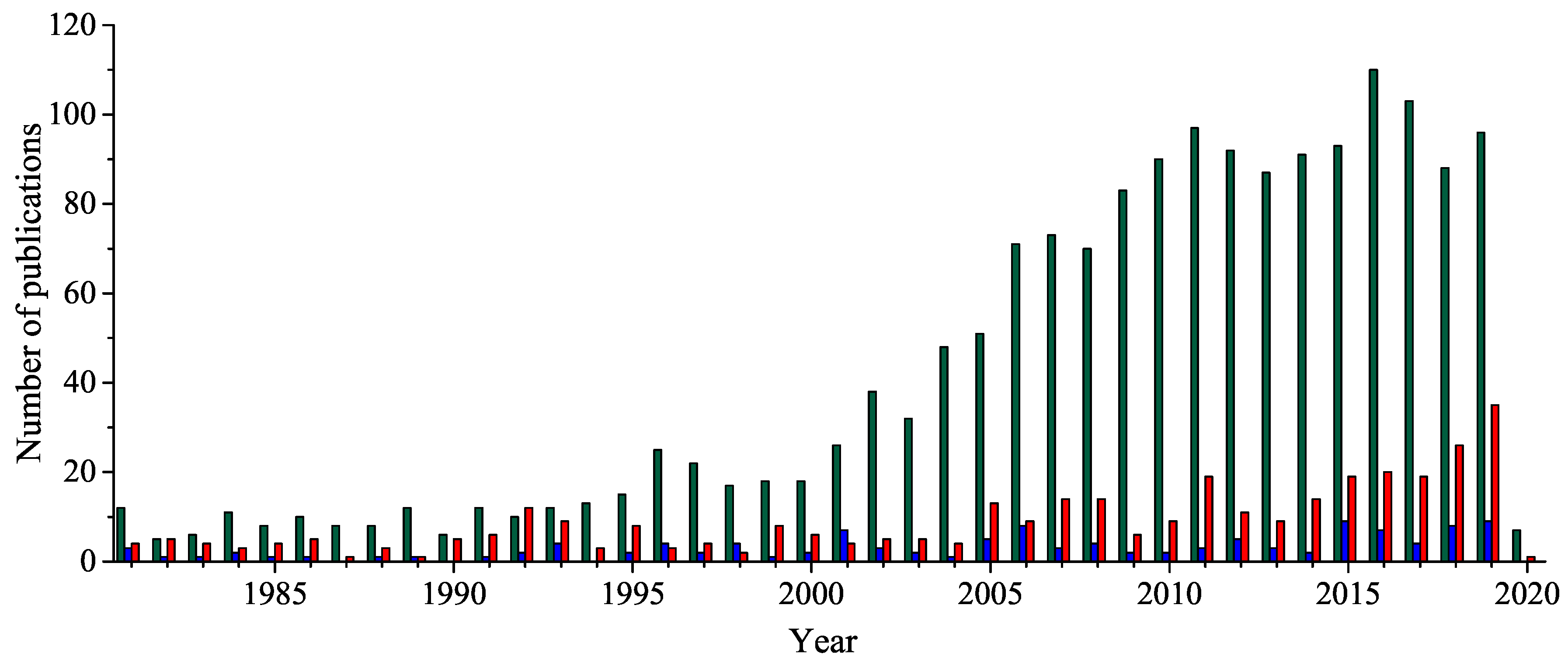
Novel 1,2,4-Oxadiazole Derivatives in Drug Discovery

 ,
,  , , , and
, , , and
Abstract
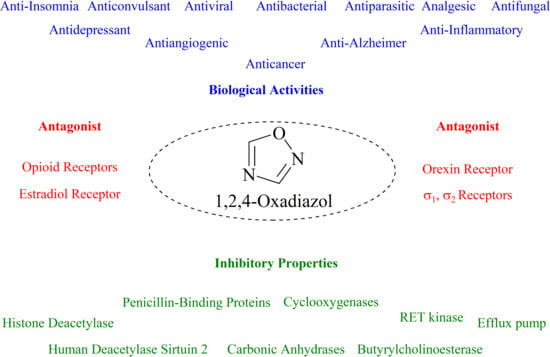
1. Introduction
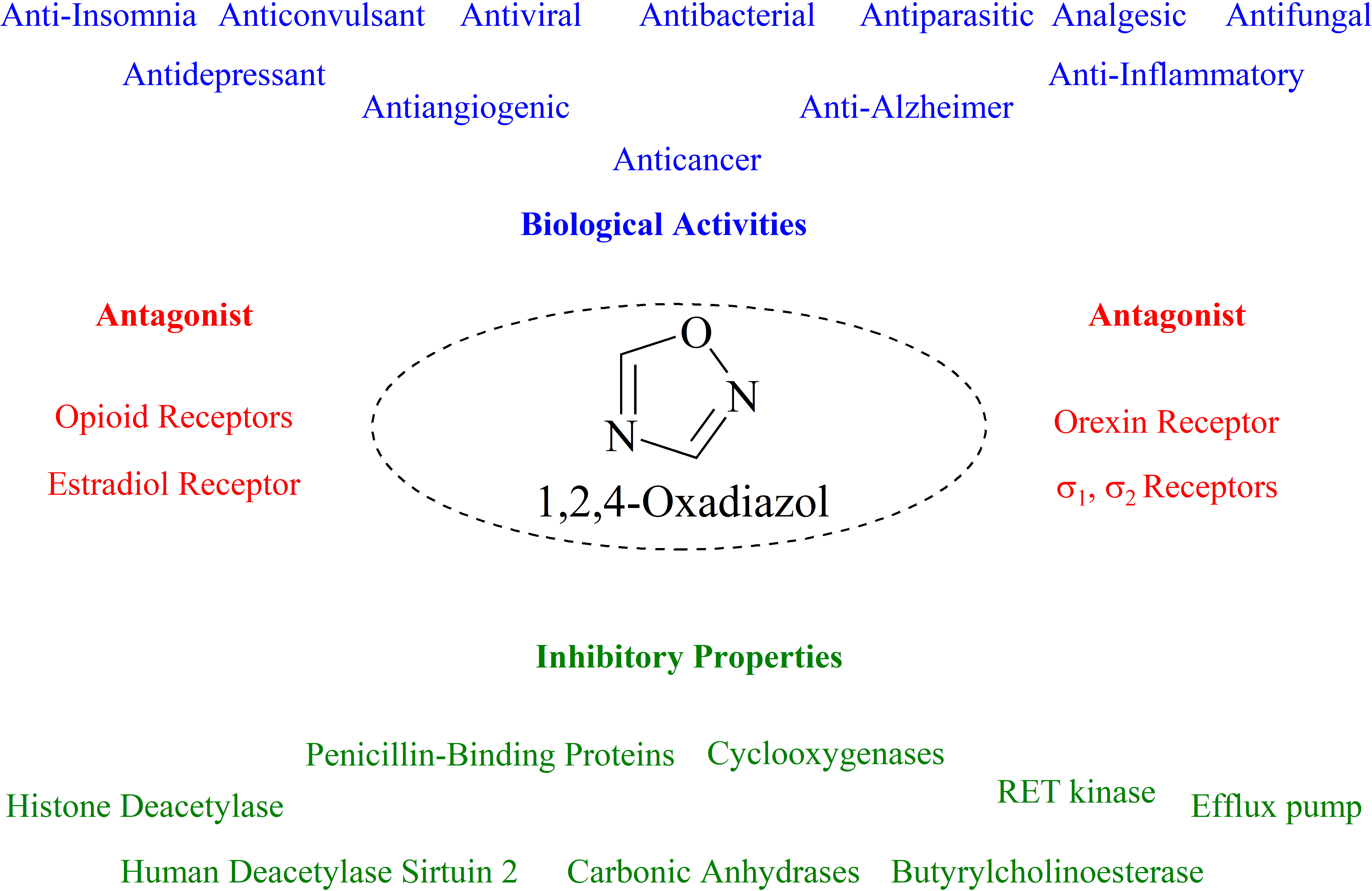
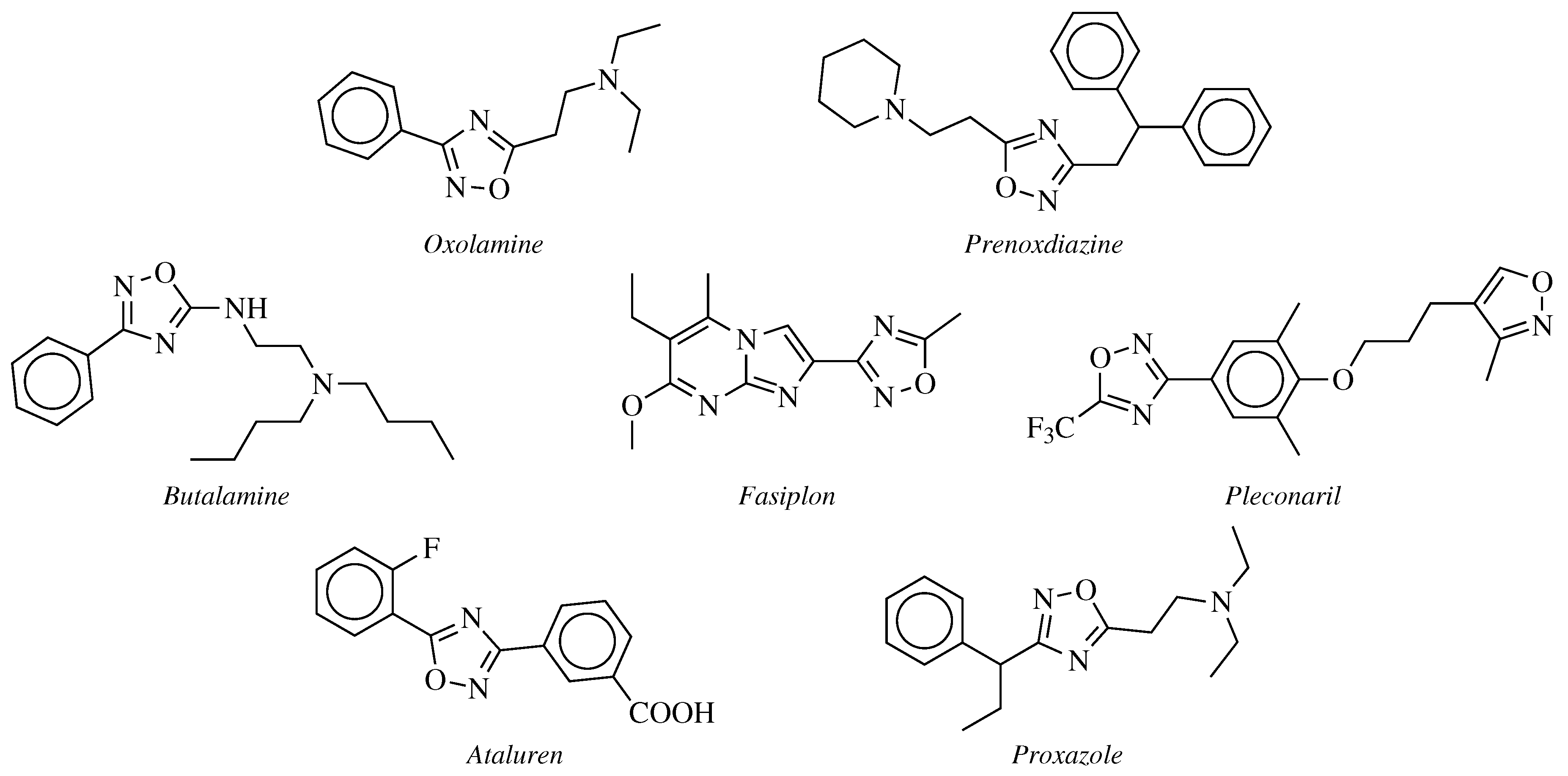
2. Historical Remarks—1,2,4-Oxadiazole
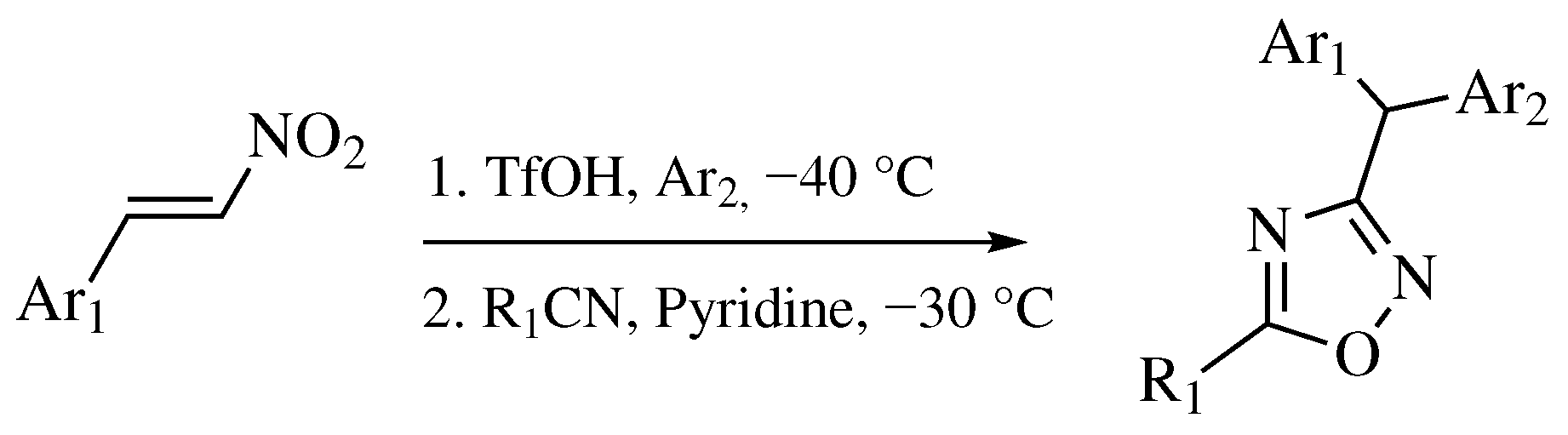
3. Methods of 1,2,4-Oxadiazole Synthesis
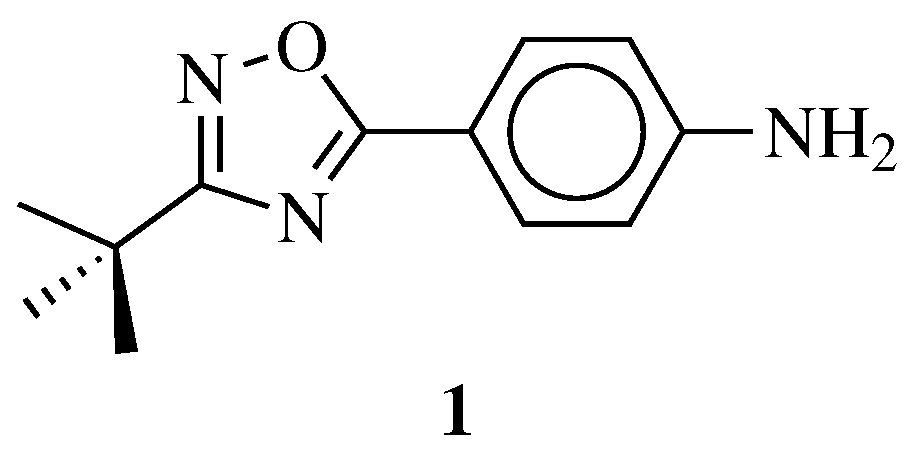
4. Anticancer Agents
5. Antimicrobial Agents
6. Anti-Inflammatory Agents
7. Anti-Allodynic Agents
8. Anticonvulsant Agents
9. Anti-Alzheimer Agents
10. Anti-Insomnia Agents
11. Other Biological Activities
12. Conclusions
Funding
Conflicts of Interest
Abbreviations
| AChE | Acetylcholinesterase |
| AD | Alzheimer Disease |
| AIDS | Acquired Immunodeficiency Syndrome |
| APAP | Acetaminophen |
| AMF | Acute Myeloid Leukemia |
| ARE | Antioxidant Responsive Element |
| BChE | Butyrylcholinoesterase |
| CCI | Chronic Constriction Injury |
| CDI | 1,1′-Carbonyldiimidazole |
| CNS | Central Nervous System |
| COX | Cyclooxygenase |
| CXCR4 | Chemokine Receptor Type 4 |
| DCC | N,N’-Dicyclohexylcarbodiimide |
| DOR | Delta-Opioid Receptor |
| DORA | Dual-Orexin Receptor Antagonist |
| EDC | 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide |
| EDG | Electron Donating Group |
| ELISA | Enzyme-Linked Immunosorbent Assay |
| ER | Estrogen Receptor |
| Et | Ethyl |
| EWG | Electron Withdrawing Group |
| FDA | Food and Drug Administration |
| GABA | gamma-Aminobutyric Acid |
| hCA | Human Carbonic Anhydrase |
| HDAC | Human Deacetylase |
| HDSirt2 | Human Deacetylase Sirtuin 2 |
| HEDMs | High Energy Density Materials |
| HIV | Human Immunodeficiency Virus |
| hRV | Human Rhinovirus |
| KOR | Kappa-Opioid Receptor |
| Me | Methyl |
| MES | Maximal Electroshock |
| MOR | Mu-Opioid Receptor |
| MPTP | 1-Methyl-4-phenyl-1,2,3,6-tetrahydropyridine |
| MRSA | Methicillin-Resistant Staphylococcus aureus |
| MWI | Microwave Irradiation |
| NAD+ | Oxidized Nicotinamide Adenine Dinucleotide |
| Nrf2 | Nuclear Factor Erythroid 2 |
| NSAIDs | NON-Steroidal Anti-Inflammatory Drugs |
| PBP2 | Penicillin-Binding Protein 2 |
| Ph | Phenyl |
| P.O. | per os |
| PROTACs | Proteolysis-targeting chimeras |
| PTP1B | Protein-Tyrosine Phosphate 1B |
| PTZ | Pentylenetetrazole |
| RT | Room Temperature |
| REM | Rapid-Eye Movement |
| RET | Rearranged During Transfection |
| SAR | Structural-Activity Relationship |
| T3P | Propylphosphonic anhydride |
| TBTU | 2-(1H-Benzotriazole-1-yl)-1,1,3,3-tetramethylaminiumtetrafluoroborate |
| TBAF | Tetra-n-butylammonium fluoride |
| TEA | Triethylamine |
| TfOH | Trifluoromethanesulfonic acid |
| THF | Tetrahydrofuran |
| VRE | Vancomycin-Resistant Enterococcus faecium |
| VRSA | Vancomycin-Resistant Staphylococcus aureus |
References
- Salahuddin; Mazumder, A.; Yar, M.S.; Mazumder, R.; Chakraborthy, G.S.; Ahsan, M.J.; Rahman, M.U. Updates on synthesis and biological activities of 1,3,4-oxadiazole: A review. Synth. Commun. 2017, 47, 1805–1847. [Google Scholar] [CrossRef]
- Bajaj, S.; Asati, V.; Singh, J.; Roy, P.P. 1,3,4-Oxadiazoles: An emerging scaffold to target growth factors, enzymes and kinases as anticancer agents. Eur. J. Med. Chem. 2015, 97, 124–141. [Google Scholar] [CrossRef]
- Bala, S.; Saini, V.; Kamboj, S.; Prasad, D.N. Review Exploring Antiinflammatory Potential of 1,3,4-Oxadiazole Derivatives as Promising Lead. Int. J. Pharm. Sci. Rev. Res. 2012, 17, 84–89. [Google Scholar]
- Khalilullah, H.J.; Ahsan, M.; Hedaitullah, M.; Khan, S.; Ahmed, B. 1,3,4-Oxadiazole: A Biologically Active Scaffold. Mini-Rev. Med. Chem. 2012, 12, 789–801. [Google Scholar] [CrossRef] [PubMed]
- Bajaj, S.; Roy, P.P.; Singh, J. 1,3,4-Oxadiazoles as Telomerase Inhibitor: Potential Anticancer Agents. Anti-Cancer Agents Med. Chem. 2018, 17, 1869–1883. [Google Scholar] [CrossRef] [PubMed]
- WebOfScience. Available online: http://www.webofknowledge.com./ (accessed on 16 December 2019).
- Wei, H.; He, C.; Zhang, J.; Shreeve, J.M. Combination of 1,2,4-Oxadiazole and 1,2,5-Oxadiazole Moieties for the Generation of High-Performance Energetic Materials. Angew. Chem. 2015, 127, 9499–9503. [Google Scholar] [CrossRef]
- Boiani, M. 1,2,5-Oxadiazole N-oxide derivatives as potential anti-cancer agents: Synthesis and biological evaluation. Part IV. Eur. J. Med. Chem. 2001, 36, 771–782. [Google Scholar] [CrossRef]
- Fershtat, L.L.; Makhova, N.N. 1,2,5-Oxadiazole-Based High-Energy-Density Materials: Synthesis and Performance. ChemPlusChem 2020, 85, 13–42. [Google Scholar] [CrossRef]
- Nguyen, M.T.; Hegarty, A.F.; Elguero, J. Can 1,2,3-Oxadiazole be Stable? Angew. Chem. Int. Ed. Engl. 1985, 24, 713–715. [Google Scholar] [CrossRef]
- Tiemann, F.; Krüger, P. Ueber Amidoxime und Azoxime. Berichte Der Dtsch. Chem. Ges. 1884, 17, 1685–1698. [Google Scholar] [CrossRef]
- Newman, H. Photochemistry of 3,5-diphenyl-1,2,4-oxadiazole II. Photolysis in protic media. Tetrahedron Lett. 1968, 9, 2421–2424. [Google Scholar] [CrossRef]
- Newman, H. Photochemistry of 3,5-diphenyl-1,2,4-oxadiazole I. Photolysis in aprotic media. Tetrahedron Lett. 1968, 9, 2417–2420. [Google Scholar] [CrossRef]
- Anderson, G.W.; Faith, H.E.; Marson, H.W.; Winnek, P.S.; Roblin, R.O. Studies in Chemotherapy. VI. Sulfanilamido Heterocycles. J. Am. Chem. Soc. 1942, 64, 2902–2905. [Google Scholar] [CrossRef]
- Silvestrini, B.; Catanese, B. Ricerche sul metabolismo del 5-beta-dietilamino-3-alfa -fenilpropil-1,2,4-oxadiazolo. Bollettino Chimico Farmaceutico 1964, 103, 447–450. [Google Scholar]
- Silvestrini, B. Un antitosse-antinfiammatorio, l’Oxolamina (Perebron). Minerva Medica 1960, 51, 4091–4094. [Google Scholar]
- Parra, M.; Hidalgo, P.; Alderete, J. New supramolecular liquid crystals induced by hydrogen bonding between pyridyl-1,2,4-oxadiazole derivatives and 2,5-thiophene dicarboxylic acid. Liq. Cryst. 2005, 32, 449–455. [Google Scholar] [CrossRef]
- Xiong, H.; Yang, H.; Lei, C.; Yang, P.; Hu, W.; Cheng, G. Combinations of furoxan and 1,2,4-oxadiazole for the generation of high performance energetic materials. Dalton Trans. 2019, 48, 14705–14711. [Google Scholar] [CrossRef]
- Yan, T.; Cheng, G.; Yang, H. 1,2,4-Oxadiazole-Bridged Polynitropyrazole Energetic Materials with Enhanced Thermal Stability and Low Sensitivity. ChemPlusChem 2019, 84, 1567–1577. [Google Scholar] [CrossRef]
- Pitasse-Santos, P.; Sueth-Santiago, V.; Lima, M. 1,2,4- and 1,3,4-Oxadiazoles as Scaffolds in the Development of Antiparasitic Agents. J. Braz. Chem. Soc. 2017, 29, 435–456. [Google Scholar] [CrossRef]
- Rosa, M.F.; Morcelli, A.C.T.; Lobo, V.S. 1,2,4-Oxadiazole: A Brief Review From The Literature About the Synthesis and Pharmacological Applications. Vis ao Acadêmica Curitiba 2015, 16, 130–157. [Google Scholar] [CrossRef]
- Coupar, I.M.; Hedges, A.; Metcalfe, H.L.; Turner, P. Effect of aminophylline, butalamine and imolamine on human isolated smooth muscle. J. Pharm. Pharmacol. 1969, 21, 474–475. [Google Scholar] [CrossRef] [PubMed]
- Rotbart, H.A.; Webster, A.D. Treatment of Potentially Life-Threatening Enterovirus Infections with Pleconaril. Clin. Infect. Dis. 2001, 32, 228–235. [Google Scholar] [CrossRef] [PubMed]
- McDonald, C.M.; Campbell, C.; Torricelli, R.E.; Finkel, R.S.; Flanigan, K.M.; Goemans, N.; Heydemann, P.; Kaminska, A.; Kirschner, J.; Muntoni, F.; et al. Ataluren in patients with nonsense mutation Duchenne muscular dystrophy (ACT DMD): A multicentre, randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2017, 390, 1489–1498. [Google Scholar] [CrossRef]
- Carbone, M.; Li, Y.; Irace, C.; Mollo, E.; Castelluccio, F.; Di Pascale, A.; Cimino, G.; Santamaria, R.; Guo, Y.W.; Gavagnin, M. Structure and Cytotoxicity of Phidianidines A and B: First Finding of 1,2,4-Oxadiazole System in a Marine Natural Product. Org. Lett. 2011, 13, 2516–2519. [Google Scholar] [CrossRef] [PubMed]
- Vitale, R.M.; Gatti, M.; Carbone, M.; Barbieri, F.; Felicità, V.; Gavagnin, M.; Florio, T.; Amodeo, P. A minimalist hybrid ligand/receptor-based pharmacophore model for CXCR4 applied to a small-library of marine natural products led to the identification of Phidianidine A as a new CXCR4 ligand exhibiting antagonist activity. ACS Chem. Biol. 2013, 8, 2762–2770. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Jiang, C.S.; Gao, L.X.; Gong, J.X.; Wang, Z.H.; Li, J.Y.; Li, J.; Li, X.W.; Guo, Y.W. Design, synthesis and in vitro activity of phidianidine B derivatives as novel PTP1B inhibitors with specific selectivity. Bioorg. Med. Chem. Lett. 2016, 26, 778–781. [Google Scholar] [CrossRef]
- Hermit, M.B.; Greenwood, J.R.; Bräuner-Osborne, H. Mutation-induced Quisqualic Acid and Ibotenic Acid Affinity at the Metabotropic Glutamate Receptor Subtype 4. J. Biol. Chem. 2004, 279, 34811–34817. [Google Scholar] [CrossRef]
- Kozikowski, A.P.; Steensma, D.; Varasi, M.; Pshenichkin, S.; Surina, E.; Wroblewski, J.T. α-substituted quisqualic acid analogs: New metabotropic glutamate receptor group II selective antagonists. Bioorg. Med. Chem. Lett. 1998, 8, 447–452. [Google Scholar] [CrossRef]
- Gangloff, A.R.; Litvak, J.; Shelton, E.J.; Sperandio, D.; Wang, V.R.; Rice, K.D. Synthesis of 3,5-disubstituted-1,2,4-oxadiazoles using tetrabutylammonium fluoride as a mild and efficient catalyst. Tetrahedron Lett. 2001, 42, 1441–1443. [Google Scholar] [CrossRef]
- Amarasinghe, K.K.; Maier, M.B.; Srivastava, A.; Gray, J.L. One-pot synthesis of 1,2,4-oxadiazoles from carboxylic acid esters and amidoximes using potassium carbonate. Tetrahedron Lett. 2006, 47, 3629–3631. [Google Scholar] [CrossRef]
- Rauf, A.; Sharma, S.; Gangal, S. An efficient, one-pot synthesis of novel 3,5-disubstituted-1,2,4- oxadiazoles from long-chain carboxylic acid derivatives. Acta Chim. Slov. 2009, 56, 369–372. [Google Scholar]
- Sureshbabu, V.V.; Hemantha, H.P.; Naik, S.A. Synthesis of 1,2,4-oxadiazole-linked orthogonally urethane-protected dipeptide mimetics. Tetrahedron Lett. 2008, 49, 5133–5136. [Google Scholar] [CrossRef]
- Augustine, J.K.; Vairaperumal, V.; Narasimhan, S.; Alagarsamy, P.; Radhakrishnan, A. Propylphosphonic anhydride (T3P®): An efficient reagent for the one-pot synthesis of 1,2,4-oxadiazoles, 1,3,4-oxadiazoles, and 1,3,4-thiadiazoles. Tetrahedron 2009, 65, 9989–9996. [Google Scholar] [CrossRef]
- Kaboudin, B.; Malekzadeh, L. Organic reactions in water: An efficient method for the synthesis of 1,2,4-oxadiazoles in water. Tetrahedron Lett. 2011, 52, 6424–6426. [Google Scholar] [CrossRef]
- de Freitas, J.J.R.; de Freitas, J.C.R.; da Silva, L.P.; de Freitas Filho, J.R.; Kimura, G.Y.; Srivastava, R.M. Microwave-induced one-pot synthesis of 4-[3-(aryl)-1,2,4-oxadiazol-5-yl]-butan-2-ones under solvent free conditions. Tetrahedron Lett. 2007, 48, 6195–6198. [Google Scholar] [CrossRef]
- Kaboudin, B.; Saadati, F. Novel method for the synthesis of 1,2,4-oxadiazoles using alumina supported ammonium fluoride under solvent-free condition. J. Heterocycl. Chem. 2005, 42, 699–701. [Google Scholar] [CrossRef]
- Rostamizadeh, S.; Ghaieni, H.R.; Aryan, R.; Amani, A.M. Clean one-pot synthesis of 1,2,4-oxadiazoles under solvent-free conditions using microwave irradiation and potassium fluoride as catalyst and solid support. Tetrahedron 2010, 66, 494–497. [Google Scholar] [CrossRef]
- Kaboudin, B.; Saadati, F. Magnesia-supported hydroxylamine hydrochloride in the presence of sodium carbonate as an efficient reagent for the synthesis of 1,2,4-oxadiazoles from nitriles. Tetrahedron Lett. 2007, 48, 2829–2832. [Google Scholar] [CrossRef]
- Adib, M.; Jahromi, A.H.; Tavoosi, N.; Mahdavi, M.; Bijanzadeh, H.R. Microwave-assisted efficient, one-pot, three-component synthesis of 3,5-disubstituted 1,2,4-oxadiazoles under solvent-free conditions. Tetrahedron Lett. 2006, 47, 2965–2967. [Google Scholar] [CrossRef]
- Rajagopalan, P. Dipolar addition reactions of nitrile oxides. VII. A new general method of synthesis of 3,5-disubstituted 1,2,4-oxadiazoles. Tetrahedron Lett. 1969, 10, 311–312. [Google Scholar] [CrossRef]
- Quadrelli, P.; Invernizzi, A.G.; Falzoni, M.; Caramella, P. Cycloadditions of nitrile oxides to amidoximes. A general synthesis of 3,5-disubstituted 1,2,4-oxadiazole-4-oxides. Tetrahedron 1997, 53, 1787–1796. [Google Scholar] [CrossRef]
- Bokach, N.A.; Khripoun, A.V.; Kukushkin, V.Y.; Haukka, M.; Pombeiro, A.J.L. A Route to 1,2,4-Oxadiazoles and Their Complexes via Platinum-Mediated 1,3-Dipolar Cycloaddition of Nitrile Oxides to Organonitriles. Inorg. Chem. 2003, 42, 896–903. [Google Scholar] [CrossRef]
- Baykov, S.; Sharonova, T.; Shetnev, A.; Rozhkov, S.; Kalinin, S.; Smirnov, A.V. The first one-pot ambient-temperature synthesis of 1,2,4-oxadiazoles from amidoximes and carboxylic acid esters. Tetrahedron 2017, 73, 945–951. [Google Scholar] [CrossRef]
- Zarei, M. A Mild and Efficient One-Pot Preparation of 1,2,4-Oxadiazoles from Nitriles and Carboxylic Acids Using Vilsmeier Reagent. ChemistrySelect 2018, 3, 11273–11276. [Google Scholar] [CrossRef]
- Vinaya, K.; Chandrashekara, G.K.; Shivaramu, P.D. One-pot synthesis of 3,5-diaryl substituted-1,2,4-oxadiazoles using gem -dibromomethylarenes. Can. J. Chem. 2019, 97, 690–696. [Google Scholar] [CrossRef]
- Golushko, A.A.; Khoroshilova, O.V.; Vasilyev, A.V. Synthesis of 1,2,4-Oxadiazoles by Tandem Reaction of Nitroalkenes with Arenes and Nitriles in the Superacid TfOH. J. Org. Chem. 2019, 84, 7495–7500. [Google Scholar] [CrossRef]
- Cai, B.G.; Chen, Z.L.; Xu, G.Y.; Xuan, J.; Xiao, W.J. [3 + 2]-Cycloaddition of 2H -Azirines with Nitrosoarenes: Visible-Light-Promoted Synthesis of 2,5-Dihydro-1,2,4-oxadiazoles. Org. Lett. 2019, 21, 4234–4238. [Google Scholar] [CrossRef]
- James, S.L.; Adams, C.J.; Bolm, C.; Braga, D.; Collier, P.; Friščić, T.; Grepioni, F.; Harris, K.D.M.; Hyett, G.; Jones, W.; et al. Mechanochemistry: Opportunities for new and cleaner synthesis. Chem. Soc. Rev. 2012, 41, 413–447. [Google Scholar] [CrossRef]
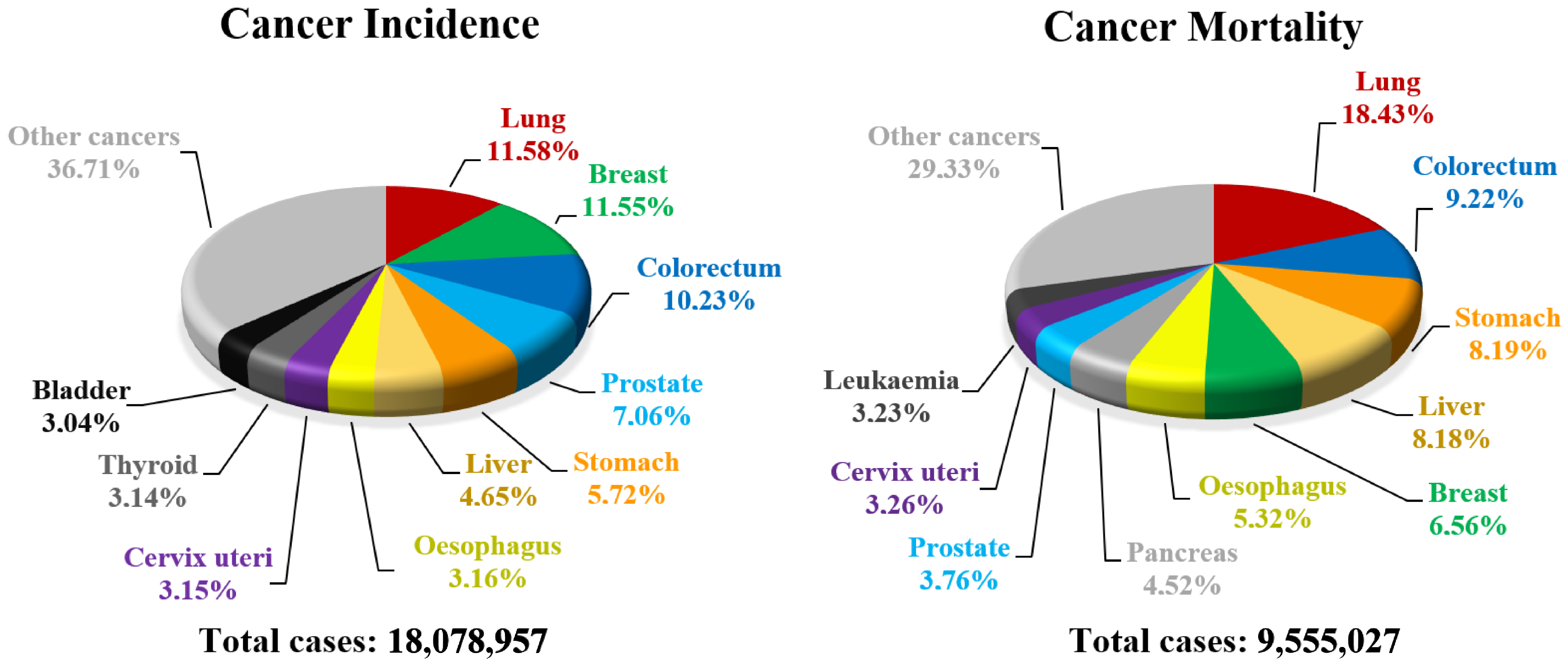
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef]
- Zhang, H.Z.; Kasibhatla, S.; Kuemmerle, J.; Kemnitzer, W.; Ollis-Mason, K.; Qiu, L.; Crogan-Grundy, C.; Tseng, B.; Drewe, J.; Cai, S.X. Discovery and Structure-Activity Relationship of 3-Aryl-5-aryl-1,2,4-oxadiazoles as a New Series of Apoptosis Inducers and Potential Anticancer Agents. J. Med. Chem. 2005, 48, 5215–5223. [Google Scholar] [CrossRef]
- Pace, A.; Buscemi, S.; Piccionello, A.P.; Pibiri, I. Recent Advances in the Chemistry of 1,2,4-Oxadiazoles. In Advances in Heterocyclic Chemistry; Academic Press Inc.: Cambridge, MA, USA, 2015; Volume 116, pp. 85–136. [Google Scholar] [CrossRef]
- Rasool, I.; Ahmad, M.; Khan, Z.A.; Mansha, A.; Maqbool, T.; Zahoor, A.F.; Aslam, S. Recent advancements in oxadiazole-based anticancer agents. Trop. J. Pharm. Res. 2017, 16, 723. [Google Scholar] [CrossRef]
- Maftei, C.V.; Fodor, E.; Jones, P.G.; Franz, M.H.; Kelter, G.; Fiebig, H.; Neda, I. Synthesis and characterization of novel bioactive 1,2,4-oxadiazole natural product analogs bearing the N-phenylmaleimide and N-phenylsuccinimide moieties. Beilstein J. Org. Chem. 2013, 9, 2202–2215. [Google Scholar] [CrossRef] [PubMed]
- Maftei, C.V.; Fodor, E.; Jones, P.G.; Daniliuc, C.G.; Franz, M.H.; Kelter, G.; Fiebig, H.H.; Tamm, M.; Neda, I. Novel 1,2,4-oxadiazoles and trifluoromethylpyridines related to natural products: Synthesis, structural analysis and investigation of their antitumor activity. Tetrahedron 2016, 72, 1185–1199. [Google Scholar] [CrossRef]
- Maftei, C.V.; Fodor, E.; Jones, P.G.; Freytag, M.; Franz, M.H.; Kelter, G.; Fiebig, H.H.; Tamm, M.; Neda, I. N -heterocyclic carbenes (NHC) with 1,2,4-oxadiazole-substituents related to natural products: Synthesis, structure and potential antitumor activity of some corresponding gold(I) and silver(I) complexes. Eur. J. Med. Chem. 2015, 101, 431–441. [Google Scholar] [CrossRef] [PubMed]
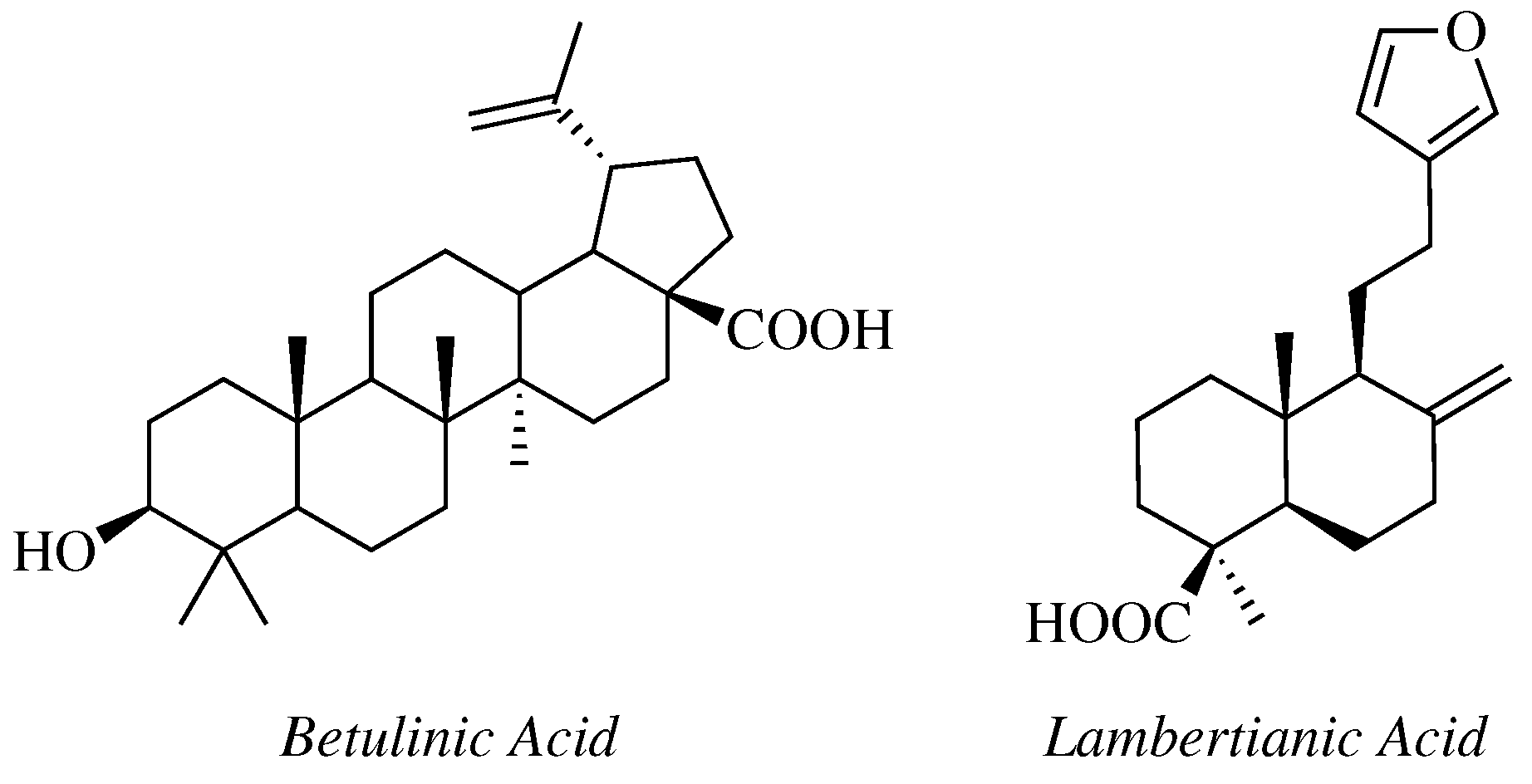
- Krishna, C.; Bhargavi, M.V.; Krupadanam, G.L.D. Design, Synthesis, and Cytotoxicity of Semisynthetic Betulinic Acid-1,2,4-Oxadiazole Amide Derivatives. Russ. J. Gen. Chem. 2018, 88, 312–318. [Google Scholar] [CrossRef]
- Challa, K.; Bhargavi, M.V.; Krupadanam, G.L.D. Design, semisynthesis and cytotoxic activity of novel ester derivatives of betulinic acid-1,2,4 oxadiazoles. J. Asian Nat. Prod. Res. 2016, 18, 1158–1168. [Google Scholar] [CrossRef]
- Hande, K. Etoposide: Four decades of development of a topoisomerase II inhibitor. Eur. J. Cancer 1998, 34, 1514–1521. [Google Scholar] [CrossRef]
- Mironov, M.E.; Pokrovsky, M.A.; Kharitonov, Y.V.; Shakirov, M.M.; Pokrovsky, A.G.; Shults, E.E. Furanolabdanoid-based 1,2,4-oxadiazoles: Synthesis and cytotoxic activity. ChemistrySelect 2016, 1, 417–424. [Google Scholar] [CrossRef]
- Guest, J.F.; Panca, M.; Sladkevicius, E.; Gough, N.; Linch, M. Cost Effectiveness of First-Line Treatment with Doxorubicin/Ifosfamide Compared to Trabectedin Monotherapy in the Management of Advanced Soft Tissue Sarcoma in Italy, Spain, and Sweden. Sarcoma 2013, 2013, 1–19. [Google Scholar] [CrossRef]
- Carvalho, C.; Santos, R.; Cardoso, S.; Correia, S.; Oliveira, P.; Santos, M.; Moreira, P. Doxorubicin: The Good, the Bad and the Ugly Effect. Curr. Med. Chem. 2009, 16, 3267–3285. [Google Scholar] [CrossRef]
- Kucukoglu, K.; Tugrak, M.; Demirtas, A.; Sakagami, H.; Gul, H.I. Synthesis and Cytotoxic Activity of (4-Substituted-benzylidene)-(3-Phenyl-1,2,4-Oxadiazol-5-YL)Methylamines. Pharm. Chem. J. 2016, 50, 234–238. [Google Scholar] [CrossRef]
- Moniot, S.; Forgione, M.; Lucidi, A.; Hailu, G.S.; Nebbioso, A.; Carafa, V.; Baratta, F.; Altucci, L.; Giacché, N.; Passeri, D.; et al. Development of 1,2,4-Oxadiazoles as Potent and Selective Inhibitors of the Human Deacetylase Sirtuin 2: Structure–Activity Relationship, X-ray Crystal Structure, and Anticancer Activity. J. Med. Chem. 2017, 60, 2344–2360. [Google Scholar] [CrossRef] [PubMed]
- Avanzo, R.E.; Padrón, J.M.; D’Accorso, N.B.; Fascio, M.L. Synthesis and in vitro antiproliferative activities of (5-aryl-1,2,4-oxadiazole-3-yl) methyl D-ribofuranosides. Bioorg. Med. Chem. Lett. 2017, 27, 3674–3677. [Google Scholar] [CrossRef] [PubMed]
- Abd el hameid, M.K.; Mohammed, M.R. Design, synthesis, and cytotoxicity screening of 5-aryl-3-(2-(pyrrolyl) thiophenyl)-1, 2, 4-oxadiazoles as potential antitumor molecules on breast cancer MCF-7 cells. Bioorg. Chem. 2019, 86, 609–623. [Google Scholar] [CrossRef]
- de Oliveira, V.N.M.; dos Santos, F.G.; Ferreira, V.P.G.; Araújo, H.M.; do Ó Pessoa, C.; Nicolete, R.; de Oliveira, R.N. Focused microwave irradiation-assisted synthesis of N-cyclohexyl-1,2,4-oxadiazole derivatives with antitumor activity. Synth. Commun. 2018, 48, 2522–2532. [Google Scholar] [CrossRef]
- Sateesh Kumar, P.; Umadevi, P. Novel Bis(1,2,4-oxadiazolyl) Fused Thiazole Derivatives: Synthesis and Anticancer Activity. Russ. J. Gen. Chem. 2018, 88, 2611–2615. [Google Scholar] [CrossRef]
- Pervaram, S.; Ashok, D.; Sarasija, M.; Reddy, C.V.R.; Sridhar, G. Synthesis and Anticancer Activity of 1,2,4-Oxadiazole Fused Benzofuran Derivatives. Russ. J. Gen. Chem. 2018, 88, 1219–1223. [Google Scholar] [CrossRef]
- Chakrapani, B.; Ramesh, V.; Pourna Chander Rao, G.; Ramachandran, D.; Madhukar Reddy, T.; Kalyan Chakravarthy, A.; Sridhar, G. Synthesis and Anticancer Evaluation of 1,2,4-Oxadiazole Linked Imidazothiadiazole Derivatives. Russ. J. Gen. Chem. 2018, 88, 1020–1024. [Google Scholar] [CrossRef]
- Srinivas, M.; Satyaveni, S.; Ram, B. Synthesis and Anticancer Activity of 1,2,4-Oxadiazol Linked Benzimidazole Derivatives. Russ. J. Gen. Chem. 2018, 88, 2653–2657. [Google Scholar] [CrossRef]
- Chiacchio, M.A.; Legnani, L.; Campisi, A.; Paola, B.; Giuseppe, L.; Iannazzo, D.; Veltri, L.; Giofrè, S.; Romeo, R. 1,2,4-Oxadiazole-5-ones as analogues of tamoxifen: Synthesis and biological evaluation. Org. Biomol. Chem. 2019, 17, 4892–4905. [Google Scholar] [CrossRef]
- Krasavin, M.; Shetnev, A.; Sharonova, T.; Baykov, S.; Tuccinardi, T.; Kalinin, S.; Angeli, A.; Supuran, C.T. Heterocyclic periphery in the design of carbonic anhydrase inhibitors: 1,2,4-Oxadiazol-5-yl benzenesulfonamides as potent and selective inhibitors of cytosolic hCA II and membrane-bound hCA IX isoforms. Bioorg. Chem. 2018, 76, 88–97. [Google Scholar] [CrossRef] [PubMed]
- Krasavin, M.; Shetnev, A.; Sharonova, T.; Baykov, S.; Kalinin, S.; Nocentini, A.; Sharoyko, V.; Poli, G.; Tuccinardi, T.; Presnukhina, S.; et al. Continued exploration of 1,2,4-oxadiazole periphery for carbonic anhydrase-targeting primary arene sulfonamides: Discovery of subnanomolar inhibitors of membrane-bound hCA IX isoform that selectively kill cancer cells in hypoxic environment. Eur. J. Med. Chem. 2019, 164, 92–105. [Google Scholar] [CrossRef] [PubMed]
- Cascioferro, S.; Attanzio, A.; Di Sarno, V.; Musella, S.; Tesoriere, L.; Cirrincione, G.; Diana, P.; Parrino, B. New 1,2,4-Oxadiazole Nortopsentin Derivatives with Cytotoxic Activity. Mar. Drugs 2019, 17, 35. [Google Scholar] [CrossRef] [PubMed]
- Polothi, R.; Raolji, G.S.B.; Kuchibhotla, V.S.; Sheelam, K.; Tuniki, B.; Thodupunuri, P. Synthesis and biological evaluation of 1,2,4-oxadiazole linked 1,3,4-oxadiazole derivatives as tubulin binding agents. Synth. Commun. 2019, 49, 1603–1612. [Google Scholar] [CrossRef]
- Yang, F.; Shan, P.; Zhao, N.; Ge, D.; Zhu, K.; Jiang, C.s.; Li, P.; Zhang, H. Development of hydroxamate-based histone deacetylase inhibitors containing 1,2,4-oxadiazole moiety core with antitumor activities. Bioorg. Med. Chem. Lett. 2019, 29, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Zhang, T.; Wu, H.; Yang, Y.; Liu, N.; Chen, A.; Li, Q.; Li, J.; Qin, L.; Jiang, B.; et al. Design and Optimization of Novel Hydroxamate-Based Histone Deacetylase Inhibitors of Bis-Substituted Aromatic Amides Bearing Potent Activities against Tumor Growth and Metastasis. J. Med. Chem. 2014, 57, 9357–9369. [Google Scholar] [CrossRef]
- Yang, Z.; Shen, M.; Tang, M.; Zhang, W.; Cui, X.; Zhang, Z.; Pei, H.; Li, Y.; Hu, M.; Bai, P.; et al. Discovery of 1,2,4-oxadiazole-Containing hydroxamic acid derivatives as histone deacetylase inhibitors potential application in cancer therapy. Eur. J. Med. Chem. 2019, 178, 116–130. [Google Scholar] [CrossRef]
- Han, M.; Li, S.; Ai, J.; Sheng, R.; Hu, Y.; Hu, Y.; Geng, M. Discovery of 4-chloro-3-(5-(pyridin-3-yl)-1,2,4-oxadiazole-3-yl)benzamides as novel RET kinase inhibitors. Bioorg. Med. Chem. Lett. 2016, 26, 5679–5684. [Google Scholar] [CrossRef]
- Avanzo, R.E.; Anesini, C.; Fascio, M.L.; Errea, M.I.; D’Accorso, N.B. 1,2,4-Triazole D-ribose derivatives: Design, synthesis and antitumoral evaluation. Eur. J. Med. Chem. 2012, 47, 104–110. [Google Scholar] [CrossRef]
- authors listed, N. Tamoxifen for early breast cancer: An overview of the randomised trials. Lancet 1998, 351, 1451–1467. [Google Scholar] [CrossRef]
- Eckermann, S.D.; Martin, A.J.; Stockier, M.R.; Simes, R.J. The benefits and costs of tamoxifen for breast cancer prevention. Aust. N. Z. J. Public Health 2003, 27, 34–40. [Google Scholar] [CrossRef] [PubMed]
- Vogel, V.G. Effects of Tamoxifen vs Raloxifene on the Risk of Developing Invasive Breast Cancer and Other Disease Outcomes: The NSABP Study of Tamoxifen and Raloxifene (STAR) P-2 Trial. JAMA 2006, 295, 2727–2741. [Google Scholar] [CrossRef] [PubMed]
- Fisher, B.; Costantino, J.P.; Wickerham, D.L.; Redmond, C.K.; Kavanah, M.; Cronin, W.M.; Vogel, V.; Robidoux, A.; Dimitrov, N.; Atkins, J.; et al. Tamoxifen for Prevention of Breast Cancer: Report of the National Surgical Adjuvant Breast and Bowel Project P-1 Study. JNCI J. Natl. Cancer Inst. 1998, 90, 1371–1388. [Google Scholar] [CrossRef] [PubMed]
- Gorin, M.B.; Day, R.; Costantino, J.P.; Fisher, B.; Redmond, C.K.; Wickerham, L.; Gomolin, J.E.; Margolese, R.G.; Mathen, M.K.; Bowman, D.M.; et al. Long-term tamoxifen citrate use and potential ocular toxicity. Am. J. Ophthalmol. 1998, 125, 493–501. [Google Scholar] [CrossRef]
- Krasavin, M.; Korsakov, M.; Dorogov, M.; Tuccinardi, T.; Dedeoglu, N.; Supuran, C.T. Probing the ‘bipolar’ nature of the carbonic anhydrase active site: Aromatic sulfonamides containing 1,3-oxazol-5-yl moiety as picomolar inhibitors of cytosolic CA I and CA II isoforms. Eur. J. Med. Chem. 2015, 101, 334–347. [Google Scholar] [CrossRef] [PubMed]
- Krasavin, M.; Korsakov, M.; Zvonaryova, Z.; Semyonychev, E.; Tuccinardi, T.; Kalinin, S.; Tanç, M.; Supuran, C.T. Human carbonic anhydrase inhibitory profile of mono- and bis-sulfonamides synthesized via a direct sulfochlorination of 3- and 4-(hetero)arylisoxazol-5-amine scaffolds. Bioorg. Med. Chem. 2017, 25, 1914–1925. [Google Scholar] [CrossRef] [PubMed]
- Supuran, C.T.; Kalinin, S.; Tanç, M.; Sarnpitak, P.; Mujumdar, P.; Poulsen, S.A.; Krasavin, M. Isoform-selective inhibitory profile of 2-imidazoline-substituted benzene sulfonamides against a panel of human carbonic anhydrases. J. Enzym. Inhib. Med. Chem. 2016, 31, 197–202. [Google Scholar] [CrossRef]
- Krasavin, M.; Korsakov, M.; Ronzhina, O.; Tuccinardi, T.; Kalinin, S.; Tanç, M.; Supuran, C.T. Primary mono- and bis-sulfonamides obtained via regiospecific sulfochlorination of N-arylpyrazoles: Inhibition profile against a panel of human carbonic anhydrases. J. Enzym. Inhib. Med. Chem. 2017, 32, 920–934. [Google Scholar] [CrossRef]
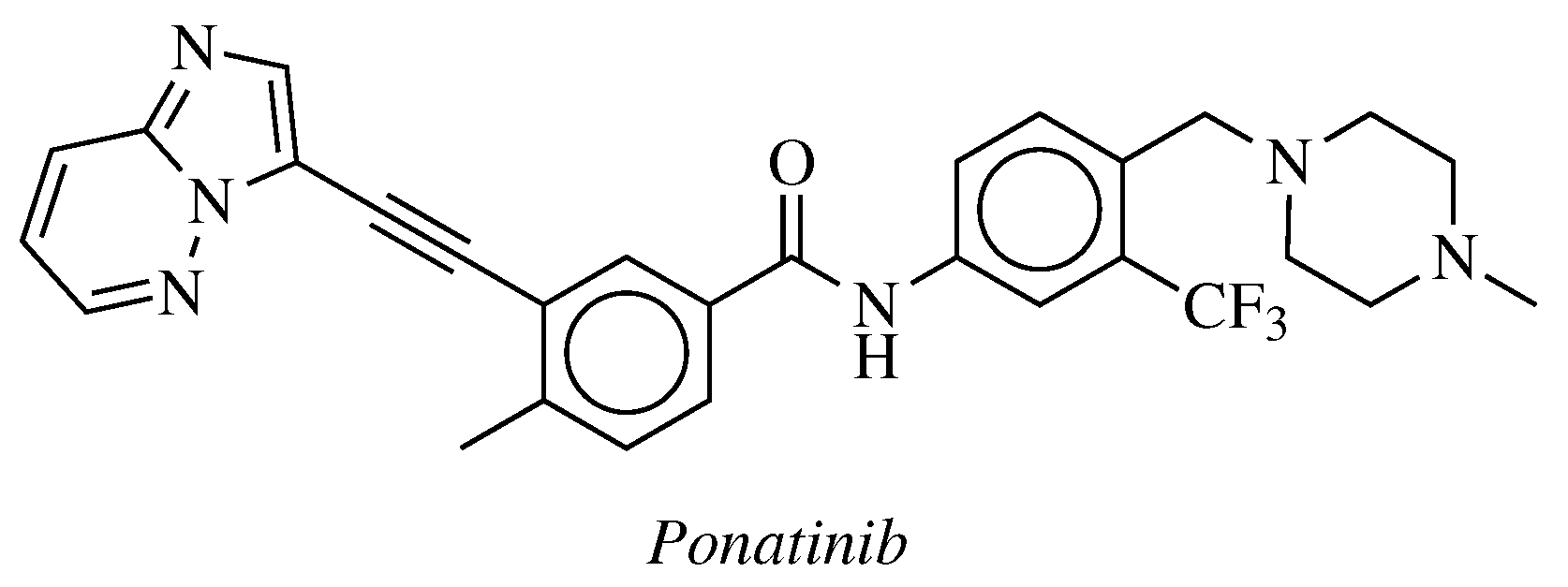
- De Falco, V.; Buonocore, P.; Muthu, M.; Torregrossa, L.; Basolo, F.; Billaud, M.; Gozgit, J.M.; Carlomagno, F.; Santoro, M. Ponatinib (AP24534) Is a Novel Potent Inhibitor of Oncogenic RET Mutants Associated With Thyroid Cancer. J. Clin. Endocrinol. Metab. 2013, 98, E811–E819. [Google Scholar] [CrossRef]
- Mologni, L.; Redaelli, S.; Morandi, A.; Plaza-Menacho, I.; Gambacorti-Passerini, C. Ponatinib is a potent inhibitor of wild-type and drug-resistant gatekeeper mutant RET kinase. Mol. Cell. Endocrinol. 2013, 377, 1–6. [Google Scholar] [CrossRef]
- Woolhouse, M.E.; Gowtage-Sequeria, S. Host Range and Emerging and Reemerging Pathogens. Emerg. Infect. Dis. 2005, 11, 1842–1847. [Google Scholar] [CrossRef] [PubMed]
- Dye, C. After 2015: Infectious diseases in a new era of health and development. Philos. Trans. R. Soc. B Biol. Sci. 2014, 369, 20130426. [Google Scholar] [CrossRef] [PubMed]
- Lozano, R.; Naghavi, M.; Foreman, K.; Lim, S.; Shibuya, K.; Aboyans, V.; Abraham, J.; Adair, T.; Aggarwal, R.; Ahn, S.Y.; et al. Global and regional mortality from 235 causes of death for 20 age groups in 1990 and 2010: A systematic analysis for the Global Burden of Disease Study 2010. Lancet 2012, 380, 2095–2128. [Google Scholar] [CrossRef]
- O’Daniel, P.I.; Peng, Z.; Pi, H.; Testero, S.A.; Ding, D.; Spink, E.; Leemans, E.; Boudreau, M.A.; Yamaguchi, T.; Schroeder, V.A.; et al. Discovery of a New Class of Non-β-lactam Inhibitors of Penicillin-Binding Proteins with Gram-Positive Antibacterial Activity. J. Am. Chem. Soc. 2014, 136, 3664–3672. [Google Scholar] [CrossRef]
- Carter, G.P.; Harjani, J.R.; Li, L.; Pitcher, N.P.; Nong, Y.; Riley, T.V.; Williamson, D.A.; Stinear, T.P.; Baell, J.B.; Howden, B.P. 1,2,4-Oxadiazole antimicrobials act synergistically with daptomycin and display rapid kill kinetics against MDR Enterococcus faecium. J. Antimicrob. Chemother. 2018, 73, 1562–1569. [Google Scholar] [CrossRef]
- Ding, D.; Boudreau, M.A.; Leemans, E.; Spink, E.; Yamaguchi, T.; Testero, S.A.; O’Daniel, P.I.; Lastochkin, E.; Chang, M.; Mobashery, S. Exploration of the structure–activity relationship of 1,2,4-oxadiazole antibiotics. Bioorg. Med. Chem. Lett. 2015, 25, 4854–4857. [Google Scholar] [CrossRef]
- Spink, E.; Ding, D.; Peng, Z.; Boudreau, M.A.; Leemans, E.; Lastochkin, E.; Song, W.; Lichtenwalter, K.; O’Daniel, P.I.; Testero, S.A.; et al. Structure–Activity Relationship for the Oxadiazole Class of Antibiotics. J. Med. Chem. 2015, 58, 1380–1389. [Google Scholar] [CrossRef]
- Leemans, E.; Mahasenan, K.V.; Kumarasiri, M.; Spink, E.; Ding, D.; O’Daniel, P.I.; Boudreau, M.A.; Lastochkin, E.; Testero, S.A.; Yamaguchi, T.; et al. Three-Dimensional QSAR Analysis and Design of New 1,2,4-Oxadiazole Antibacterials. Bioorg. Med. Chem. Lett. 2016, 26, 1011–1015. [Google Scholar] [CrossRef]
- Xiao, Q.; Vakulenko, S.; Chang, M.; Mobashery, S. Mutations in mmpL and in the Cell Wall Stress Stimulon Contribute to Resistance to Oxadiazole Antibiotics in Methicillin-Resistant Staphylococcus aureus. Antimicrob. Agents Chemother. 2014, 58, 5841–5847. [Google Scholar] [CrossRef]
- Janardhanan, J.; Chang, M.; Mobashery, S. The oxadiazole antibacterials. Curr. Opin. Microbiol. 2016, 33, 13–17. [Google Scholar] [CrossRef]
- Ceballos, S.; Kim, C.; Ding, D.; Mobashery, S.; Chang, M.; Torres, C. Activities of Oxadiazole Antibacterials against Staphylococcus aureus and Other Gram-Positive Bacteria. Antimicrob. Agents Chemother. 2018, 62. [Google Scholar] [CrossRef] [PubMed]
- Krolenko, K.Y.; Vlasov, S.V.; Zhuravel, I.A. Synthesis and antimicrobial activity of 5-(1H-1,2,3-triazol-4-yl)-1,2,4-oxadiazole derivatives. Chem. Heterocycl. Compd. 2016, 52, 823–830. [Google Scholar] [CrossRef]
- Cunha, F.; Nogueira, J.; de Aguiar, A. Synthesis and Antibacterial Evaluation of 3,5-Diaryl-1,2,4-oxadiazole Derivatives. J. Braz. Chem. Soc. 2018, 29, 2405–2416. [Google Scholar] [CrossRef]
- Shi, G.; He, X.; Shang, Y.; Xiang, L.; Yang, C.; Han, G.; Du, B. Synthesis of 3′,4′-Diaryl-4′H -spiro[indoline-3,5′-[1′,2′,4′]oxadiazol]-2-ones via DMAP-catalyzed Domino Reactions and Their Antibacterial Activity. Chin. J. Chem. 2016, 34, 901–909. [Google Scholar] [CrossRef]
- Shetnev, A.; Baykov, S.; Kalinin, S.; Belova, A.; Sharoyko, V.; Rozhkov, A.; Zelenkov, L.; Tarasenko, M.; Sadykov, E.; Korsakov, M.; et al. 1,2,4-Oxadiazole/2-Imidazoline Hybrids: Multi-target-directed Compounds for the Treatment of Infectious Diseases and Cancer. Int. J. Mol. Sci. 2019, 20, 1699. [Google Scholar] [CrossRef] [PubMed]
- Tarasenko, M.; Sidneva, V.; Belova, A.; Romanycheva, A.; Sharonova, T.; Baykov, S.; Shetnev, A.; Kofanov, E.; Kuznetsov, M. An efficient synthesis and antimicrobial evaluation of 5-alkenyl- and 5-styryl-1,2,4-oxadiazoles. Arkivoc 2018, 2018, 458–470. [Google Scholar] [CrossRef]
- Atmaram Upare, A.; Gadekar, P.K.; Sivaramakrishnan, H.; Naik, N.; Khedkar, V.M.; Sarkar, D.; Choudhari, A.; Mohana Roopan, S. Design, synthesis and biological evaluation of (E)-5-styryl-1,2,4-oxadiazoles as anti-tubercular agents. Bioorg. Chem. 2019, 86, 507–512. [Google Scholar] [CrossRef]
- Shruthi, T.; Eswaran, S.; Shivarudraiah, P.; Narayanan, S.; Subramanian, S. Synthesis, antituberculosis studies and biological evaluation of new quinoline derivatives carrying 1,2,4-oxadiazole moiety. Bioorg. Med. Chem. Lett. 2019, 29, 97–102. [Google Scholar] [CrossRef]
- dos Santos Filho, J.M.; de Queiroz e Silva, D.M.A.; Macedo, T.S.; Teixeira, H.M.P.; Moreira, D.R.M.; Challal, S.; Wolfender, J.L.; Queiroz, E.F.; Soares, M.B.P. Conjugation of N-acylhydrazone and 1,2,4-oxadiazole leads to the identification of active antimalarial agents. Bioorg. Med. Chem. 2016, 24, 5693–5701. [Google Scholar] [CrossRef]
- Kim, J.; Shin, J.S.; Ahn, S.; Han, S.B.; Jung, Y.S. 3-Aryl-1,2,4-oxadiazole Derivatives Active Against Human Rhinovirus. ACS Med. Chem. Lett. 2018, 9, 667–672. [Google Scholar] [CrossRef]
- Rozenski, J.; De Ranter, C.J.; Verplanken, H. Quantitative Structure-Activity Relationships for Antimicrobial Nitroheterocyclic Drugs. Quant. Struct.-Act. Relatsh. 1995, 14, 134–141. [Google Scholar] [CrossRef]
- Haynes, K.M.; Abdali, N.; Jhawar, V.; Zgurskaya, H.I.; Parks, J.M.; Green, A.T.; Baudry, J.; Rybenkov, V.V.; Smith, J.C.; Walker, J.K. Identification and Structure–Activity Relationships of Novel Compounds that Potentiate the Activities of Antibiotics in Escherichia coli. J. Med. Chem. 2017, 60, 6205–6219. [Google Scholar] [CrossRef] [PubMed]
- Cardona, P.J. Understanding Tuberculosis—New Approaches to Fighting against Drug Resistance; InTech: London, UK, 2012. [Google Scholar] [CrossRef]
- dos Santos Filho, J.M.; Leite, A.C.L.; de Oliveira, B.G.; Moreira, D.R.M.; Lima, M.S.; Soares, M.B.P.; Leite, L.F.C. Design, synthesis and cruzain docking of 3-(4-substituted-aryl)-1,2,4-oxadiazole-N-acylhydrazones as anti-Trypanosoma cruzi agents. Bioorg. Med. Chem. 2009, 17, 6682–6691. [Google Scholar] [CrossRef] [PubMed]
- dos Santos Filho, J.M.; Moreira, D.R.M.; de Simone, C.A.; Ferreira, R.S.; McKerrow, J.H.; Meira, C.S.; Guimarães, E.T.; Soares, M.B.P. Optimization of anti-Trypanosoma cruzi oxadiazoles leads to identification of compounds with efficacy in infected mice. Bioorg. Med. Chem. 2012, 20, 6423–6433. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Jung, Y.K.; Kim, C.; Shin, J.S.; Scheers, E.; Lee, J.Y.; Han, S.B.; Lee, C.K.; Neyts, J.; Ha, J.D.; et al. A Novel Series of Highly Potent Small Molecule Inhibitors of Rhinovirus Replication. J. Med. Chem. 2017, 60, 5472–5492. [Google Scholar] [CrossRef] [PubMed]
- Navarro-González, J.F.; Mora-Fernández, C.; de Fuentes, M.M.; García-Pérez, J. Inflammatory molecules and pathways in the pathogenesis of diabetic nephropathy. Nat. Rev. Nephrol. 2011, 7, 327–340. [Google Scholar] [CrossRef]
- Mantovani, A.; Allavena, P.; Sica, A.; Balkwill, F. Cancer-related inflammation. Nature 2008, 454, 436–444. [Google Scholar] [CrossRef]
- Vitale, P.; Panella, A.; Scilimati, A.; Perrone, M.G. COX-1 Inhibitors: Beyond Structure Toward Therapy. Med. Res. Rev. 2016, 36, 641–671. [Google Scholar] [CrossRef]
- Yatam, S.; Gundla, R.; Jadav, S.S.; reddy Pedavenkatagari, N.; Chimakurthy, J.; Rani B, N.; Kedam, T. Focused library design and synthesis of 2-mercapto benzothiazole linked 1,2,4-oxadiazoles as COX-2/5-LOX inhibitors. J. Mol. Struct. 2018, 1159, 193–204. [Google Scholar] [CrossRef]
- Yatam, S.; Jadav, S.S.; Gundla, R.; Gundla, K.P.; Reddy, G.M.; Ahsan, M.J.; Chimakurthy, J. Design, Synthesis and Biological Evaluation of 2 (((5-aryl-1,2,4-oxadiazol-3-yl)methyl)thio)benzo[d]oxazoles: New Antiinflammatory and Antioxidant Agents. ChemistrySelect 2018, 3, 10305–10310. [Google Scholar] [CrossRef]
- Vijaya Bhargavi, M.; Shashikala, P.; Sumakanth, M.; Krishna, C. Synthesis, Molecular Docking, Analgesic, and Anti-Inflammatory Activities of New 1,2,4-Oxadiazolo-Sulfonamides. Russ. J. Gen. Chem. 2018, 88, 804–811. [Google Scholar] [CrossRef]
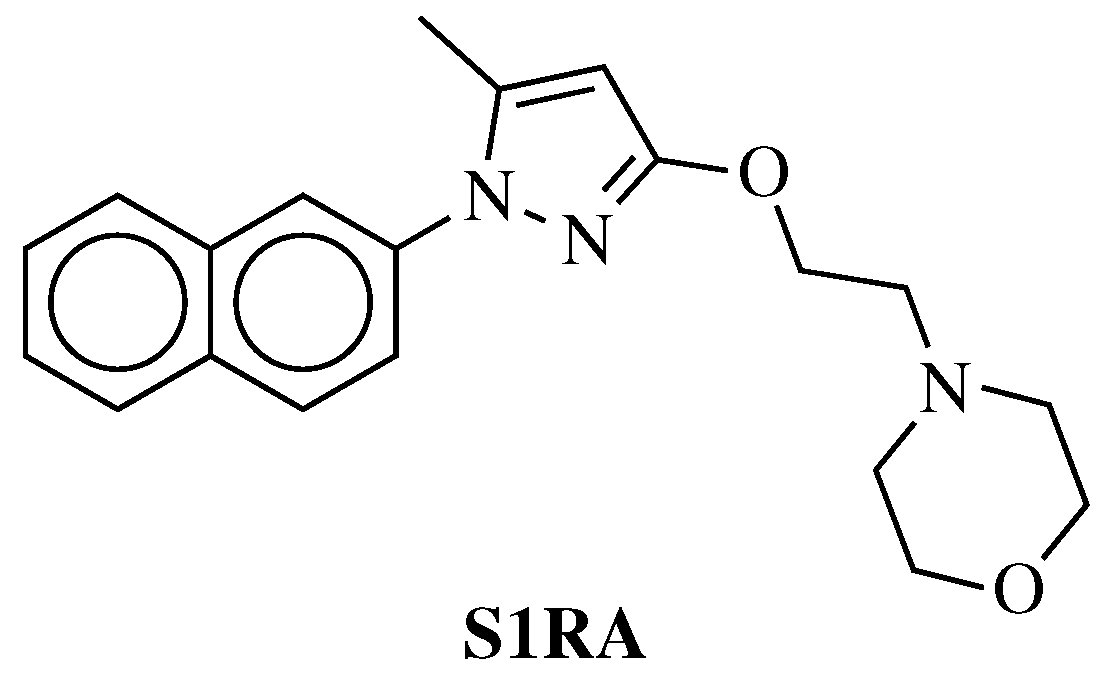
- Cao, X.; Yao, Z.; Dou, F.; Zhang, Y.; Qiu, Y.; Zhao, S.; Xu, X.; Liu, X.; Liu, B.; Chen, Y.; et al. Synthesis and Biological Evaluation of Sigma-1 (σ1) Receptor Ligands Based on Phenyl-1,2,4-oxadiazole Derivatives. Chem. Biodivers. 2019, 16, e1800599. [Google Scholar] [CrossRef] [PubMed]
- Mohammadi-Khanaposhtani, M.; Shabani, M.; Faizi, M.; Aghaei, I.; Jahani, R.; Sharafi, Z.; Shamsaei Zafarghandi, N.; Mahdavi, M.; Akbarzadeh, T.; Emami, S.; et al. Design, synthesis, pharmacological evaluation, and docking study of new acridone-based 1,2,4-oxadiazoles as potential anticonvulsant agents. Eur. J. Med. Chem. 2016, 112, 91–98. [Google Scholar] [CrossRef] [PubMed]
- Mohammadi-Khanaposhtani, M.; Saeedi, M.; Zafarghandi, N.S.; Mahdavi, M.; Sabourian, R.; Razkenari, E.K.; Alinezhad, H.; Khanavi, M.; Foroumadi, A.; Shafiee, A.; et al. Potent acetylcholinesterase inhibitors: Design, synthesis, biological evaluation, and docking study of acridone linked to 1,2,3-triazole derivatives. Eur. J. Med. Chem. 2015, 92, 799–806. [Google Scholar] [CrossRef]
- Zhang, J.; Li, J.C.; Song, J.L.; Cheng, Z.Q.; Sun, J.Z.; Jiang, C.S. Synthesis and evaluation of coumarin/1,2,4-oxadiazole hybrids as selective BChE inhibitors with neuroprotective activity. J. Asian Nat. Prod. Res. 2019, 21, 1090–1103. [Google Scholar] [CrossRef]
- Brotschi, C.; Roch, C.; Gatfield, J.; Treiber, A.; Williams, J.T.; Sifferlen, T.; Heidmann, B.; Jenck, F.; Bolli, M.H.; Boss, C. Oxadiazole Derivatives as Dual Orexin Receptor Antagonists: Synthesis, Structure–Activity Relationships, and Sleep-Promoting Properties in Rats. ChemMedChem 2019, 14, 1257–1270. [Google Scholar] [CrossRef]
- Van Zee, A. The Promotion and Marketing of OxyContin: Commercial Triumph, Public Health Tragedy. Am. J. Public Health 2009, 99, 221–227. [Google Scholar] [CrossRef]
- Ji, R.R.; Xu, Z.Z.; Gao, Y.J. Emerging targets in neuroinflammation-driven chronic pain. Nat. Rev. Drug Discov. 2014, 13, 533–548. [Google Scholar] [CrossRef]
- Huang, Y.S.; Lu, H.L.; Zhang, L.J.; Wu, Z. Sigma-2 Receptor Ligands and Their Perspectives in Cancer Diagnosis and Therapy. Med. Res. Rev. 2014, 34, 532–566. [Google Scholar] [CrossRef]
- Hayashi, T.; Su, T.P. σ-1 Receptor Ligands Potential in the Treatment of Neuropsychiatric Disorders. CNS Drugs 2004, 18, 269–284. [Google Scholar] [CrossRef]
- Lan, Y.; Chen, Y.; Cao, X.; Zhang, J.; Wang, J.; Xu, X.; Qiu, Y.; Zhang, T.; Liu, X. Synthesis and Biological Evaluation of Novel Sigma-1 Receptor Antagonists Based on Pyrimidine Scaffold As Agents for Treating Neuropathic Pain. J. Med. Chem. 2014, 57, 10404–10423. [Google Scholar] [CrossRef] [PubMed]
- Cao, X.; Chen, Y.; Zhang, Y.; Lan, Y.; Zhang, J.; Xu, X.; Qiu, Y.; Zhao, S.; Liu, X.; Liu, B.F.; et al. Synthesis and Biological Evaluation of Novel σ1 Receptor Ligands for Treating Neuropathic Pain: 6-Hydroxypyridazinones. J. Med. Chem. 2016, 59, 2942–2961. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, E.M.; Coulter, D.A. Mechanisms of epileptogenesis: A convergence on neural circuit dysfunction. Nat. Rev. Neurosci. 2013, 14, 337–349. [Google Scholar] [CrossRef] [PubMed]
- Löscher, W. New visions in the pharmacology of anticonvulsion. Eur. J. Pharmacol. 1998, 342, 1–13. [Google Scholar] [CrossRef]
- Cramer, J.A.; Mintzer, S.; Wheless, J.; Mattson, R.H. Adverse effects of antiepileptic drugs: A brief overview of important issues. Expert Rev. Neurother. 2010, 10, 885–891. [Google Scholar] [CrossRef] [PubMed]
- Querfurth, H.W.; LaFerla, F.M. Alzheimer’s Disease. N. Engl. J. Med. 2010, 362, 329–344. [Google Scholar] [CrossRef]
- Sterling, J.; Herzig, Y.; Goren, T.; Finkelstein, N.; Lerner, D.; Goldenberg, W.; Miskolczi, I.; Molnar, S.; Rantal, F.; Tamas, T.; et al. Novel Dual Inhibitors of AChE and MAO Derived from Hydroxy Aminoindan and Phenethylamine as Potential Treatment for Alzheimer’s Disease. J. Med. Chem. 2002, 45, 5260–5279. [Google Scholar] [CrossRef]
- Jiang, C.S.; Fu, Y.; Zhang, L.; Gong, J.X.; Wang, Z.Z.; Xiao, W.; Zhang, H.Y.; Guo, Y.W. Synthesis and biological evaluation of novel marine-derived indole-based 1,2,4-oxadiazoles derivatives as multifunctional neuroprotective agents. Bioorg. Med. Chem. Lett. 2015, 25, 216–220. [Google Scholar] [CrossRef]
- Mei, W.; Ji, S.; Xiao, W.; Wang, X.D.; Jiang, C.S.; Ma, W.Q.; Zhang, H.Y.; Gong, J.; Guo, Y. Synthesis and biological evaluation of benzothiazol-based 1,3,4-oxadiazole derivatives as amyloid β-targeted compounds against Alzheimer’s disease. Monatshefte Für Chem. Chem. Mon. 2017, 148, 1807–1815. [Google Scholar] [CrossRef]
- Ge, L.; Guyatt, G.; Tian, J.; Pan, B.; Chang, Y.; Chen, Y.; Li, H.; Zhang, J.; Li, Y.; Ling, J.; et al. Insomnia and risk of mortality from all-cause, cardiovascular disease, and cancer: Systematic review and meta-analysis of prospective cohort studies. Sleep Med. Rev. 2019, 48, 101215. [Google Scholar] [CrossRef]
- de Lecea, L.; Kilduff, T.S.; Peyron, C.; Gao, X.B.; Foye, P.E.; Danielson, P.E.; Fukuhara, C.; Battenberg, E.L.F.; Gautvik, V.T.; Bartlett, F.S.; et al. The hypocretins: Hypothalamus-specific peptides with neuroexcitatory activity. Proc. Natl. Acad. Sci. USA 1998, 95, 322–327. [Google Scholar] [CrossRef]
- Sakurai, T.; Amemiya, A.; Ishii, M.; Matsuzaki, I.; Chemelli, R.M.; Tanaka, H.; Williams, S.; Richardson, J.A.; Kozlowski, G.P.; Wilson, S.; et al. Orexins and Orexin Receptors: A Family of Hypothalamic Neuropeptides and G Protein-Coupled Receptors that Regulate Feeding Behavior. Cell 1998, 92, 573–585. [Google Scholar] [CrossRef]
- Brisbare-Roch, C.; Dingemanse, J.; Koberstein, R.; Hoever, P.; Aissaoui, H.; Flores, S.; Mueller, C.; Nayler, O.; van Gerven, J.; de Haas, S.L.; et al. Promotion of sleep by targeting the orexin system in rats, dogs and humans. Nat. Med. 2007, 13, 150–155. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, Y.; Naoe, Y.; Terauchi, T.; Ozaki, F.; Doko, T.; Takemura, A.; Tanaka, T.; Sorimachi, K.; Beuckmann, C.T.; Suzuki, M.; et al. Discovery of (1R,2S)-2-{[(2,4-Dimethylpyrimidin-5-yl)oxy]methyl}-2-(3-fluorophenyl)-N-(5-fluoropyridin-2-yl)cycloprop anecarboxamide (E2006): A Potent and Efficacious Oral Orexin Receptor Antagonist. J. Med. Chem. 2015, 58, 4648–4664. [Google Scholar] [CrossRef]
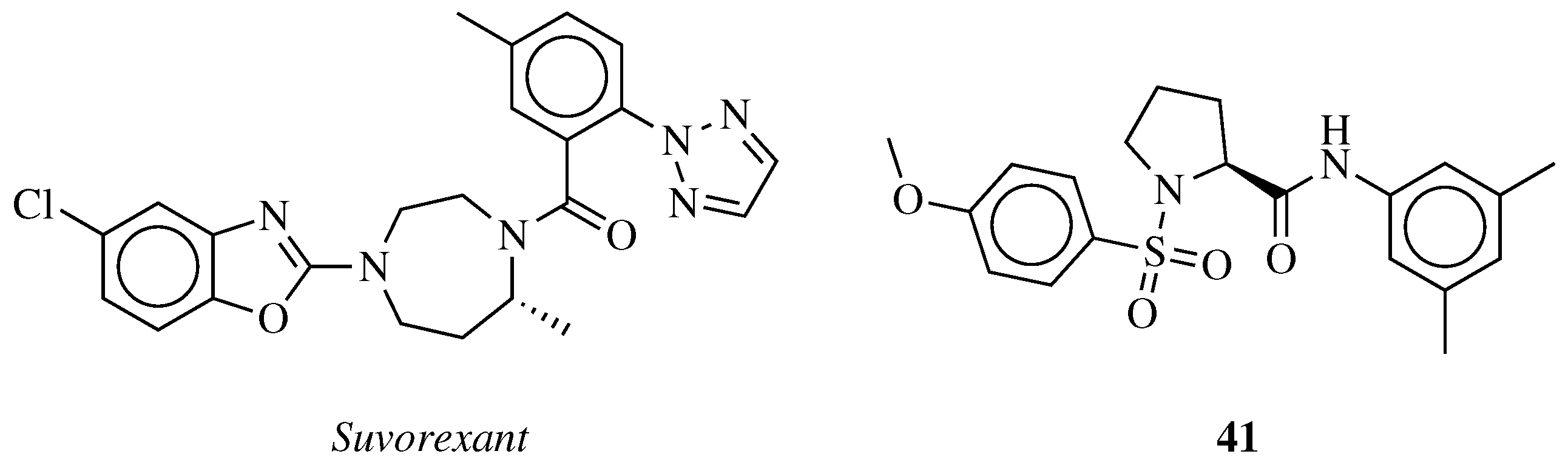
- Dubey, A.; Handu, S.; Mediratta, P. Suvorexant: The first orexin receptor antagonist to treat insomnia. J. Pharmacol. Pharmacother. 2015, 6, 118. [Google Scholar] [CrossRef] [PubMed]
- Jacobson, L.H.; Callander, G.E.; Hoyer, D. Suvorexant for the treatment of insomnia. Expert Rev. Clin. Pharmacol. 2014, 7, 711–730. [Google Scholar] [CrossRef] [PubMed]
- Boss, C.; Roch-Brisbare, C.; Steiner, M.A.; Treiber, A.; Dietrich, H.; Jenck, F.; von Raumer, M.; Sifferlen, T.; Brotschi, C.; Heidmann, B.; et al. Structure-Activity Relationship, Biological, and Pharmacological Characterization of the Proline Sulfonamide ACT-462206: A Potent, Brain-Penetrant Dual Orexin 1/Orexin 2 Receptor Antagonist. ChemMedChem 2014, 9, 2486–2496. [Google Scholar] [CrossRef]
- Heidmann, B.; Gatfield, J.; Roch, C.; Treiber, A.; Tortoioli, S.; Brotschi, C.; Williams, J.T.; Bolli, M.H.; Abele, S.; Sifferlen, T.; et al. Discovery of Highly Potent Dual Orexin Receptor Antagonists via a Scaffold-Hopping Approach. ChemMedChem 2016, 11, 2132–2146. [Google Scholar] [CrossRef]
- Sifferlen, T.; Boller, A.; Chardonneau, A.; Cottreel, E.; Gatfield, J.; Treiber, A.; Roch, C.; Jenck, F.; Aissaoui, H.; Williams, J.T.; et al. Substituted pyrrolidin-2-ones: Centrally acting orexin receptor antagonists promoting sleep. Part 2. Bioorg. Med. Chem. Lett. 2015, 25, 1884–1891. [Google Scholar] [CrossRef]
- Van’t Veer, A.; Carlezon, W.A. Role of kappa-opioid receptors in stress and anxiety-related behavior. Psychopharmacology 2013, 229, 435–452. [Google Scholar] [CrossRef]
- Clark, S.D.; Abi-Dargham, A. The Role of Dynorphin and the Kappa Opioid Receptor in the Symptomatology of Schizophrenia: A Review of the Evidence. Biol. Psychiatry 2019, 86, 502–511. [Google Scholar] [CrossRef] [PubMed]
- Tejeda, H.A.; Bonci, A. Dynorphin/Kappa-opioid receptor control of dopamine dynamics: Implications for negative affective states and psychiatric disorders. Brain Res. 2019, 1713, 91–101. [Google Scholar] [CrossRef] [PubMed]
- Guerrero, M.; Urbano, M.; Kim, E.K.; Gamo, A.M.; Riley, S.; Abgaryan, L.; Leaf, N.; Van Orden, L.J.; Brown, S.J.; Xie, J.Y.; et al. Design and Synthesis of a Novel and Selective Kappa Opioid Receptor (KOR) Antagonist (BTRX-335140). J. Med. Chem. 2019, 62, 1761–1780. [Google Scholar] [CrossRef]
- Urbano, M.; Guerrero, M.; Rosen, H.; Roberts, E. Antagonists of the kappa opioid receptor. Bioorg. Med. Chem. Lett. 2014, 24, 2021–2032. [Google Scholar] [CrossRef] [PubMed]
- Kensler, T.W.; Wakabayashi, N.; Biswal, S. Cell Survival Responses to Environmental Stresses Via the Keap1-Nrf2-ARE Pathway. Annu. Rev. Pharmacol. Toxicol. 2007, 47, 89–116. [Google Scholar] [CrossRef] [PubMed]
- Dai, X.; Yan, X.; Wintergerst, K.A.; Cai, L.; Keller, B.B.; Tan, Y. Nrf2: Redox and Metabolic Regulator of Stem Cell State and Function. Trends Mol. Med. 2020, 26, 185–200. [Google Scholar] [CrossRef]
- Jaramillo, M.C.; Zhang, D.D. The emerging role of the Nrf2-Keap1 signaling pathway in cancer. Genes Dev. 2013, 27, 2179–2191. [Google Scholar] [CrossRef]
- Lu, M.C.; Ji, J.A.; Jiang, Z.Y.; You, Q.D. The Keap1-Nrf2-ARE Pathway As a Potential Preventive and Therapeutic Target: An Update. Med. Res. Rev. 2016, 36, 924–963. [Google Scholar] [CrossRef]
- Xu, L.L.; Zhu, J.F.; Xu, X.L.; Zhu, J.; Li, L.; Xi, M.Y.; Jiang, Z.Y.; Zhang, M.Y.; Liu, F.; Lu, M.C.; et al. Discovery and Modification of in Vivo Active Nrf2 Activators with 1,2,4-Oxadiazole Core: Hits Identification and Structure–Activity Relationship Study. J. Med. Chem. 2015, 58, 5419–5436. [Google Scholar] [CrossRef]
- Jiang, Z.Y.; Lu, M.C.; Xu, L.L.; Yang, T.T.; Xi, M.Y.; Xu, X.L.; Guo, X.K.; Zhang, X.J.; You, Q.D.; Sun, H.P. Discovery of Potent Keap1–Nrf2 Protein–Protein Interaction Inhibitor Based on Molecular Binding Determinants Analysis. J. Med. Chem. 2014, 57, 2736–2745. [Google Scholar] [CrossRef]
- Xi, M.Y.; Sun, Z.Y.; Sun, H.P.; Jia, J.M.; Jiang, Z.Y.; Tao, L.; Ye, M.; Yang, X.; Wang, Y.J. Synthesis and bioevaluation of a series of α-pyrone derivatives as potent activators of Nrf2/ARE pathway (part I). Eur. J. Med. Chem. 2013, 66, 364–371. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.L.; Wu, Y.F.; Wang, L.; Li, C.C.; Li, L.; Di, B.; You, Q.D.; Jiang, Z.Y. Structure-activity and structure-property relationships of novel Nrf2 activators with a 1,2,4-oxadiazole core and their therapeutic effects on acetaminophen (APAP)-induced acute liver injury. Eur. J. Med. Chem. 2018, 157, 1376–1394. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.L.; Wu, Y.F.; Yan, F.; Li, C.C.; Dai, Z.; You, Q.D.; Jiang, Z.Y.; Di, B. 5-(3,4-Difluorophenyl)-3-(6-methylpyridin-3-yl)-1,2,4-oxadiazole (DDO-7263), a novel Nrf2 activator targeting brain tissue, protects against MPTP-induced subacute Parkinson’s disease in mice by inhibiting the NLRP3 inflammasome and protects PC12 cells aga. Free Radic. Biol. Med. 2019, 134, 288–303. [Google Scholar] [CrossRef] [PubMed]
- Coleman, K.G.; Crews, C.M. Proteolysis-Targeting Chimeras: Harnessing the Ubiquitin-Proteasome System to Induce Degradation of Specific Target Proteins. Annu. Rev. Cancer Biol. Online 2018, 2, 41–58. [Google Scholar] [CrossRef]
- Tong, M.L.; Chen, X.M. Modern Inorganic Synthetic Chemistry, 2nd ed.; Elsevier B.V.: Amsterdam, The Netherlands, 2017; p. 808. [Google Scholar]
- Kozak, W.; Demkowicz, S.; Daśko, M.; Rachon, J.; Rak, J. Modifications at the C(5) position of pyrimidine nucleosides. Russ. Chem. Rev. 2020, 89, 281–310. [Google Scholar] [CrossRef]




















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
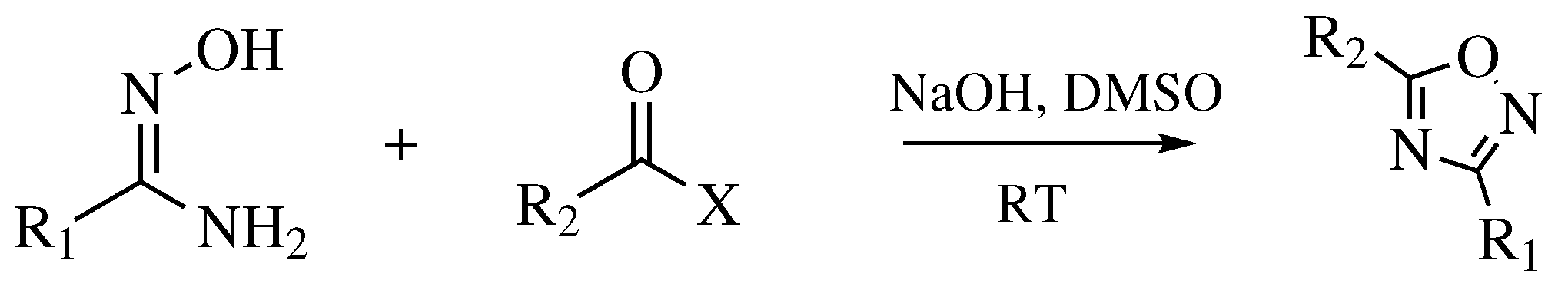{kind=link}
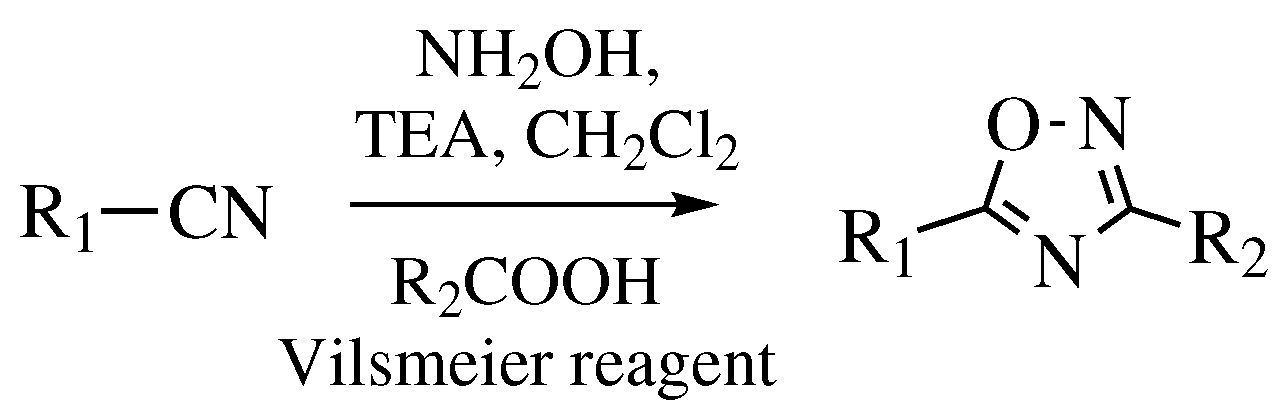{kind=link}
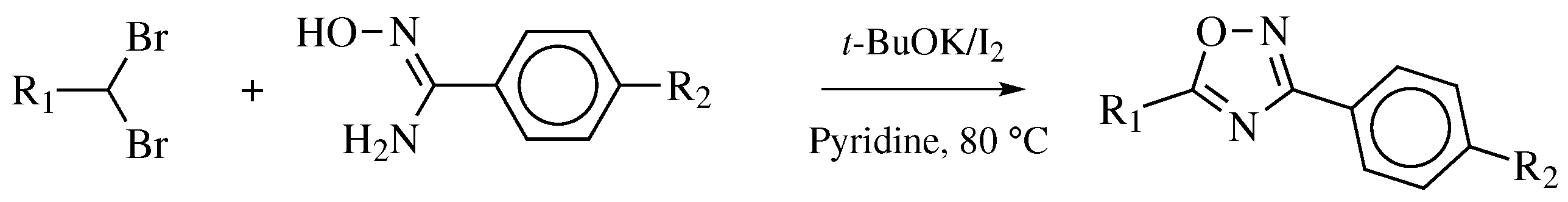{kind=link}
{kind=link}
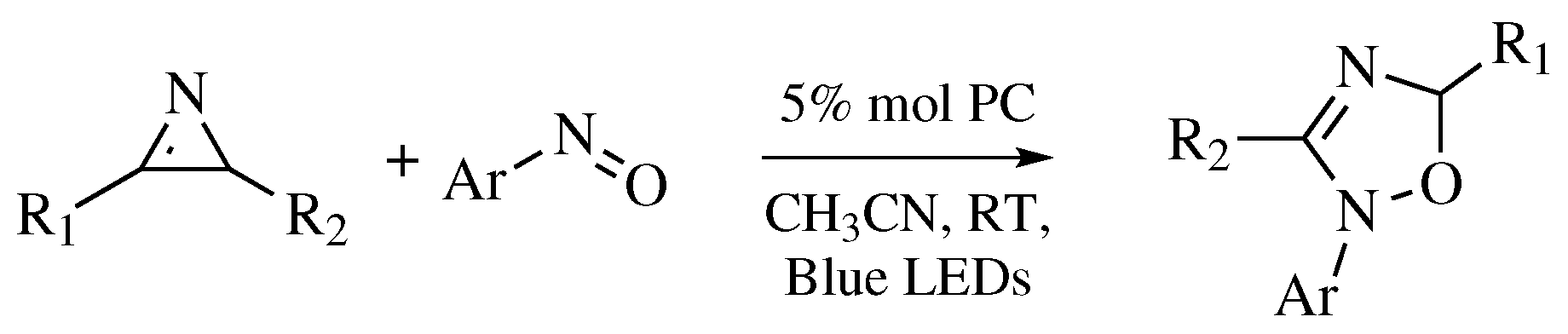{kind=link}
| Starting Material | Reagents | Substituents | Conditions | Products | Advantages and Limitations | Ref. | |
|---|---|---|---|---|---|---|---|
| Entry 1 |  |  | R1, R2 = methyl or phenyl. | Solvent-free; Melting. |  | Low yields; Long reaction time; Difficult purification; Presence of by-products. | [11] |
| Entry 2 |  |  | R1 = phenyl, 2-methylphenyl, 3-methylphenyl, 4-methylphenyl, 4-nitrophenyl or methyl; R2 = methyl, phenyl, 2-nitrophenyl, 3-nitrophenyl, 4-nitrophenyl, benzyl, methoxy, chloromethyl, t-butyl or trifluoromethyl. | THF; RT; TBAF as catalyst. |  | Short to long reaction time (1–72 h); RT; Poor to excellent yields (<5% to 98%). | [30] |
| Entry 3 |  |  | X = methoxy, ethoxy or Cl; R1 = methyl, chloromethyl, phenyl or 4-methylphenyl; R2 = benzyl, 4-methylbenzyl, 4-methoxybenzyl or t-butylpropionate. | Toluene; Reflux; K2CO3. |  | Good yields (50–95% for esters and 70–79% for acyl chlorides); Moderate to short reaction time (∼12 h for esters and ∼2 h for acyl chlorides); Easy work-up. | [31,32] |
| Entry 4 |  |  | R1 = H, t-butyl, 4-t-butylphenyl, 2,4-dichlorophenyl, 4-bromophenyl, 4-methyl-1,2,3-thiadiazol-5-yl or 3-methyl-4-nitrophenyl; R2 = 4-bromophenyl, N-Boc-azetidine-3-yl,1-naphthyl, N-Boc-aminoethyl or 5-methyl-3-thiophen-2-yl. | TEA; T3P; ∼80 °C. |  | Excellent yields (87–97%); Short to moderate reaction time (0.5–6 h); Easy work-up; Expensive activating agent (T3P). | [34] |
| Entry 5 |  |  | R1 = phenyl, 4-chlorophenyl, 4-bromophenyl, 2,4-dichlorophenyl, 4-methoxybenzyl or cyclohexyl; R2 = phenyl, 4-methylphenyl or n-pentyl. | H2O; Reflux; 12 h. |  | Low to excellent yields (35–93%); Moderate reaction time (12 h); Catalyst-free; Organic solvent-free; Aqueous medium. | [35] |
| Entry 6 |  |  | X = hydroxy, methoxy, ethoxy or Cl; R1 = phenyl, o-, m-, p-tolyl, 4-chlorophenyl, 3-bromophenyl, 4-bromophenyl, 4-nitrophenyl or 4-methoxyphenyl; R2 = 3-oxo-1-butyl, phenyl, 2,4-dichlorophenyl, 4-chlorobenzyl or cyclohexyl. | Solvent-free; NH4F/Al2O4 as a catalyst; MWI; ∼10 min. |  | Moderate to excellent yields (40–90%); Remarkably short reaction time (∼10 min); Organic solvent-free; Simply work-up; Absence of by-products. | [36,37] |
| Entry 7 | R1-CN | Step I: NH2OH·HCl MWI; Step II: R2COCl or R2CHO; MWI. | R1 = phenyl, 4-chlorophenyl, 4-bromophenyl, 4-methylphenyl, and many others (see Ref.); R2 = phenyl, 4-methoxyphenyl, 3-nitrophenyl, and many others (see Ref). | Step I: Solvent-free; CH3COOH, MgO or Na2CO3 as catalyst; MWI; Step II: Solvent-free; MWI. |  | Usually excellent yields (>90%); One-pot procedure; Cheap catalysts; Remarkably short reaction time (∼2–10 min); Organic-solvent-free; None by-products; Easy work-up. | [37,38,39,40] |
| Entry 8 |  | R1-CN | R1 = 2,4,6-trimethyl or 2,4,6-trimethoxy; R2 = methyl or ethyl. | Step I: [PtCl4-R’2(CN)]2 (R’ = CH3, CH3CH2, PhCH2), CH3CN, CH2Cl2; Step II: Pyridine, CH2Cl2. |  | Low yields; Long reaction time (up to 72 h); Poor solubility of Pt compounds; Difficult purification. | [43] |
| General Structure | Substituents | The Most Active Derivatives | Activity | Ref. |
 | R1 = H, NH2 and other (see Ref.); R2 = H or phenyl. |  | IC50 values of 2.76 and 9.27 M against OVXF 899 and PXF 1752 cancer cell lines, respectively. | [55] |
 | X = Cl or Br; R1 = methyl, benzyl, 2-pyridinyl or anthracen-9-ylmethyl. |  | IC50 values of 3 nM against LXFA 629 and MAXF 401 cancer cell lines, respectively. | [56] |
 | X = O or NH; R1 = phenyl, benzyl, 2-chlorophenyl, 4-fluorophenyl, 2-mehylphenyl, 4-bromophenyl, 4-methylphenyl, 4-methoxyphenyl, 4-pyridinyl, 2-methoxyphenyl, 2-benzyloxyphenyl or 3-pyridinyl. |  | IC50 values between 26.1–34.3 M against Colo 205, Hep G2 and Hela cell lines. | [57,58] |
 | R1 = methyl, chloromethyl or phenyl. |  | GI50 values of 0.08 (5a) and 0.34 (5b) M against CEM-13 cell line. | [60] |
 | R1 = H, Br, Cl, F, methoxy or NH2. |  | CC50 values of 137.3, 79.0 and 140.3 M against Ca9-22 cell line, respectively. | [63] |
 | R1 = H, 2-chloro, 3-chloro, 4-chloro, 4-nitro, 4-methyl, 4-methoxy, 4-trifluoromethyl, 2-bromo, 3-bromo, 4-bromo or 4-fluoro; R2 = N(CH3)2, N(C2H5)2, pyrrolidine-1-yl, azepan-1-yl, morpholin-1-yl, thiomorpholine-1-yl, N-methylpiperazin-1-yl, N-phenylpiperazin-1-yl, 3-bromopropan-1-yl or 3-chloropropan-1-yl. |  | 80% of death of NB4, K562 and MDA-MB-231 cancer cell lines at 25 (7a) and 10 (7b) M. | [64] |
 | R1 = H or NH2; R2 = isopropylidene or cyclopentylidene; R3 = 4-nitrophenyl, 4-chlorophenyl or 3,4,5-trimethylphenyl. |  | GI50 of 4.5 M against WiDr cancer cell line. | [65] |
 | R1 = CH3 or —(CH2)4—; R2 = H, Cl, Br, methyl or methoxy. |  | IC50 values of 0.48 (9a), 0.78 (9b), 0.19 (9c) M against MCF-7 cancer cell line. | [66] |
 | R1 = H, 3-methyl, 4-methyl, 3-bromo, 4-methoxy, 4-trifluoromethyl, 4-chloro, 4-bromo or 4-fluoro. |  | IC50 values between 13.6–48.37 M against HCT-116, PC-3, SNB-19, B16F10, L929 cell lines. | [67] |
 | R1 = H, 3,4,5-trimethoxy, 4-methoxy, 4-chloro, 4-bromo, 4-fluoro, 4-trifluoromethyl, 4-nitro, 4-cyano or 4-methyl. |  | IC50 values in a range from 0.11 to 2.09 M against MCF-7, A375 and HT-29 cancer cell lines. | [68] |
 | R1 = H, 3,4,5-trimethoxy, 4-methoxy, 4-chloro, 4-bromo, 4-fluoro, 4-trifluoromethyl, 4-nitro, 4-cyano or 4-methyl. |  | IC50 values between 0.011–1.89 M against A549, MCF-7, A375 and HT-29 cancer cell lines. | [69] |
 | R1 = H, 3,4,5-trimethoxy, 4-methoxy, 4-chloro, 4-bromo, 4-fluoro, 4-trifluoromethyl, 4-nitro, 3-nitro or 4-methyl. |  | IC50 values in a range of 0.11–1.47 M against A375, MCF-7 and ACHN cancer cell lines. | [70] |
 | R1 = H, 3,4,5-trimethoxy, 4-methoxy, 4-chloro, 4-bromo, 4-fluoro, 4-trifluoromethyl, 4-nitro, 4-cyano or 4-methyl. |  | IC50 values between 0.12–2.78 M against MCF-7, A549 and A375 cancer cell lines. | [71] |
 | R1 = methyl, phenyl, 4-fluorophenyl, benzyl or 4-methoxbenzyl; R2 = phenyl, 9-phenanthryl or 4-pyridinyl; R3 = 4-nitrophenyl, 4-chlorophenyl, 4-trifluoromethylphenyl or 4-fluorophenyl. |  | IC50 value of 10.38 M toward MCF-7 cancer cell line. | [72] |
 | R1 = methyl, phenyl, 2-methoxyphenyl, 3-methoxyphenyl, 4-t-butylphenyl, 4-methylphenyl, 2-methoxyphenyl, 3-methoxyphenyl, cyclopropyl, 2-pyridinyl, 3-pyridinyl, 4-pyridinyl, 2-thienyl, 3-thienyl, 4-cyanophenyl, 2-fluorophenyl, 3-fluorophenyl, 4-fluorophenyl, 2-chlorophenyl or 3,4-dichlorophenyl; Ar1 = p-phenylene, m-phenylene, p-methoxyphenylene or 2,4-thienyl. |  | Ki value of 89 pm and 0.75 nm (hCA IX and hCA II, respectively) for 16a in CO2 hydration stopped-flow biochemical assay. 16b showed high selectivity toward PANC-1 cancer cell line. | [73,74] |
 | R1 = H, F, Cl, Br or methoxy; R2 = H, F or Br. |  | IC50 values of 0.65 (17a) and 2.41 M (17b) against MCF-7 cancer cell line. | [75] |
 | R1 = H, 3,4,5-trimethoxy, 4-methoxy, 4-chloro, 4-bromo, 4-fluoro, 4-nitro, 3-nitro, 4-cyano or 4-trifluoromethyl. |  | IC50 values in a range of 0.45–2.11 M against MCF-7, A549, MDA-MB-231 cancer cell lines. | [76] |
 | X, Y = N, O or O, N; n = 5 or 6; R1 = H, 2-methyl, 4-methyl, 4-methoxy, 2-fluoro, 3-fluoro, 4-fluoro, 4-bromo or 4-nitro. |  | IC50 values of 8.2, 10.5, 12.1 nM (20a, 20b, 20c, respectively) toward HDAC-1. | [77,78] |
 | R1 = H, 4-methyl, 3-methyl, 2-fluoro, 4-fluoro, 2,4-difluoro, 2-chloro, 4-cyano, 4-trifluoromethyl or 2-chloro-4-fluoro. |  | IC50 values of 1.8, 3.6 and 3.0 nM against HDAC-1, -2 and -3, respectively. | [79] |
 | R1 = 3-pyridinyl, 4-pyridinyl, 4-methoxy-3-pyridinyl, 5-(2-methoxyethoxy)-3-pyridinyl, 5-morpholin-3-pyridinyl or 5-(1-methyl-1H-pyrazol-3-yl)-3-pyridinyl. |  | IC50 value of 7.3 nM against RET enzyme in ELISA assay. | [80] |
| General Structure | Substituents | The Most Active Derivatives | Activity | Ref. |
|---|---|---|---|---|
 | R1 = H, OH, OCH3, NH2, NHAc, NH3Cl, NHMs, NH-nBu, NH-tBu, NHCOPh, NH-iPr, PO3H2, PO(OEt)2, SO2NH2, CONH2, COOH, COOCH3 F, Cl, Br, I, NO2, ethynyl or CN; Ar1 = phenyl, benzyl, 2-pyrole, 3-pyridyl, 4-pyridyl, 5-indole, 3-pyrrazole, 2-imidazole and many others (see Ref.); Ar2, Ar3 = p-phenylene, 6-indole, 2-pyridyl, 6-chromene, carbazole, N-phenylpiperazine, N-phenylmorpholine and many others (see Ref.); X = NH, CH2, O, CO, NBn, SO or SO2. |  | MIC50 values <4 g/mL against over 210 diverse, MRSA and VRE strains. | [96,98,99,103] |
 | X = NH or none; R1 = H, 3-chloro-4-fluorophenyl, 2-chlorophenyl, 2-ethyl, 4-ethyl, 5-bromo-2-fluorophenyl or 2-methylpyridin-5-yl. |  | Grown inhibition zone within 20–25 mm against S. aureus, B. subtilis, E. coli, P. vulgaris, P. aeruginosa, C. albicans. | [104] |
 | R1 = H, 2-chloro or 3-chloro; X = CH or N; R2 = H, 2-nitro, 2-chloro, 3-bromo, 2-chloro-5-nitro, 2-bromo, 3-nitro, 2-iodo, 3,5-dinitro, 4-nitro or 2-hydroxy. |  | MIC value of 60 M against E. coli. | [105] |
 | R1 = H, F, Cl, Br, I, methyl, ethyl, methoxy or iPr; R2 = H, methyl, methoxy, iPr, F, Cl, Br or I; R3 = H, F, Cl, Br, nitro, iPr, OBn, methoxy, ethoxy or CN. |  | MIC value of 64 g/mL against S. epidermidis. | [106] |
 | R1 = H or methyl; Ar1 = p-phenylene or m-phenylene; R2 = methyl, cyclopropyl, 2-thienyl, 2-chlorophenyl, 3-chlorophenyl, 3,4-dichlorophenyl, 4-ethylphenyl, 4-t-butylphenyl, 4-methylphenyl, 3,4,-dimethylphenyl and many others (see Ref.). |  | MIC values in a range 8–16 g/mL toward S. aureus, B. subtilis, E. coli, P. fluorescent. | [107] |
 | R1 = phenyl, 4-mehtoxyphenyl, 4-chlorophenyl, 3-methylthienyl or 2-pyridinyl; R2, R3 = H, methyl, phenyl, 4-chlorophenyl, 4-methoxyphenyl, 3,4,-dimethoxyphenyl or 2,3-dimethoxyphenyl. |  | MIC value of 0.68 mM against S. aureus. | [108] |
 | R1 = phenyl, 4-methylphenyl, 4-methoxyphenyl, 4-methylthiophenyl, 2-chlorophenyl, 4-chlorophenyl, 2-3-dichlorophenyl, 3,4-dichlorophenyl, 4-fluorophenyl, 4-bromophenyl, 4-hydroksyphenyl, 2-bromo-4-fluorophenyl, 4-cyanophenyl, 4-pyridinyl, 1-napthyl and others (see Ref.). |  | IC50 value of 0.045 g/mL against M. tuberculosis (H37Ra). | [109] |
 | R1 = 4-pyridyl, 3-pyridinyl or 3,5-difluorophenyl; R2 = 3,5-dimethoxyphenyl, 3,5-difluorophenyl, 3-cyanophenyl, 2,3-dimethylphenyl, cyclopentyl or 4-izopropylphenyl. |  | MIC value of 0.5 g/mL against M. tuberculosis (H37Ra). | [110] |
 | R1 = H, F, Cl, Br, methyl, nitro, methoxy or hydroxy; R2 = 4-hydroksy-3-methoxyphenyl, 2-styryl, ferrocene or 5-benzo[1,3]dioxole. |  | IC50 value of 0.02 M against P. falciparum. In vivo studies failed—none in vivo activity. | [111] |
 | R1 = Me, Et, cyclopropyl, iPr, CF3, iBu or CH2OCH3; Ar1 = p-phenylene, p-2-methylphenylene, p-2,6-dimethylphenylene, 2,5-pyridinyl or 3-methylbenzothiophene |  | IC50 values of 66.0, 22.0 and 3.7 nM against hRV-B14, hRV-A21 and hRV-A71, respectively. | [112] |
| General Structure | Substituents | The Most Active Derivatives | Activity | Ref. |
|---|---|---|---|---|
| ANTI-INFLAMMATORY ACTIVITY | ||||
 | R1 = NO2, CF3, F, Cl, COOH, COOCH3, CON(CH3)OCH3, CONH(t-Bu) and others (see Ref.). |  | IC50 value of 5.0 M against COX-2. | [122] |
 | R1 = 2-fluorophenyl, 4-fluorophenyl, 2,4-difluorophenyl, 2,5-difluorophenyl, 2-chlorophenyl, 3-chlorophenyl, 4-chlorophenyl, 2,4-dichlorophenyl, 4-nitrophenyl, 4-trifluoromethylphenyl, 4-methylphenyl, 3-pyridinyl or 5-thiazole. |  | IC50 value of 4.83 M against COX-2. | [123] |
 | R1 = H, 4-methyl, 4-methoxy or 4-chloro; R2 = 4-fluorophenyl, 4-methylphenyl, 4-chloro, 4-methoxyphenyl or 2-chlorophenyl. |  | 55% inhibition of acute inflammation (3 h after injection at 40 mg/kg). | [124] |
| ANTI-ALLODYNIC ACTIVITY | ||||
 | R1 = 2-napthyl, 4-methylphenyl, 4-chlorophenyl, 4-fluorophenyl, 2,3-dichlorophenyl, 2,4-dichlorophenyl, 2,5-dichlorophenyl, 3,5-dichlorophenyl, 3,4-dichlorophenyl, 3-chloro-4-fluorophenyl or 3,4-difluorophenyl; n = 2 or 3; R2 = N-morpholine, N-piperidine, 4-methyl-N-piperdine, 3,5-dimethyl-N-piperidine, N-piperidin-4-one, N-methyl-N-piperazine, N-ethyl-N-piperazine, N-pyrrolidine, N-dimethylamine or N-diethylamine. |  | Ki values of 0.28 nM and 164 nM for (σ1 and σ2, respectively). | [125] |
| ANTICONVULSANT ACTIVITY | ||||
 | R1 = H, Cl, methoxy, Br, methyl or ethyl; R2 = methyl, Cl or methoxy. |  | ED50 values of 2.08 and 3.71 mg/kg in PTZ and MES, respectively. | [126] |
 | R1 = H, methyl or phenyl; R2 = methyl, Cl or Br. |  | 100% of seizures protection (in an assay with mice at 7 mg/kg dose in a MES test). | [127] |
| ANTI-ALZHEIMER ACTIVITY | ||||
 | R1 = phenyl, 2-methylphenyl, 3-methylphenyl, 4-methylphenyl, 3-methoxyphenyl, 4-methoxyphenyl, 4-N,N-diethylaniline, 4-ethynylphenyl, 2-fluorophenyl, 3-fluorophenyl, 4-fluorophenyl, 4-chlorophenyl, 4-bromophenyl, 2-nitrphenyl, 3-nitrophenyl, 4-nitrophenyl and others (see Ref.). |  | IC50 values of 8.2 and 77.6 M against BChE and AChE, respectively. | [128] |
| ANTI-INSOMNIA ACTIVITY | ||||
 | R1 = H, Cl, methyl, F or methoxy; R2 = H, methyl, methoxy, Cl, F or OCF3; Ar1 = piperazine, diethylamine, ethylamine, pyrrolidine or azetidine and others (see Ref.). |  | Decreases the time spent in active-wake and increases time spent in non-REM and REM sleep (−24%, +14.3% and +35.2%, respectively, at 100 mg/kg). | [129] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Biernacki, K.; Daśko, M.; Ciupak, O.; Kubiński, K.; Rachon, J.; Demkowicz, S. Novel 1,2,4-Oxadiazole Derivatives in Drug Discovery. Pharmaceuticals 2020, 13, 111. https://doi.org/10.3390/ph13060111
Biernacki K, Daśko M, Ciupak O, Kubiński K, Rachon J, Demkowicz S. Novel 1,2,4-Oxadiazole Derivatives in Drug Discovery. Pharmaceuticals. 2020; 13(6):111. https://doi.org/10.3390/ph13060111
Chicago/Turabian StyleBiernacki, Karol, Mateusz Daśko, Olga Ciupak, Konrad Kubiński, Janusz Rachon, and Sebastian Demkowicz. 2020. "Novel 1,2,4-Oxadiazole Derivatives in Drug Discovery" Pharmaceuticals 13, no. 6: 111. https://doi.org/10.3390/ph13060111
APA StyleBiernacki, K., Daśko, M., Ciupak, O., Kubiński, K., Rachon, J., & Demkowicz, S. (2020). Novel 1,2,4-Oxadiazole Derivatives in Drug Discovery. Pharmaceuticals, 13(6), 111. https://doi.org/10.3390/ph13060111