Therapeutic Targeting of Pancreatic Cancer via EphA2 Dimeric Agonistic Agents



Abstract

1. Introduction
2. Results and Discussion
2.1. Synthesis and Characterization of Dimeric and Tetrameric 12-Mers Targeting the EphA2-LBD
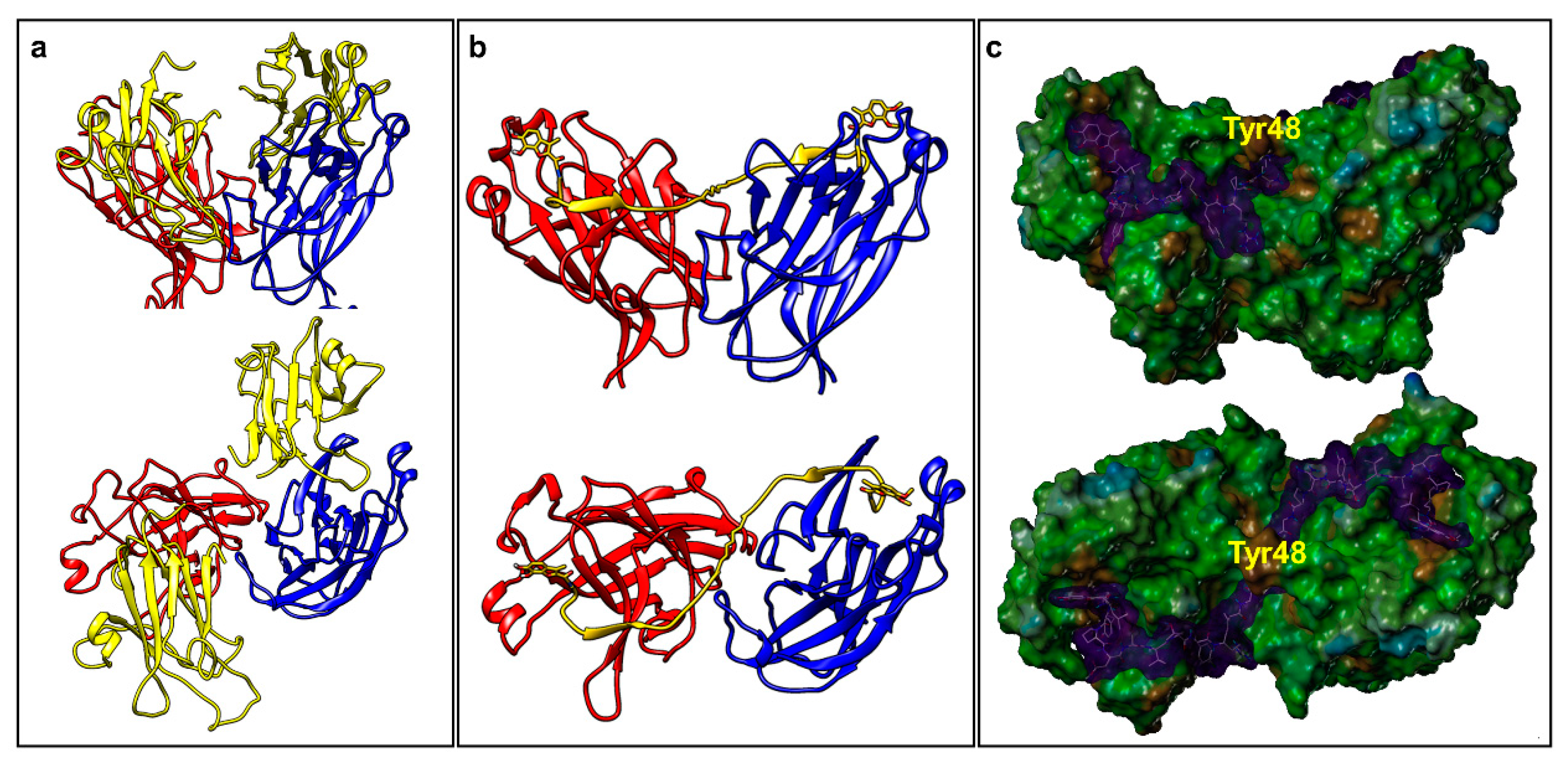
2.2. Dimeric Agents May Promote Dimerization of EphA2-LBD
2.3. Dimeric and Tetrameric Compounds Are Potent Agonists of EphA2 Signaling
2.4. Cell Migration Studies
3. Materials and Methods
3.1. Synthetic Chemistry
3.2. Molecular Modeling and In Vitro Studies
3.3. Cell Lines, Cell Culture, and Antibodies
3.4. Establishment of an EphA2 Knocked-Out Pancreatic Cancer Cell Line
3.5. Cell Migration Assays
3.6. Immunoblotting Assays
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Caparello, C.; Meijer, L.L.; Garajova, I.; Falcone, A.; Le Large, T.Y.; Funel, N.; Kazemier, G.; Peters, G.J.; Vasile, E.; Giovannetti, E. FOLFIRINOX and translational studies: Towards personalized therapy in pancreatic cancer. World J. Gastroenterol. 2016, 22, 6987–7005. [Google Scholar] [CrossRef]
- Saif, M.W. U.S. Food and Drug Administration approves paclitaxel protein-bound particles (Abraxane(R)) in combination with gemcitabine as first-line treatment of patients with metastatic pancreatic cancer. JOP 2013, 14, 686–688. [Google Scholar] [CrossRef]
- Borazanci, E.; Von Hoff, D.D. Nab-paclitaxel and gemcitabine for the treatment of patients with metastatic pancreatic cancer. Expert Rev. Gastroenterol. Hepatol. 2014, 8, 739–747. [Google Scholar] [CrossRef]
- Von Hoff, D.D.; Ervin, T.; Arena, F.P.; Chiorean, E.G.; Infante, J.; Moore, M.; Seay, T.; Tjulandin, S.A.; Ma, W.W.; Saleh, M.N.; et al. Increased survival in pancreatic cancer with nab-paclitaxel plus gemcitabine. N. Engl. J. Med. 2013, 369, 1691–1703. [Google Scholar] [CrossRef]
- Walker-Daniels, J.; Coffman, K.; Azimi, M.; Rhim, J.S.; Bostwick, D.G.; Snyder, P.; Kerns, B.J.; Waters, D.J.; Kinch, M.S. Overexpression of the EphA2 tyrosine kinase in prostate cancer. Prostate 1999, 41, 275–280. [Google Scholar] [CrossRef]
- Ogawa, K.; Pasqualini, R.; Lindberg, R.A.; Kain, R.; Freeman, A.L.; Pasquale, E.B. The ephrin-A1 ligand and its receptor, EphA2, are expressed during tumor neovascularization. Oncogene 2000, 19, 6043–6052. [Google Scholar] [CrossRef]
- Zelinski, D.P.; Zantek, N.D.; Stewart, J.C.; Irizarry, A.R.; Kinch, M.S. EphA2 overexpression causes tumorigenesis of mammary epithelial cells. Cancer Res. 2001, 61, 2301–2306. [Google Scholar] [PubMed]
- Coffman, K.T.; Hu, M.; Carles-Kinch, K.; Tice, D.; Donacki, N.; Munyon, K.; Kifle, G.; Woods, R.; Langermann, S.; Kiener, P.A.; et al. Differential EphA2 epitope display on normal versus malignant cells. Cancer Res. 2003, 63, 7907–7912. [Google Scholar] [PubMed]
- Duxbury, M.S.; Ito, H.; Zinner, M.J.; Ashley, S.W.; Whang, E.E. Ligation of EphA2 by Ephrin A1-Fc inhibits pancreatic adenocarcinoma cellular invasiveness. Biochem. Biophys. Res. Commun 2004, 320, 1096–1102. [Google Scholar] [CrossRef] [PubMed]
- Duxbury, M.S.; Ito, H.; Zinner, M.J.; Ashley, S.W.; Whang, E.E. EphA2: A determinant of malignant cellular behavior and a potential therapeutic target in pancreatic adenocarcinoma. Oncogene 2004, 23, 1448–1456. [Google Scholar] [CrossRef]
- Saito, T.; Masuda, N.; Miyazaki, T.; Kanoh, K.; Suzuki, H.; Shimura, T.; Asao, T.; Kuwano, H. Expression of EphA2 and E-cadherin in colorectal cancer: Correlation with cancer metastasis. Oncol. Rep. 2004, 11, 605–611. [Google Scholar] [CrossRef] [PubMed]
- Ireton, R.C.; Chen, J. EphA2 receptor tyrosine kinase as a promising target for cancer therapeutics. Curr. Cancer Drug Targets 2005, 5, 149–157. [Google Scholar] [CrossRef] [PubMed]
- Landen, C.N.; Kinch, M.S.; Sood, A.K. EphA2 as a target for ovarian cancer therapy. Expert Opin. Ther. Targets 2005, 9, 1179–1187. [Google Scholar] [CrossRef] [PubMed]
- Wykosky, J.; Gibo, D.M.; Stanton, C.; Debinski, W. EphA2 as a novel molecular marker and target in glioblastoma multiforme. Mol. Cancer Res. 2005, 3, 541–551. [Google Scholar] [CrossRef]
- Abraham, S.; Knapp, D.W.; Cheng, L.; Snyder, P.W.; Mittal, S.K.; Bangari, D.S.; Kinch, M.; Wu, L.; Dhariwal, J.; Mohammed, S.I. Expression of EphA2 and Ephrin A-1 in carcinoma of the urinary bladder. Clin. Cancer Res. 2006, 12, 353–360. [Google Scholar] [CrossRef]
- Lin, Y.G.; Han, L.Y.; Kamat, A.A.; Merritt, W.M.; Landen, C.N.; Deavers, M.T.; Fletcher, M.S.; Urbauer, D.L.; Kinch, M.S.; Sood, A.K. EphA2 overexpression is associated with angiogenesis in ovarian cancer. Cancer 2007, 109, 332–340. [Google Scholar] [CrossRef]
- Margaryan, N.V.; Strizzi, L.; Abbott, D.E.; Seftor, E.A.; Rao, M.S.; Hendrix, M.J.; Hess, A.R. EphA2 as a promoter of melanoma tumorigenicity. Cancer Biol. Ther. 2009, 8, 279–288. [Google Scholar] [CrossRef]
- Pasquale, E.B. Eph receptors and ephrins in cancer: Bidirectional signalling and beyond. Nat. Rev. Cancer 2010, 10, 165–180. [Google Scholar] [CrossRef]
- Trinidad, E.M.; Zapata, A.G.; Alonso-Colmenar, L.M. Eph-ephrin bidirectional signaling comes into the context of lymphocyte transendothelial migration. Cell Adhes. Migr. 2010, 4, 363–367. [Google Scholar] [CrossRef]
- Takahashi, Y.; Itoh, M.; Nara, N.; Tohda, S. Effect of EPH-ephrin signaling on the growth of human leukemia cells. Anticancer. Res. 2014, 34, 2913–2918. [Google Scholar]
- Quinn, B.A.; Lee, N.A.; Kegelman, T.P.; Bhoopathi, P.; Emdad, L.; Das, S.K.; Pellecchia, M.; Sarkar, D.; Fisher, P.B. The Quest for an Effective Treatment for an Intractable Cancer: Established and Novel Therapies for Pancreatic Adenocarcinoma. Adv. Cancer Res. 2015, 127, 283–306. [Google Scholar] [CrossRef] [PubMed]
- Koshikawa, N.; Minegishi, T.; Kiyokawa, H.; Seiki, M. Specific detection of soluble EphA2 fragments in blood as a new biomarker for pancreatic cancer. Cell Death Dis. 2017, 8, e3134. [Google Scholar] [CrossRef] [PubMed]
- Markosyan, N.; Li, J.; Sun, Y.H.; Richman, L.P.; Lin, J.H.; Yan, F.; Quinones, L.; Sela, Y.; Yamazoe, T.; Gordon, N.; et al. Tumor cell-intrinsic EPHA2 suppresses anti-tumor immunity by regulating PTGS2 (COX-2). J. Clin. Investig. 2019, 130, 3594–3609. [Google Scholar] [CrossRef] [PubMed]
- Quinn, B.A.; Wang, S.; Barile, E.; Das, S.K.; Emdad, L.; Sarkar, D.; De, S.K.; Morvaridi, S.K.; Stebbins, J.L.; Pandol, S.J.; et al. Therapy of pancreatic cancer via an EphA2 receptor-targeted delivery of gemcitabine. Oncotarget 2016, 7, 17103–17110. [Google Scholar] [CrossRef]
- Wang, S.; Placzek, W.J.; Stebbins, J.L.; Mitra, S.; Noberini, R.; Koolpe, M.; Zhang, Z.; Dahl, R.; Pasquale, E.B.; Pellecchia, M. Novel targeted system to deliver chemotherapeutic drugs to EphA2-expressing cancer cells. J. Med. Chem. 2012, 55, 2427–2436. [Google Scholar] [CrossRef]
- Salem, A.F.; Wang, S.; Billet, S.; Chen, J.F.; Udompholkul, P.; Gambini, L.; Baggio, C.; Tseng, H.R.; Posadas, E.M.; Bhowmick, N.A.; et al. Reduction of Circulating Cancer Cells and Metastases in Breast-Cancer Models by a Potent EphA2-Agonistic Peptide-Drug Conjugate. J. Med. Chem. 2018, 61, 2052–2061. [Google Scholar] [CrossRef]
- Wu, B.; Wang, S.; De, S.K.; Barile, E.; Quinn, B.A.; Zharkikh, I.; Purves, A.; Stebbins, J.L.; Oshima, R.G.; Fisher, P.B.; et al. Design and Characterization of Novel EphA2 Agonists for Targeted Delivery of Chemotherapy to Cancer Cells. Chem. Biol. 2015, 22, 876–887. [Google Scholar] [CrossRef]
- Barile, E.; Wang, S.; Das, S.K.; Noberini, R.; Dahl, R.; Stebbins, J.L.; Pasquale, E.B.; Fisher, P.B.; Pellecchia, M. Design, synthesis and bioevaluation of an EphA2 receptor-based targeted delivery system. ChemMedChem 2014, 9, 1403–1412. [Google Scholar] [CrossRef]
- Wang, S.; Noberini, R.; Stebbins, J.L.; Das, S.; Zhang, Z.; Wu, B.; Mitra, S.; Billet, S.; Fernandez, A.; Bhowmick, N.A.; et al. Targeted delivery of paclitaxel to EphA2-expressing cancer cells. Clin. Cancer Res. 2013, 19, 128–137. [Google Scholar] [CrossRef]
- Gambini, L.; Salem, A.F.; Udompholkul, P.; Tan, X.F.; Baggio, C.; Shah, N.; Aronson, A.; Song, J.; Pellecchia, M. Structure-Based Design of Novel EphA2 Agonistic Agents with Nanomolar Affinity in Vitro and in Cell. ACS Chem. Biol. 2018, 13, 2633–2644. [Google Scholar] [CrossRef]
- Himanen, J.P.; Yermekbayeva, L.; Janes, P.W.; Walker, J.R.; Xu, K.; Atapattu, L.; Rajashankar, K.R.; Mensinga, A.; Lackmann, M.; Nikolov, D.B.; et al. Architecture of Eph receptor clusters. Proc. Natl. Acad. Sci. USA 2010, 107, 10860–10865. [Google Scholar] [CrossRef] [PubMed]
- Koolpe, M.; Dail, M.; Pasquale, E.B. An ephrin mimetic peptide that selectively targets the EphA2 receptor. J. Biol. Chem. 2002, 277, 46974–46979. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Soler, M.; Petersen Gehring, M.; Lechtenberg, B.C.; Zapata-Mercado, E.; Hristova, K.; Pasquale, E.B. Engineering nanomolar peptide ligands that differentially modulate EphA2 receptor signaling. J. Biol. Chem. 2019, 294, 8791–8805. [Google Scholar] [CrossRef]
- Duggineni, S.; Mitra, S.; Lamberto, I.; Han, X.; Xu, Y.; An, J.; Pasquale, E.B.; Huang, Z. Design and Synthesis of Potent Bivalent Peptide Agonists Targeting the EphA2 Receptor. ACS Med. Chem. Lett. 2013, 4. [Google Scholar] [CrossRef]
- Singh, D.R.; Kanvinde, P.; King, C.; Pasquale, E.B.; Hristova, K. The EphA2 receptor is activated through induction of distinct, ligand-dependent oligomeric structures. Commun. Biol. 2018, 1, 15. [Google Scholar] [CrossRef] [PubMed]
- Himanen, J.P.; Chumley, M.J.; Lackmann, M.; Li, C.; Barton, W.A.; Jeffrey, P.D.; Vearing, C.; Geleick, D.; Feldheim, D.A.; Boyd, A.W.; et al. Repelling class discrimination: ephrin-A5 binds to and activates EphB2 receptor signaling. Nat. Neurosci. 2004, 7, 501–509. [Google Scholar] [CrossRef] [PubMed]
- Ansuini, H.; Meola, A.; Gunes, Z.; Paradisi, V.; Pezzanera, M.; Acali, S.; Santini, C.; Luzzago, A.; Mori, F.; Lazzaro, D.; et al. Anti-EphA2 Antibodies with Distinct In Vitro Properties Have Equal In Vivo Efficacy in Pancreatic Cancer. J. Oncol. 2009, 2009, 951917. [Google Scholar] [CrossRef] [PubMed]
- Yuan, W.J.; Ge, J.; Chen, Z.K.; Wu, S.B.; Shen, H.; Yang, P.; Hu, B.; Zhang, G.W.; Chen, Z.H. Over-expression of EphA2 and EphrinA-1 in human gastric adenocarcinoma and its prognostic value for postoperative patients. Dig. Dis. Sci. 2009, 54, 2410–2417. [Google Scholar] [CrossRef]
- Mitra, S.; Duggineni, S.; Koolpe, M.; Zhu, X.; Huang, Z.; Pasquale, E.B. Structure-activity relationship analysis of peptides targeting the EphA2 receptor. Biochemistry 2010, 49, 6687–6695. [Google Scholar] [CrossRef]
- Tandon, M.; Vemula, S.V.; Mittal, S.K. Emerging strategies for EphA2 receptor targeting for cancer therapeutics. Expert Opin. Ther. Targets 2011, 15, 31–51. [Google Scholar] [CrossRef]
- Incerti, M.; Tognolini, M.; Russo, S.; Pala, D.; Giorgio, C.; Hassan-Mohamed, I.; Noberini, R.; Pasquale, E.B.; Vicini, P.; Piersanti, S.; et al. Amino acid conjugates of lithocholic acid as antagonists of the EphA2 receptor. J. Med. Chem. 2013, 56, 2936–2947. [Google Scholar] [CrossRef] [PubMed]
- Russo, S.; Incerti, M.; Tognolini, M.; Castelli, R.; Pala, D.; Hassan-Mohamed, I.; Giorgio, C.; De Franco, F.; Gioiello, A.; Vicini, P.; et al. Synthesis and structure-activity relationships of amino acid conjugates of cholanic acid as antagonists of the EphA2 receptor. Molecules 2013, 18, 13043–13060. [Google Scholar] [CrossRef] [PubMed]
- Tognolini, M.; Incerti, M.; Pala, D.; Russo, S.; Castelli, R.; Hassan-Mohamed, I.; Giorgio, C.; Lodola, A. Target hopping as a useful tool for the identification of novel EphA2 protein-protein antagonists. ChemMedChem 2014, 9, 67–72. [Google Scholar] [CrossRef] [PubMed]
- Hasegawa, J.; Sue, M.; Yamato, M.; Ichikawa, J.; Ishida, S.; Shibutani, T.; Kitamura, M.; Wada, T.; Agatsuma, T. Novel anti-EPHA2 antibody, DS-8895a for cancer treatment. Cancer Biol. Ther. 2016, 17, 1158–1167. [Google Scholar] [CrossRef] [PubMed]
- Giorgio, C.; Incerti, M.; Corrado, M.; Rusnati, M.; Chiodelli, P.; Russo, S.; Callegari, D.; Ferlenghi, F.; Ballabeni, V.; Barocelli, E.; et al. Pharmacological evaluation of new bioavailable small molecules targeting Eph/ephrin interaction. Biochem. Pharmacol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Lodola, A.; Giorgio, C.; Incerti, M.; Zanotti, I.; Tognolini, M. Targeting Eph/ephrin system in cancer therapy. Eur. J. Med. Chem. 2017, 142, 152–162. [Google Scholar] [CrossRef] [PubMed]
- Festuccia, C.; Gravina, G.L.; Giorgio, C.; Mancini, A.; Pellegrini, C.; Colapietro, A.; Delle Monache, S.; Maturo, M.G.; Sferra, R.; Chiodelli, P.; et al. UniPR1331, a small molecule targeting Eph/ephrin interaction, prolongs survival in glioblastoma and potentiates the effect of antiangiogenic therapy in mice. Oncotarget 2018, 9, 24347–24363. [Google Scholar] [CrossRef]
- Petty, A.; Idippily, N.; Bobba, V.; Geldenhuys, W.J.; Zhong, B.; Su, B.; Wang, B. Design and synthesis of small molecule agonists of EphA2 receptor. Eur. J. Med. Chem. 2018, 143, 1261–1276. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ID | Sequence | IC50 (nM) (DELFIA) |
|---|---|---|
| 123B9 | (4F,3ClPhOCH2CO)SAYPDSVP(Nle)(Hsr)S-CONH2 | 6500 ± 1700, n = 2 |
| YSA | H2N-YSAYPDSVPMMS-CONH2 | 16200 ± 800, n = 14 |
| 135B12 | H2N-YSAYPDSVPFRP-CONH2 | 1600 ± 200, n = 16 |
| 135C11 (dimer of 135B12) | (H2N-YSAYPDSVPFRPG)2-K-CONH2 | 700 ± 100, n = 4 |
| 135C12 (dimer of 135B12) | (H2N-YSAYPDSVPFRP-βAla)2-K-CONH2 | 300 ± 100, n = 3 |
| 135D1 (dimer of 135B12) | (H2N-YSAYPDSVPFRP-GABA)2-K-CONH2 | 400 ± 200, n = 3 |
| 135E2 | (4F,3Cl-PhOCH2CO)SAYPDSVPFRP-CONH2 | 3100 ± 600, n = 3 |
| 135G3 | (4F,3Cl-PhOCH2CO)SAYPDSV(Hyp)(4Cl-Phe)RP-CONH2 | 600 ± 100, n = 6 |
| 135G4 (dimer of 135G3) | ((4F,3Cl-PhOCH2CO)SAYPDSV(Hyp)(4Cl-Phe)RPG)2-K-CONH2 | 130 ± 2 n = 1 |
| 135H11 | (3-CH3,6,7-OCH3,Benzofuranoic acid)LA(4-CH3-Tyr)PDA V(Hyp)(4Cl-Phe)RP-CONH2 | 130 ± 1, n = 4 |
| 135H12 (dimer of 135H11) | ((3-CH3,6,7-OCH3,Benzofuranoic acid)LA(4-CH3-Tyr)PDAV(Hyp)(4Cl-Phe) RPG)2-K-CONH2 | 150 ± 60, n = 3 |
| 135I1 (tetramer of 135H11) | {[(3-CH3,6,7-OCH3,Benzofuranoic acid)LA(4-CH3-Tyr)PDAV(Hyp)(4Cl-Phe)RPG]2-K-K}2-K-CONH2 | 60 ± 10, n = 2 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Salem, A.F.; Gambini, L.; Udompholkul, P.; Baggio, C.; Pellecchia, M. Therapeutic Targeting of Pancreatic Cancer via EphA2 Dimeric Agonistic Agents. Pharmaceuticals 2020, 13, 90. https://doi.org/10.3390/ph13050090
Salem AF, Gambini L, Udompholkul P, Baggio C, Pellecchia M. Therapeutic Targeting of Pancreatic Cancer via EphA2 Dimeric Agonistic Agents. Pharmaceuticals. 2020; 13(5):90. https://doi.org/10.3390/ph13050090
Chicago/Turabian StyleSalem, Ahmed F., Luca Gambini, Parima Udompholkul, Carlo Baggio, and Maurizio Pellecchia. 2020. "Therapeutic Targeting of Pancreatic Cancer via EphA2 Dimeric Agonistic Agents" Pharmaceuticals 13, no. 5: 90. https://doi.org/10.3390/ph13050090
APA StyleSalem, A. F., Gambini, L., Udompholkul, P., Baggio, C., & Pellecchia, M. (2020). Therapeutic Targeting of Pancreatic Cancer via EphA2 Dimeric Agonistic Agents. Pharmaceuticals, 13(5), 90. https://doi.org/10.3390/ph13050090