherosclerosis: The Atherosclerosis Risk in Communities (ARIC) Study. JAMA 1998, 279, 119–124. [Google Scholar] [CrossRef]
- Bhat, V.M.; Cole, J.W.; Sorkin, J.D.; Wozniak, M.A.; Malarcher, A.M.; Giles, W.H.; Stern, B.J.; Kittner, S.J. Dose-Response Relationship Between Cigarette Smoking and Risk of Ischemic Stroke in Young Women. Stroke 2008, 39, 2439–2443. [Google Scholar] [CrossRef]
- Patanavanich, R.; Glantz, S.A. Smoking Is Associated with COVID-19 Progression: A Meta-analysis. Nicotine Tob. Res. 2020. [Google Scholar] [CrossRef]
- Sopori, M. Effects of cigarette smoke on the immune system. Nat. Rev. Immunol. 2002, 2, 372–377. [Google Scholar] [CrossRef]
- Geng, Y.; Savage, S.M.; Razani-Boroujerdi, S.; Sopori, M.L. Effects of nicotine on the immune response. II. Chronic nicotine treatment induces T cell anergy. J. Immunol. 1996, 156, 2384–2390. [Google Scholar]
- Zhou, Z.; Chen, P.; Peng, H. Are healthy smokers really healthy? Tob. Induc. Dis. 2016, 14, 35. [Google Scholar] [CrossRef]
- Vardavas, C.I.; Nikitara, K. COVID-19 and smoking: A systematic review of the evidence. Tob. Induc. Dis. 2020, 18, 20. [Google Scholar] [CrossRef]
- Abbruscato, T.J.; Lopez, S.P.; Mark, K.S.; Hawkins, B.T.; Davis, T.P. Nicotine and Cotinine Modulate Cerebral Microvascular Permeability and Protein Expression of ZO-1 through Nicotinic Acetylcholine Receptors Expressed on Brain Endothelial Cells. J. Pharm. Sci. 2002, 91, 2525–2538. [Google Scholar] [CrossRef]
- Hutamekalin, P.; Farkas, A.E.; Orbók, A.; Wilhelm, I.; Nagyőszi, P.; Veszelka, S.; Deli, M.A.; Buzás, K.; Hunyadi-Gulyás, É.; Medzihradszky, K.F.; et al. Effect of nicotine and polyaromtic hydrocarbons on cerebral endothelial cells. Cell Biol. Int. 2008, 32, 198–209. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Gaztelumendi, A.; Alvehus, M.; Andersson, T.; Jacobsson, S.O. Comparison of the effects of nicotine upon the transcellular electrical resistance and sucrose permeability of human ECV304/rat C6 co-cultures and human CaCo(2) cells. Toxicol. Lett. 2011, 207, 1–6. [Google Scholar] [CrossRef]
- Hawkins, B.T.; Abbruscato, T.J.; Egleton, R.D.; Brown, R.C.; Huber, J.D.; Campos, C.R.; Davis, T.P. Nicotine increases in vivo blood-brain barrier permeability and alters cerebral microvascular tight junction protein distribution. Brain Res. 2004, 1027, 48–58. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.-H.; Chen, S.H.-M.; Jong, A.; Zhou, Z.Y.; Li, W.; Suzuki, K.; Huang, S.-H. Enhanced Escherichia coli invasion of human brain microvascular endothelial cells is associated with alterations in cytoskeleton induced by nicotine. Cell. Microbiol. 2002, 4, 503–514. [Google Scholar] [CrossRef]
- Mazzone, P.; Tierney, W.; Hossain, M.; Puvenna, V.; Janigro, D.; Cucullo, L. Pathophysiological impact of cigarette smoke exposure on the cerebrovascular system with a focus on the blood-brain barrier: Expanding the awareness of smoking toxicity in an underappreciated area. Int. J. Environ. Res. Public Health 2010, 7, 4111–4126. [Google Scholar] [CrossRef] [PubMed]
- Bradford, S.T.; Stamatovic, S.M.; Dondeti, R.S.; Keep, R.F.; Andjelkovic, A.V. Nicotine aggravates the brain postischemic inflammatory response. Am. J. Physiol. Heart Circ. Physiol. 2011, 300, H1518–H1529. [Google Scholar] [CrossRef] [PubMed]
- Blomster, J.I.; Zoungas, S.; Chalmers, J.; Li, Q.; Chow, C.K.; Woodward, M.; Mancia, G.; Poulter, N.; Williams, B.; Harrap, S.; et al. The Relationship between Alcohol Consumption and Vascular Complications and Mortality in Individuals With Type 2 Diabetes. Diabetes Care 2014, 37, 1353–1359. [Google Scholar] [CrossRef]
- Jones, A.; McMillan, M.R.; Jones, R.W.; Kowalik, G.T.; Steeden, J.A.; Pruessner, J.C.; Taylor, A.M.; Deanfield, J.E.; Muthurangu, V. Habitual alcohol consumption is associated with lower cardiovascular stress responses—A novel explanation for the known cardiovascular benefits of alcohol? Stress 2013, 16, 369–376. [Google Scholar] [CrossRef]
- Perry, P.J.; Argo, T.R.; Barnett, M.J.; Liesveld, J.L.; Liskow, B.; Hernan, J.M.; Trnka, M.G.; Brabson, M.A. The Association of Alcohol-Induced Blackouts and Grayouts to Blood Alcohol Concentrations. J. Forensic Sci. 2006, 51, 896–899. [Google Scholar] [CrossRef]
- Liu, J.; Lewohl, J.M.; Harris, R.A.; Iyer, V.R.; Dodd, P.R.; Randall, P.K.; Mayfield, R.D. Patterns of gene expression in the frontal cortex discriminate alcoholic from nonalcoholic individuals. Neuropsychopharmacology 2006, 31, 1574–1582. [Google Scholar] [CrossRef] [PubMed]
- Blednov, Y.A.; Benavidez, J.M.; Geil, C.; Perra, S.; Morikawa, H.; Harris, R.A. Activation of inflammatory signaling by lipopolysaccharide produces a prolonged increase of voluntary alcohol intake in mice. Brain Behav. Immun. 2011, 25 (Suppl. 1), S92–S105. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Mao, X.; Chang, S.L. Altered gene expression in the spleen of adolescent rats following high ethanol concentration binge drinking. Int. J. Clin. Exp. Med. 2011, 4, 252–257. [Google Scholar] [PubMed]
- Wang, H.J.; Zakhari, S.; Jung, M.K. Alcohol, inflammation, and gut-liver-brain interactions in tissue damage and disease development. World J. Gastroenterol. 2010, 16, 1304–1313. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Connaghan, K.P.; Wei, Y.; Yang, Z.; Li, M.D.; Chang, S.L. Involvement of the Hippocampus in Binge Ethanol-Induced Spleen Atrophy in Adolescent Rats. Alcohol. Clin. Exp. Res. 2016, 40, 1489–1500. [Google Scholar] [CrossRef] [PubMed]
- Lachenmeier, D.W.; Rehm, J. Comparative risk assessment of alcohol, tobacco, cannabis and other illicit drugs using the margin of exposure approach. Sci. Rep. 2015, 5, 8126. [Google Scholar] [CrossRef]
- Lewis, S.M.; Williams, A.; Eisenbarth, S.C. Structure and function of the immune system in the spleen. Sci. Immunol. 2019, 4, eaau6085. [Google Scholar] [CrossRef]
- Singh, A.K.; Jiang, Y.; Gupta, S.; Benlhabib, E. Effects of chronic ethanol drinking on the blood–brain barrier and ensuing neuronal toxicity in alcohol-preferring rats subjected to intraperitoneal LPS injection. Alcohol Alcohol. 2007, 42, 385–399. [Google Scholar] [CrossRef] [PubMed]
- Haorah, J.; Knipe, B.; Gorantla, S.; Zheng, J.; Persidsky, Y. Alcohol-induced blood-brain barrier dysfunction is mediated via inositol 1,4,5-triphosphate receptor (IP3R)-gated intracellular calcium release. J. Neurochem. 2007, 100, 324–336. [Google Scholar] [CrossRef]
- Haorah, J.; Knipe, B.; Leibhart, J.; Ghorpade, A.; Persidsky, Y. Alcohol-induced oxidative stress in brain endothelial cells causes blood-brain barrier dysfunction. J. Leukoc. Biol. 2005, 78, 1223–1232. [Google Scholar] [CrossRef]
- Aggarwal, S.K.; Carter, G.T.; Sullivan, M.D.; ZumBrunnen, C.; Morrill, R.; Mayer, J.D. Medicinal use of cannabis in the United States: Historical perspectives, current trends, and future directions. J. Opioid. Manag. 2009, 5, 153–168. [Google Scholar] [CrossRef]
- Robson, P.J. Therapeutic potential of cannabinoid medicines. Drug Test. Anal. 2014, 6, 24–30. [Google Scholar] [CrossRef] [PubMed]
- Pertwee, R.G. Pharmacological actions of cannabinoids. Handb. Exp. Pharmacol. 2005, 1–51. [Google Scholar] [CrossRef]
- Miller, A.M.; Stella, N. CB2 receptor-mediated migration of immune cells: It can go either way. Br. J. Pharmacol. 2008, 153, 299–308. [Google Scholar] [CrossRef]
- Pacher, P.; Steffens, S.; Haskó, G.; Schindler, T.H.; Kunos, G. Cardiovascular effects of marijuana and synthetic cannabinoids: The good, the bad, and the ugly. Nat. Rev. Cardiol. 2018, 15, 151–166. [Google Scholar] [CrossRef]
- Cabral, G.A.; Jamerson, M. Marijuana use and brain immune mechanisms. Int. Rev. Neurobiol. 2014, 118, 199–230. [Google Scholar] [CrossRef]
- Archie, S.R.; Cucullo, L. Harmful Effects of Smoking Cannabis: A Cerebrovascular and Neurological Perspective. Front. Pharmacol. 2019, 10. [Google Scholar] [CrossRef] [PubMed]
- Tan, W.C.; Lo, C.; Jong, A.; Xing, L.; Fitzgerald, M.J.; Vollmer, W.M.; Buist, S.A.; Sin, D.D. Marijuana and chronic obstructive lung disease: A population-based study. CMAJ 2009, 180, 814–820. [Google Scholar] [CrossRef]
- Tashkin, D.P. Does smoking marijuana increase the risk of chronic obstructive pulmonary disease? CMAJ 2009, 180, 797–798. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Wang, B.; Kovalchuk, A.; Li, D.; Ilnytskyy, Y.; Kovalchuk, I.; Kovalchuk, O. In Search of Preventative Strategies: Novel Anti-Inflammatory High-CBD Cannabis Sativa Extracts Modulate ACE2 Expression in COVID-19 Gateway Tissues. Preprints 2020. [Google Scholar] [CrossRef]
- Katz, N.; Mazer, N.A. The impact of opioids on the endocrine system. Clin. J. Pain 2009, 25, 170–175. [Google Scholar] [CrossRef]
- Plein, L.M.; Rittner, H.L. Opioids and the immune system—Friend or foe. Br. J. Pharmacol. 2018, 175, 2717–2725. [Google Scholar] [CrossRef] [PubMed]
- Holden, J.E.; Jeong, Y.; Forrest, J.M. The endogenous opioid system and clinical pain management. AACN Clin. Issues 2005, 16, 291–301. [Google Scholar] [CrossRef] [PubMed]
- Trigo, J.M.; Martin-Garcia, E.; Berrendero, F.; Robledo, P.; Maldonado, R. The endogenous opioid system: A common substrate in drug addiction. Drug Alcohol Depend. 2010, 108, 183–194. [Google Scholar] [CrossRef] [PubMed]
- House, S.D.; Mao, X.; Wu, G.; Espinelli, D.; Li, W.X.; Chang, S.L. Chronic morphine potentiates the inflammatory response by disrupting interleukin-1beta modulation of the hypothalamic-pituitary-adrenal axis. J. Neuroimmunol. 2001, 118, 277–285. [Google Scholar] [CrossRef]
- Ocasio, F.M.; Jiang, Y.; House, S.D.; Chang, S.L. Chronic morphine accelerates the progression of lipopolysaccharide-induced sepsis to septic shock. J. Neuroimmunol. 2004, 149, 90–100. [Google Scholar] [CrossRef]
- Afonso, L.; Mohammad, T.; Thatai, D. Crack whips the heart: A review of the cardiovascular toxicity of cocaine. Am. J. Cardiol. 2007, 100, 1040–1043. [Google Scholar] [CrossRef]
- Phillips, K.; Luk, A.; Soor, G.S.; Abraham, J.R.; Leong, S.; Butany, J. Cocaine cardiotoxicity: A review of the pathophysiology, pathology, and treatment options. Am. J. Cardiovasc. Drugs 2009, 9, 177–196. [Google Scholar] [CrossRef]
- Schwartz, B.G.; Rezkalla, S.; Kloner, R.A. Cardiovascular effects of cocaine. Circulation 2010, 122, 2558–2569. [Google Scholar] [CrossRef]
- Pramanik, P. Cocaine Cardiac Toxicity: Revisited. In Cardiotoxicity; IntechOpen: Rijeka, Croatia, 2018. [Google Scholar]
- Graziani, M.; Antonilli, L.; Togna, A.R.; Grassi, M.C.; Badiani, A.; Saso, L. Cardiovascular and Hepatic Toxicity of Cocaine: Potential Beneficial Effects of Modulators of Oxidative Stress. Oxidative Med. Cell. Longev. 2016, 2016, 8408479. [Google Scholar] [CrossRef]
- Wood, D.M.; Dargan, P.I.; Hoffman, R.S. Management of cocaine-induced cardiac arrhythmias due to cardiac ion channel dysfunction. Clin. Toxicol. 2009, 47, 14–23. [Google Scholar] [CrossRef] [PubMed]
- Calatayud, J.; González, Á. History of the Development and Evolution of Local Anesthesia since the Coca Leaf. Anesthesiol. J. Am. Soc. Anesthesiol. 2003, 98, 1503–1508. [Google Scholar] [CrossRef] [PubMed]
- Premkumar, L.S. Selective Potentiation of L-Type Calcium Channel Currents by Cocaine in Cardiac Myocytes. Mol. Pharmacol. 1999, 56, 1138–1142. [Google Scholar] [CrossRef] [PubMed]
- Lange, R.A.; Cigarroa, R.G.; Yancy, C.W., Jr.; Willard, J.E.; Popma, J.J.; Sills, M.N.; McBride, W.; Kim, A.S.; Hillis, L.D. Cocaine-induced coronary-artery vasoconstriction. N. Engl. J. Med. 1989, 321, 1557–1562. [Google Scholar] [CrossRef] [PubMed]
- Fischman, M.W.; Schuster, C.R.; Resnekov, L.; Shick, J.F.; Krasnegor, N.A.; Fennell, W.; Freedman, D.X. Cardiovascular and subjective effects of intravenous cocaine administration in humans. Arch. Gen. Psychiatry 1976, 33, 983–989. [Google Scholar] [CrossRef] [PubMed]
- Flores, E.D.; Lange, R.A.; Cigarroa, R.G.; Hillis, L.D. Effect of cocaine on coronary artery dimensions in atherosclerotic coronary artery disease: Enhanced vasoconstriction at sites of significant stenoses. J. Am. Coll. Cardiol. 1990, 16, 74–79. [Google Scholar] [CrossRef]
- Havranek, E.P.; Nademanee, K.; Grayburn, P.A.; Eichhorn, E.J. Endothelium-dependent vasorelaxation is impaired in cocaine arteriopathy. J. Am. Coll. Cardiol. 1996, 28, 1168–1174. [Google Scholar] [CrossRef]
- Vita, J.A.; Treasure, C.B.; Fish, R.D.; Yeung, A.C.; Vekshtein, V.I.; Ganz, P.; Selwyn, A.P. Endothelial dysfunction leads to increased coronary constriction to catecholamines in patients with early atherosclerosis. J. Am. Coll. Cardiol. 1990, 15, A158. [Google Scholar] [CrossRef]
- Kelly, R.F.; Sompalli, V.; Sattar, P.; Khankari, K. Increased TIMI frame counts in cocaine users: A case for increased microvascular resistance in the absence of epicardial coronary disease or spasm. Clin. Cardiol. 2003, 26, 319–322. [Google Scholar] [CrossRef]
- Weber, J.E.; Hollander, J.E.; Murphy, S.A.; Braunwald, E.; Gibson, C.M. Quantitative comparison of coronary artery flow and myocardial perfusion in patients with acute myocardial infarction in the presence and absence of recent cocaine use. J. Thromb. Thrombolysis 2002, 14, 239–245. [Google Scholar] [CrossRef]
- Ritz, M.C.; Lamb, R.J.; Goldberg, S.R.; Kuhar, M.J. Cocaine receptors on dopamine transporters are related to self-administration of cocaine. Science 1987, 237, 1219–1223. [Google Scholar] [CrossRef] [PubMed]
- Sharkey, J.; Ritz, M.C.; Schenden, J.A.; Hanson, R.C.; Kuhar, M.J. Cocaine inhibits muscarinic cholinergic receptors in heart and brain. J. Pharmacol. Exp. Ther. 1988, 246, 1048–1052. [Google Scholar] [PubMed]
- Flynn, D.D.; Vaishnav, A.A.; Mash, D.C. Interactions of cocaine with primary and secondary recognition sites on muscarinic receptors. Mol. Pharmacol. 1992, 41, 736–742. [Google Scholar] [PubMed]
- Sharkey, J.; Glen, K.A.; Wolfe, S.; Kuhar, M.J. Cocaine binding at sigma receptors. Eur. J. Pharmacol. 1988, 149, 171–174. [Google Scholar] [CrossRef]
- Goeders, N.E. The HPA axis and cocaine reinforcement. Psychoneuroendocrinology 2002, 27, 13–33. [Google Scholar] [CrossRef]
- Halpern, J.H.; Sholar, M.B.; Glowacki, J.; Mello, N.K.; Mendelson, J.H.; Siegel, A.J. Diminished interleukin-6 response to proinflammatory challenge in men and women after intravenous cocaine administration. J. Clin. Endocrinol. Metab. 2003, 88, 1188–1193. [Google Scholar] [CrossRef]
- Barroso-Moguel, R.; Villeda-Hernandez, J.; Mendez-Armenta, M.; Rios, C. Brain capillary lesions produced by cocaine in rats. Toxicol. Lett. 1997, 92, 9–14. [Google Scholar] [CrossRef]
- Sharma, H.S.; Muresanu, D.; Sharma, A.; Patnaik, R. Chapter 11—Cocaine-Induced Breakdown of the Blood–Brain Barrier and Neurotoxicity. In International Review of Neurobiology; Academic Press: Cambridge, MA, USA, 2009; Volume 88, pp. 297–334. [Google Scholar]
- Yang, Y.; Yao, H.; Lu, Y.; Wang, C.; Buch, S. Cocaine Potentiates Astrocyte Toxicity Mediated by Human Immunodeficiency Virus (HIV-1) Protein gp120. PLoS ONE 2010, 5, e13427. [Google Scholar] [CrossRef]
- Yao, H.; Kim, K.; Duan, M.; Hayashi, T.; Guo, M.; Morgello, S.; Prat, A.; Wang, J.; Su, T.-P.; Buch, S. Cocaine Hijacks σ1 Receptor to Initiate Induction of Activated Leukocyte Cell Adhesion Molecule: Implication for Increased Monocyte Adhesion and Migration in the CNS. J. Neurosci. 2011, 31, 5942–5955. [Google Scholar] [CrossRef]
- Fiala, M.; Eshleman, A.J.; Cashman, J.; Lin, J.; Lossinsky, A.S.; Suarez, V.; Yang, W.; Zhang, J.; Popik, W.; Singer, E.; et al. Cocaine increases human immunodeficiency virus type 1 neuroinvasion through remodeling brain microvascular endothelial cells. J. Neurovirology 2005, 11, 281–291. [Google Scholar] [CrossRef]
- Dhillon, N.K.; Peng, F.; Bokhari, S.; Callen, S.; Shin, S.-H.; Zhu, X.; Kim, K.-J.; Buch, S.J. Cocaine-mediated Alteration in Tight Junction Protein Expression and Modulation of CCL2/CCR2 Axis Across the Blood-Brain Barrier: Implications for HIV-Dementia. J. Neuroimmune Pharmacol. 2008, 3, 52–56. [Google Scholar] [CrossRef] [PubMed]
- Gandhi, N.; Saiyed, Z.M.; Napuri, J.; Samikkannu, T.; Reddy, P.V.B.; Agudelo, M.; Khatavkar, P.; Saxena, S.K.; Nair, M.P.N. Interactive role of human immunodeficiency virus type 1 (HIV-1) clade-specific Tat protein and cocaine in blood-brain barrier dysfunction: Implications for HIV-1-associated neurocognitive disorder. J. Neurovirology 2010, 16, 294–305. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.W.; Hennig, B.; Fiala, M.; Kim, K.S.; Toborek, M. Cocaine activates redox-regulated transcription factors and induces TNF-alpha expression in human brain endothelial cells. Brain Res. 2001, 920, 125–133. [Google Scholar] [CrossRef]
- Cadet, J.L.; Krasnova, I.N. Chapter 5—Molecular Bases of Methamphetamine-Induced Neurodegeneration. In International Review of Neurobiology; Hari Shanker, S., Ed.; Academic Press: Cambridge, MA, USA, 2009; Volume 88, pp. 101–119. [Google Scholar]
- Cadet, J.L.; Krasnova, I.N. Interactions of HIV and Methamphetamine: Cellular and Molecular Mechanisms of Toxicity Potentiation. Neurotox. Res. 2007, 12, 181–204. [Google Scholar] [CrossRef]
- Sekine, Y.; Minabe, Y.; Ouchi, Y.; Takei, N.; Iyo, M.; Nakamura, K.; Suzuki, K.; Tsukada, H.; Okada, H.; Yoshikawa, E.; et al. Association of dopamine transporter loss in the orbitofrontal and dorsolateral prefrontal cortices with methamphetamine-related psychiatric symptoms. Am. J. Psychiatry 2003, 160, 1699–1701. [Google Scholar] [CrossRef]
- Volkow, N.D.; Chang, L.; Wang, G.J.; Fowler, J.S.; Leonido-Yee, M.; Franceschi, D.; Sedler, M.J.; Gatley, S.J.; Hitzemann, R.; Ding, Y.S.; et al. Association of dopamine transporter reduction with psychomotor impairment in methamphetamine abusers. Am. J. Psychiatry 2001, 158, 377–382. [Google Scholar] [CrossRef]
- Zahniser, N.R.; Sorkin, A. Trafficking of dopamine transporters in psychostimulant actions. Semin. Cell Dev. Biol. 2009, 20, 411–417. [Google Scholar] [CrossRef]
- Kahlig, K.M.; Lute, B.J.; Wei, Y.; Loland, C.J.; Gether, U.; Javitch, J.A.; Galli, A. Regulation of dopamine transporter trafficking by intracellular amphetamine. Mol. Pharmacol. 2006, 70, 542–548. [Google Scholar] [CrossRef]
- Rothman, R.B.; Baumann, M.H. Monoamine transporters and psychostimulant drugs. Eur. J. Pharmacol. 2003, 479, 23–40. [Google Scholar] [CrossRef]
- Volkow, N.D.; Chang, L.; Wang, G.J.; Fowler, J.S.; Ding, Y.S.; Sedler, M.; Logan, J.; Franceschi, D.; Gatley, J.; Hitzemann, R.; et al. Low level of brain dopamine D2 receptors in methamphetamine abusers: Association with metabolism in the orbitofrontal cortex. Am. J. Psychiatry 2001, 158, 2015–2021. [Google Scholar] [CrossRef]
- Sekine, Y.; Ouchi, Y.; Takei, N.; Yoshikawa, E.; Nakamura, K.; Futatsubashi, M.; Okada, H.; Minabe, Y.; Suzuki, K.; Iwata, Y.; et al. Brain serotonin transporter density and aggression in abstinent methamphetamine abusers. Arch. Gen. Psychiatry 2006, 63, 90–100. [Google Scholar] [CrossRef] [PubMed]
- Moszczynska, A.; Fitzmaurice, P.; Ang, L.; Kalasinsky, K.S.; Schmunk, G.A.; Peretti, F.J.; Aiken, S.S.; Wickham, D.J.; Kish, S.J. Why is parkinsonism not a feature of human methamphetamine users? Brain J. Neurol. 2004, 127, 363–370. [Google Scholar] [CrossRef]
- Wilson, J.M.; Kalasinsky, K.S.; Levey, A.I.; Bergeron, C.; Reiber, G.; Anthony, R.M.; Schmunk, G.A.; Shannak, K.; Haycock, J.W.; Kish, S.J. Striatal dopamine nerve terminal markers in human, chronic methamphetamine users. Nat. Med. 1996, 2, 699–703. [Google Scholar] [CrossRef] [PubMed]
- Sulzer, D.; Sonders, M.S.; Poulsen, N.W.; Galli, A. Mechanisms of neurotransmitter release by amphetamines: A review. Prog. Neurobiol. 2005, 75, 406–433. [Google Scholar] [CrossRef] [PubMed]
- Won, S.; Hong, R.A.; Shohet, R.V.; Seto, T.B.; Parikh, N.I. Methamphetamine-associated cardiomyopathy. Clin. Cardiol. 2013, 36, 737–742. [Google Scholar] [CrossRef] [PubMed]
- Gagliano, H.; Andero, R.; Armario, A.; Nadal, R. Repeated amphetamine administration in rats revealed consistency across days and a complete dissociation between locomotor and hypothalamic-pituitary-adrenal axis effects of the drug. Psychopharmacol. Berl 2009, 207, 447–459. [Google Scholar] [CrossRef]
- Rotllant, D.; Nadal, R.; Armario, A. Differential effects of stress and amphetamine administration on Fos-like protein expression in corticotropin releasing factor-neurons of the rat brain. Dev. Neurobiol. 2007, 67, 702–714. [Google Scholar] [CrossRef]
- Williams, M.T.; Schaefer, T.L.; Furay, A.R.; Ehrman, L.A.; Vorhees, C.V. Ontogeny of the adrenal response to (+)-methamphetamine in neonatal rats: The effect of prior drug exposure. Stress 2006, 9, 153–163. [Google Scholar] [CrossRef][Green Version]
- Doyle, J.R.; Yamamoto, B.K. Serotonin 2 receptor modulation of hyperthermia, corticosterone, and hippocampal serotonin depletions following serial exposure to chronic stress and methamphetamine. Psychoneuroendocrinology 2010, 35, 629–633. [Google Scholar] [CrossRef][Green Version]
- Zuloaga, D.G.; Jacobskind, J.S.; Raber, J. Methamphetamine and the hypothalamic-pituitary-adrenal axis. Front. Neurosci. 2015, 9, 178. [Google Scholar] [CrossRef]
- Parrott, A.C.; Montgomery, C.; Wetherell, M.A.; Downey, L.A.; Stough, C.; Scholey, A.B. MDMA, cortisol, and heightened stress in recreational ecstasy users. Behav. Pharmacol. 2014, 25, 458–472. [Google Scholar] [CrossRef] [PubMed]
- Silva, A.P.; Martins, T.; Baptista, S.; Gonçalves, J.; Agasse, F.; Malva, J.O. Brain injury associated with widely abused amphetamines: Neuroinflammation, neurogenesis and blood-brain barrier. Curr. Drug Abus. Rev. 2010, 3, 239–254. [Google Scholar] [CrossRef] [PubMed]
- Ohta, K.; Mori, M.; Yoritaka, A.; Okamoto, K.; Kishida, S. Delayed ischemic stroke associated with methamphetamine use. J. Emerg. Med. 2005, 28, 165–167. [Google Scholar] [CrossRef] [PubMed]
- Rothrock, J.F.; Rubenstein, R.; Lyden, P.D. Ischemic stroke associated with methamphetamine inhalation. Neurology 1988, 38, 589–592. [Google Scholar] [CrossRef] [PubMed]
- Polesskaya, O.; Silva, J.; Sanfilippo, C.; Desrosiers, T.; Sun, A.; Shen, J.; Feng, C.; Polesskiy, A.; Deane, R.; Zlokovic, B.; et al. Methamphetamine causes sustained depression in cerebral blood flow. Brain Res. 2011, 1373, 91–100. [Google Scholar] [CrossRef]
- Chung, Y.A.; Peterson, B.S.; Yoon, S.J.; Cho, S.N.; Chai, S.; Jeong, J.; Kim, D.J. In vivo evidence for long-term CNS toxicity, associated with chronic binge use of methamphetamine. Drug Alcohol Depend. 2010, 111, 155–160. [Google Scholar] [CrossRef]
- Dietrich, J.B. Alteration of blood-brain barrier function by methamphetamine and cocaine. Cell Tissue Res. 2009, 336, 385–392. [Google Scholar] [CrossRef]
- Reiner, B.C.; Keblesh, J.P.; Xiong, H. Methamphetamine abuse, HIV infection, and neurotoxicity. Int. J. Physiol. Pathophysiol. Pharmacol. 2009, 1, 162–179. [Google Scholar]
- Turowski, P.; Kenny, B.-A. The blood-brain barrier and methamphetamine: Open sesame? Front. Neurosci. 2015, 9, 156. [Google Scholar] [CrossRef]
- Asanuma, M.; Miyazaki, I.; Higashi, Y.; Tsuji, T.; Ogawa, N. Specific gene expression and possible involvement of inflammation in methamphetamine-induced neurotoxicity. Ann. N. Y. Acad. Sci. 2004, 1025, 69–75. [Google Scholar] [CrossRef]
- Kousik, S.M.; Napier, T.C.; Carvey, P.M. The effects of psychostimulant drugs on blood brain barrier function and neuroinflammation. Front. Pharmacol. 2012, 3, 121. [Google Scholar] [CrossRef]
- Nath, A.; Maragos, W.F.; Avison, M.J.; Schmitt, F.A.; Berger, J.R. Acceleration of HIV dementia with methamphetamine and cocaine. J. Neurovirol. 2001, 7, 66–71. [Google Scholar] [CrossRef] [PubMed]
- Letendre, S.L.; Cherner, M.; Ellis, R.J.; Marquie-Beck, J.; Gragg, B.; Marcotte, T.; Heaton, R.K.; McCutchan, J.A.; Grant, I. The effects of hepatitis C, HIV, and methamphetamine dependence on neuropsychological performance: Biological correlates of disease. Aids 2005, 19 (Suppl. 3), S72–S78. [Google Scholar] [CrossRef] [PubMed]
- Schep, L.J.; Slaughter, R.J.; Beasley, D.M. The clinical toxicology of metamfetamine. Clin. Toxicol. Phila 2010, 48, 675–694. [Google Scholar] [CrossRef] [PubMed]
- Sharma, H.S.; Ali, S.F. Acute Administration of 3,4-Methylenedioxymethamphetamine Induces Profound Hyperthermia, Blood–Brain Barrier Disruption, Brain Edema Formation, and Cell Injury. Ann. N. Y. Acad. Sci. 2008, 1139, 242–258. [Google Scholar] [CrossRef]
- Monks, T.J.; Jones, D.C.; Bai, F.; Lau, S.S. The role of metabolism in 3,4-(+)-methylenedioxyamphetamine and 3,4-(+)-methylenedioxymethamphetamine (ecstasy) toxicity. Ther. Drug Monit. 2004, 26, 132–136. [Google Scholar] [CrossRef]
- Gonçalves, J.; Martins, T.; Ferreira, R.; Milhazes, N.; Borges, F.; Ribeiro, C.F.; Malva, J.O.; Macedo, T.R.; Silva, A.P. Methamphetamine-Induced Early Increase of IL-6 and TNF-α mRNA Expression in the Mouse Brain. Ann. N. Y. Acad. Sci. 2008, 1139, 103–111. [Google Scholar] [CrossRef]
- Lai, Y.-T.; Tsai, Y.-P.N.; Cherng, C.G.; Ke, J.-J.; Ho, M.-C.; Tsai, C.-W.; Yu, L. Lipopolysaccharide mitagates methamphetamine-induced striatal dopamine depletion via modulating local TNF-α and dopamine transporter expression. J. Neural Transm. 2009, 116, 405–415. [Google Scholar] [CrossRef]
- Gonçalves, J.; Baptista, S.; Martins, T.; Milhazes, N.; Borges, F.; Ribeiro, C.F.; Malva, J.O.; Silva, A.P. Methamphetamine-induced neuroinflammation and neuronal dysfunction in the mice hippocampus: Preventive effect of indomethacin. Eur. J. Neurosci. 2010, 31, 315–326. [Google Scholar] [CrossRef]
- Torres, E.; Gutierrez-Lopez, M.D.; Mayado, A.; Rubio, A.; O’Shea, E.; Colado, M.I. Changes in interleukin-1 signal modulators induced by 3,4-methylenedioxymethamphetamine (MDMA): Regulation by CB2 receptors and implications for neurotoxicity. J. Neuroinflammation 2011, 8, 53. [Google Scholar] [CrossRef]
- Yamamoto, B.K.; Bankson, M.G. Amphetamine Neurotoxicity: Cause and Consequence of Oxidative Stress. Crit. Rev. Neurobiol. 2005, 17, 87–118. [Google Scholar] [CrossRef] [PubMed]
- Sanders, J.M.; Monogue, M.L.; Jodlowski, T.Z.; Cutrell, J.B. Pharmacologic Treatments for Coronavirus Disease 2019 (COVID-19): A Review. JAMA 2020. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Zhou, Q.; Li, Y.; Garner, L.V.; Watkins, S.P.; Carter, L.J.; Smoot, J.; Gregg, A.C.; Daniels, A.D.; Jervey, S.; et al. Research and Development on Therapeutic Agents and Vaccines for COVID-19 and Related Human Coronavirus Diseases. ACS Cent. Sci. 2020, 6, 315–331. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Hou, Y.; Shen, J.; Huang, Y.; Martin, W.; Cheng, F. Network-based drug repurposing for novel coronavirus 2019-nCoV/SARS-CoV-2. Cell Discov. 2020, 6, 14. [Google Scholar] [CrossRef] [PubMed]
- Mahévas, M.; Tran, V.-T.; Roumier, M.; Chabrol, A.; Paule, R.; Guillaud, C.; Fois, E.; Lepeule, R.; Szwebel, T.-A.; Lescure, F.-X.; et al. Clinical efficacy of hydroxychloroquine in patients with covid-19 pneumonia who require oxygen: Observational comparative study using routine care data. BMJ 2020, 369, m1844. [Google Scholar] [CrossRef]
- Wang, M.; Cao, R.; Zhang, L.; Yang, X.; Liu, J.; Xu, M.; Shi, Z.; Hu, Z.; Zhong, W.; Xiao, G. Remdesivir and chloroquine effectively inhibit the recently emerged novel coronavirus (2019-nCoV) in vitro. Cell Res. 2020, 30, 269–271. [Google Scholar] [CrossRef]
- Mehra, M.R.; Desai, S.S.; Ruschitzka, F.; Patel, A.N. RETRACTED: Hydroxychloroquine or chloroquine with or without a macrolide for treatment of COVID-19: A multinational registry analysis. Lancet 2020. [Google Scholar] [CrossRef]
- World Health Organization. “Solidarity” Clinical Trial for COVID-19 Treatments. Available online: https://www.who.int/emergencies/diseases/novel-coronavirus-2019/global-research-on-novel-coronavirus-2019-ncov/solidarity-clinical-trial-for-covid-19-treatments (accessed on 29 June 2020).
- U.S. Food & Drug Administration. Coronavirus (COVID-19) Update: FDA Revokes Emergency Use Authorization for Chloroquine and Hydroxychloroquine. Available online: https://www.fda.gov/news-events/press-announcements/coronavirus-covid-19-update-fda-revokes-emergency-use-authorization-chloroquine-and (accessed on 29 June 2020).
- Hung, I.F.-N.; Lung, K.-C.; Tso, E.Y.-K.; Liu, R.; Chung, T.W.-H.; Chu, M.-Y.; Ng, Y.-Y.; Lo, J.; Chan, J.; Tam, A.R.; et al. Triple combination of interferon beta-1b, lopinavir–ritonavir, and ribavirin in the treatment of patients admitted to hospital with COVID-19: An open-label, randomised, phase 2 trial. Lancet 2020. [Google Scholar] [CrossRef]
- Cao, B.; Wang, Y.; Wen, D.; Liu, W.; Wang, J.; Fan, G.; Ruan, L.; Song, B.; Cai, Y.; Wei, M.; et al. A Trial of Lopinavir–Ritonavir in Adults Hospitalized with Severe Covid-19. N. Engl. J. Med. 2020, 382, 1787–1799. [Google Scholar] [CrossRef]
- Khalili, J.S.; Zhu, H.; Mak, N.S.A.; Yan, Y.; Zhu, Y. Novel coronavirus treatment with ribavirin: Groundwork for an evaluation concerning COVID-19. J. Med. Virol. 2020, 92, 740–746. [Google Scholar] [CrossRef]
- Al-Motawa, M.; Abbas, H.; Wijten, P.; de la Fuente, A.; Xue, M.; Rabbani, N.; Thornalley, P.J. Vulnerabilities of the SARS-CoV-2 virus to proteotoxicity—Opportunity for repurposed chemotherapy of COVID-19 infection. bioRxiv 2020. [Google Scholar] [CrossRef]
- Hu, K.; Guan, W.J.; Bi, Y.; Zhang, W.; Li, L.; Zhang, B.; Liu, Q.; Song, Y.; Li, X.; Duan, Z.; et al. Efficacy and safety of Lianhuaqingwen capsules, a repurposed Chinese herb, in patients with coronavirus disease 2019: A multicenter, prospective, randomized controlled trial. Phytomedicine 2020. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Shi, P.-Y.; Li, H.; Zhou, J. Broad Spectrum Antiviral Agent Niclosamide and Its Therapeutic Potential. ACS Infect. Dis. 2020. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Wei, J.; Zou, L.; Jiang, T.; Wang, G.; Chen, L.; Huang, L.; Meng, F.; Huang, L.; Wang, N.; et al. Ruxolitinib in treatment of severe coronavirus disease 2019 (COVID-19): A multicenter, single-blind, randomized controlled trial. J. Allergy Clin. Immunol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Coomes, E.A.; Haghbayan, H. Favipiravir, an antiviral for COVID-19? J. Antimicrob. Chemother. 2020, 75, 2013–2014. [Google Scholar] [CrossRef] [PubMed]
- Beigel, J.H.; Tomashek, K.M.; Dodd, L.E.; Mehta, A.K.; Zingman, B.S.; Kalil, A.C.; Hohmann, E.; Chu, H.Y.; Luetkemeyer, A.; Kline, S.; et al. Remdesivir for the Treatment of Covid-19—Preliminary Report. N. Engl. J. Med. 2020. [Google Scholar] [CrossRef]
- Grein, J.; Ohmagari, N.; Shin, D.; Diaz, G.; Asperges, E.; Castagna, A.; Feldt, T.; Green, G.; Green, M.L.; Lescure, F.X.; et al. Compassionate Use of Remdesivir for Patients with Severe Covid-19. N. Engl. J. Med. 2020. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, D.; Du, G.; Du, R.; Zhao, J.; Jin, Y.; Fu, S.; Gao, L.; Cheng, Z.; Lu, Q.; et al. Remdesivir in adults with severe COVID-19: A randomised, double-blind, placebo-controlled, multicentre trial. Lancet 2020, 395, 1569–1578. [Google Scholar] [CrossRef]
- Ledford, H. Coronavirus breakthrough: Dexamethasone is first drug shown to save lives. Nature 2020. [Google Scholar] [CrossRef]
- Gao, Y.; Yan, L.; Huang, Y.; Liu, F.; Zhao, Y.; Cao, L.; Wang, T.; Sun, Q.; Ming, Z.; Zhang, L.; et al. Structure of the RNA-dependent RNA polymerase from COVID-19 virus. Science 2020, eabb7498. [Google Scholar] [CrossRef]
- Gordon, C.J.; Tchesnokov, E.P.; Feng, J.Y.; Porter, D.P.; Gotte, M. The antiviral compound remdesivir potently inhibits RNA-dependent RNA polymerase from Middle East respiratory syndrome coronavirus. J. Biol. Chem. 2020, 295, 4773–4779. [Google Scholar] [CrossRef] [PubMed]
- Yin, W.; Mao, C.; Luan, X.; Shen, D.D.; Shen, Q.; Su, H.; Wang, X.; Zhou, F.; Zhao, W.; Gao, M.; et al. Structural basis for inhibition of the RNA-dependent RNA polymerase from SARS-CoV-2 by remdesivir. Science 2020. [Google Scholar] [CrossRef] [PubMed]
- Jin, Z.; Du, X.; Xu, Y.; Deng, Y.; Liu, M.; Zhao, Y.; Zhang, B.; Li, X.; Zhang, L.; Peng, C.; et al. Structure of M(pro) from COVID-19 virus and discovery of its inhibitors. Nature 2020. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Lin, D.; Sun, X.; Curth, U.; Drosten, C.; Sauerhering, L.; Becker, S.; Rox, K.; Hilgenfeld, R. Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved alpha-ketoamide inhibitors. Science 2020, 368, 409–412. [Google Scholar] [CrossRef] [PubMed]
- Ul Qamar, M.T.; Alqahtani, S.M.; Alamri, M.A.; Chen, L.-L. Structural basis of SARS-CoV-2 3CLpro and anti-COVID-19 drug discovery from medicinal plants. J. Pharm. Anal. 2020. [Google Scholar] [CrossRef]
- Gentile, D.; Patamia, V.; Scala, A.; Sciortino, M.T.; Piperno, A.; Rescifina, A. Putative Inhibitors of SARS-CoV-2 Main Protease from A Library of Marine Natural Products: A Virtual Screening and Molecular Modeling Study. Mar. Drugs 2020, 18, 225. [Google Scholar] [CrossRef]
- Dai, W.; Zhang, B.; Jiang, X.-M.; Su, H.; Li, J.; Zhao, Y.; Xie, X.; Jin, Z.; Peng, J.; Liu, F.; et al. Structure-based design of antiviral drug candidates targeting the SARS-CoV-2 main protease. Science 2020, 368, 1331–1335. [Google Scholar] [CrossRef]
- Wu, Y.; Wang, F.; Shen, C.; Peng, W.; Li, D.; Zhao, C.; Li, Z.; Li, S.; Bi, Y.; Yang, Y.; et al. A non-competing pair of human neutralizing antibodies block COVID-19 virus binding to its receptor ACE2. medRxiv 2020. [Google Scholar] [CrossRef]
- Shi, R.; Shan, C.; Duan, X.; Chen, Z.; Liu, P.; Song, J.; Song, T.; Bi, X.; Han, C.; Wu, L.; et al. A human neutralizing antibody targets the receptor binding site of SARS-CoV-2. Nature 2020. [Google Scholar] [CrossRef]
- Rogers, T.F.; Zhao, F.; Huang, D.; Beutler, N.; Burns, A.; He, W.-t.; Limbo, O.; Smith, C.; Song, G.; Woehl, J.; et al. Isolation of potent SARS-CoV-2 neutralizing antibodies and protection from disease in a small animal model. Science 2020, eabc7520. [Google Scholar] [CrossRef]
- Ju, B.; Zhang, Q.; Ge, J.; Wang, R.; Sun, J.; Ge, X.; Yu, J.; Shan, S.; Zhou, B.; Song, S.; et al. Human neutralizing antibodies elicited by SARS-CoV-2 infection. Nature 2020. [Google Scholar] [CrossRef] [PubMed]
- Wec, A.Z.; Wrapp, D.; Herbert, A.S.; Maurer, D.P.; Haslwanter, D.; Sakharkar, M.; Jangra, R.K.; Dieterle, M.E.; Lilov, A.; Huang, D.; et al. Broad neutralization of SARS-related viruses by human monoclonal antibodies. Science 2020, eabc7424. [Google Scholar] [CrossRef] [PubMed]
- Chi, X.; Yan, R.; Zhang, J.; Zhang, G.; Zhang, Y.; Hao, M.; Zhang, Z.; Fan, P.; Dong, Y.; Yang, Y.; et al. A neutralizing human antibody binds to the N-terminal domain of the Spike protein of SARS-CoV-2. Science 2020. [Google Scholar] [CrossRef]
- Lei, C.; Qian, K.; Li, T.; Zhang, S.; Fu, W.; Ding, M.; Hu, S. Neutralization of SARS-CoV-2 spike pseudotyped virus by recombinant ACE2-Ig. Nat. Commun. 2020, 11, 2070. [Google Scholar] [CrossRef] [PubMed]
- Wrapp, D.; De Vlieger, D.; Corbett, K.S.; Torres, G.M.; Wang, N.; Van Breedam, W.; Roose, K.; van Schie, L.; Team, V.-C.C.-R.; Hoffmann, M.; et al. Structural Basis for Potent Neutralization of Betacoronaviruses by Single-Domain Camelid Antibodies. Cell 2020, 181, 1004–1015.e1015. [Google Scholar] [CrossRef] [PubMed]
- Hansen, J.; Baum, A.; Pascal, K.E.; Russo, V.; Giordano, S.; Wloga, E.; Fulton, B.O.; Yan, Y.; Koon, K.; Patel, K.; et al. Studies in humanized mice and convalescent humans yield a SARS-CoV-2 antibody cocktail. Science 2020, eabd0827. [Google Scholar] [CrossRef]
- Brouwer, P.J.M.; Caniels, T.G.; van der Straten, K.; Snitselaar, J.L.; Aldon, Y.; Bangaru, S.; Torres, J.L.; Okba, N.M.A.; Claireaux, M.; Kerster, G.; et al. Potent neutralizing antibodies from COVID-19 patients define multiple targets of vulnerability. Science 2020, eabc5902. [Google Scholar] [CrossRef]
- Baum, A.; Fulton, B.O.; Wloga, E.; Copin, R.; Pascal, K.E.; Russo, V.; Giordano, S.; Lanza, K.; Negron, N.; Ni, M.; et al. Antibody cocktail to SARS-CoV-2 spike protein prevents rapid mutational escape seen with individual antibodies. Science 2020, eabd0831. [Google Scholar] [CrossRef]
- Hotez, P.J.; Corry, D.B.; Bottazzi, M.E. COVID-19 vaccine design: The Janus face of immune enhancement. Nat. Rev. Immunol. 2020, 20, 347–348. [Google Scholar] [CrossRef]
- World Health Organization. Draft Landscape of COVID-19 Candidate Vaccines. Available online: https://www.who.int/publications/m/item/draft-landscape-of-covid-19-candidate-vaccines (accessed on 19 June 2020).
- Thanh Le, T.; Andreadakis, Z.; Kumar, A.; Gómez Román, R.; Tollefsen, S.; Saville, M.; Mayhew, S. The COVID-19 vaccine development landscape. Nat. Rev. Drug Discov. 2020, 19, 305–306. [Google Scholar] [CrossRef]
- Wang, F.; Kream, R.M.; Stefano, G.B. An Evidence Based Perspective on mRNA-SARS-CoV-2 Vaccine Development. Med. Sci. Monit. 2020, 26, e924700. [Google Scholar] [CrossRef]
- Jackson, L.A.; Anderson, E.J.; Rouphael, N.G.; Roberts, P.C.; Makhene, M.; Coler, R.N.; McCullough, M.P.; Chappell, J.D.; Denison, M.R.; Stevens, L.J.; et al. An mRNA Vaccine against SARS-CoV-2—Preliminary Report. N. Engl. J. Med. 2020. [Google Scholar] [CrossRef]
- Zhu, F.-C.; Li, Y.-H.; Guan, X.-H.; Hou, L.-H.; Wang, W.-J.; Li, J.-X.; Wu, S.-P.; Wang, B.-S.; Wang, Z.; Wang, L.; et al. Safety, tolerability, and immunogenicity of a recombinant adenovirus type-5 vectored COVID-19 vaccine: A dose-escalation, open-label, non-randomised, first-in-human trial. Lancet 2020. [Google Scholar] [CrossRef]
- Feldmann, M.; Maini, R.N.; Woody, J.N.; Holgate, S.T.; Winter, G.; Rowland, M.; Richards, D.; Hussell, T. Trials of anti-tumour necrosis factor therapy for COVID-19 are urgently needed. Lancet 2020, 395, 1407–1409. [Google Scholar] [CrossRef]
- Conti, P.; Ronconi, G.; Caraffa, A.; Gallenga, C.E.; Ross, R.; Frydas, I.; Kritas, S.K. Induction of pro-inflammatory cytokines (IL-1 and IL-6) and lung inflammation by Coronavirus-19 (COVI-19 or SARS-CoV-2): Anti-inflammatory strategies. J. Biol. Regul. Homeost. Agents 2020, 34. [Google Scholar] [CrossRef]
- Thevarajan, I.; Nguyen, T.H.O.; Koutsakos, M.; Druce, J.; Caly, L.; van de Sandt, C.E.; Jia, X.; Nicholson, S.; Catton, M.; Cowie, B.; et al. Breadth of concomitant immune responses prior to patient recovery: A case report of non-severe COVID-19. Nat. Med. 2020, 26, 453–455. [Google Scholar] [CrossRef]
- Grifoni, A.; Weiskopf, D.; Ramirez, S.I.; Mateus, J.; Dan, J.M.; Moderbacher, C.R.; Rawlings, S.A.; Sutherland, A.; Premkumar, L.; Jadi, R.S.; et al. Targets of T cell responses to SARS-CoV-2 coronavirus in humans with COVID-19 disease and unexposed individuals. Cell 2020. [Google Scholar] [CrossRef] [PubMed]
- Braun, J.; Loyal, L.; Frentsch, M.; Wendisch, D.; Georg, P.; Kurth, F.; Hippenstiel, S.; Dingeldey, M.; Kruse, B.; Fauchere, F.; et al. Presence of SARS-CoV-2 reactive T cells in COVID-19 patients and healthy donors. medRxiv 2020. [Google Scholar] [CrossRef]
- Long, Q.-X.; Tang, X.-J.; Shi, Q.-L.; Li, Q.; Deng, H.-J.; Yuan, J.; Hu, J.-L.; Xu, W.; Zhang, Y.; Lv, F.-J.; et al. Clinical and immunological assessment of asymptomatic SARS-CoV-2 infections. Nat. Med. 2020. [Google Scholar] [CrossRef]
- Iwasaki, A.; Yang, Y. The potential danger of suboptimal antibody responses in COVID-19. Nat. Rev. Immunol. 2020, 20, 339–341. [Google Scholar] [CrossRef]
- Kulkarni, R. Antibody-Dependent Enhancement of Viral Infections. Dyn. Immune Act. Viral Dis. 2019, 9–41. [Google Scholar] [CrossRef]
- Green, T.C.; Bratberg, J.; Finnell, D.S. Opioid use disorder and the COVID 19 pandemic: A call to sustain regulatory easements and further expand access to treatment. Subst. Abus. 2020, 41, 147–149. [Google Scholar] [CrossRef] [PubMed]
- Kissler, S.M.; Tedijanto, C.; Goldstein, E.; Grad, Y.H.; Lipsitch, M. Projecting the transmission dynamics of SARS-CoV-2 through the postpandemic period. Science 2020, eabb5793. [Google Scholar] [CrossRef] [PubMed]
- Qiu, J. Covert coronavirus infections could be seeding new outbreaks. Nature 2020. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Comorbid Conditions | Case Fatality (%) | Mortality (per 100,000 Population) |
|---|---|---|
| Overall | 2.3 | 150 |
| None | 0.9 | 50 |
| Hypertension | 6.0 | 380 |
| Diabetes | 7.3 | 450 |
| Cardiovascular diseases | 10.5 | 680 |
| Chronic respiratory diseases | 6.3 | 400 |
| Cancer | 5.6 | 360 |
| Comorbid Conditions | Not Hospitalize N (%) | Hospitalized N (%) | No Critical Illness N (%) | Critical Illness N (%) |
|---|---|---|---|---|
| Total cases | 2104 | 1999 | 932 | 650 |
| Tobacco use (current or former) | 358 (19.5) | 520 (26.0) | 237 (25.5) | 173 (26.6) |
| Obesity (BMI ≥ 30) | 304 (14.4) | 796 (39.8) | 378 (40.6) | 260 (40.0) |
| Cardiovascular conditions | 344 (16.3) | 891 (44.6) | 391 (42.0) | 306 (47.1) |
| Hypertension | 241 (11.5) | 742 (37.1) | 320 (34.3) | 257 (39.5) |
| Diabetes | 111 (5.3) | 503 (25.2) | 213 (22.9) | 176 (27.1) |
| Asthma or COPD | 106 (5.0) | 206 (10.3) | 91 (9.8) | 71 (10.9) |
| Substance | Target System | Pathology | COVID-19 |
|---|---|---|---|
| Tobacco (Nicotine) | Respiratory system | Main cause of COPD [10,11,82] | Increased severity and mortality [13,81] |
| Immune system | Immune suppression, Decreased CD8+ T-cells [83,84] | Higher infection rate [85] | |
| Increased inflammatory cytokines (TNFα, IL-1β, IL-18) and chemokines (CCL2, CCL8, and CXC3CL1); decreased anti-inflammatory factors, Bcl6, IL-10, and CCL25 [92] | Increased inflammatory cytokines and chemokines, TNFα, IL-1β, IL-6 [61,62,63] | ||
| CNS | BBB leakage through loss of tight junction proteins [86,87,88,89] | Endotheliitis and CNS infection [53,55,56,57] | |
| Alcohol | Immune system | Increased proinflammatory cytokines, IL-1β and IL-6, and chemokine CCL-2 [98] | Increased inflammatory cytokines and chemokines, TNFα, IL-1β, IL-6 [61,62,63] |
| Spleen atrophy [100]. | Impaired production of antibodies and lymphocytes [102] | ||
| CNS | Increased BBB permeability through cytoskeletal and tight junction remodeling [103,104,105]. | Endotheliitis and CNS infection [53,55,56,57] | |
| Marijuana (THC, CBD) | Respiratory system | Enhanced COPD with tobacco [113,114] | Increased severity and mortality [13,81] |
| Immune system | Immunosuppression; reduced antibody response and T lymphocyte activities; reduced migration of macrophage [111] | Increased infection and reduced viral response and clearance [111] | |
| Opioids (heroine, fentanyl, morphine) | Respiratory system | Respiratory depression [14,15,119] | Increased severity and mortality [14,15] |
| Immune system | Desensitizing HPA axis; inhibiting glucocorticoid release, increased IL-1β; neuroinflammation [120] | Increased opportunistic infections, excessive inflammatory response [117,118,121] | |
| Cocaine | Cardiovascular system | Cardiac arrhythmias and acute MI; oxygen imbalance; microvascular diseases and thrombosis [122,123,124,125,126,127,129,130,131,132] | Increased severity and mortality [12,37,38] |
| Immune system | Stimulating HPA axis; immunosuppression; defects in antibody formation, lymphocyte proliferation, macrophage and NK activation [141,142] | High incidence of viral infection [142] | |
| CNS | Increased BBB permeability due to loss of tight junction proteins; rearrangement of cytoskeleton structure [143,144,145,146] | Endotheliitis and CNS infection [53,55,56,57] | |
| Amphetamine, METH, MDMA | Cardiovascular system | Hypertension, tachycardia, and cardiomyopathy leading to pulmonary edema [16,163]; Ischemic stroke and hypoxia; restricted blood flow [171,172,173,174] | Increased severity and mortality [12,37,38] |
| Immune system | Altered HPA axis, impairing GR and MR expression, immunosuppression [164,165,166,167] | Increased infection rate, depression, anxiety/despair [168,169] | |
| Increased expression of TNFα, IL-1β, and IL-6; neuroinflammation [185,186,187,188] | Excessive inflammatory response [61,62,63] | ||
| CNS | BBB damage due to loss of tight junction protein; edema [175,176,177,178,179] | Endotheliitis and CNS infection [53,55,56,57,180,181,182] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wei, Y.; Shah, R. Substance Use Disorder in the COVID-19 Pandemic: A Systematic Review of Vulnerabilities and Complications. Pharmaceuticals 2020, 13, 155. https://doi.org/10.3390/ph13070155
Wei Y, Shah R. Substance Use Disorder in the COVID-19 Pandemic: A Systematic Review of Vulnerabilities and Complications. Pharmaceuticals. 2020; 13(7):155. https://doi.org/10.3390/ph13070155
Chicago/Turabian StyleWei, Yufeng, and Rameen Shah. 2020. "Substance Use Disorder in the COVID-19 Pandemic: A Systematic Review of Vulnerabilities and Complications" Pharmaceuticals 13, no. 7: 155. https://doi.org/10.3390/ph13070155
APA StyleWei, Y., & Shah, R. (2020). Substance Use Disorder in the COVID-19 Pandemic: A Systematic Review of Vulnerabilities and Complications. Pharmaceuticals, 13(7), 155. https://doi.org/10.3390/ph13070155