Experimental Evaluation of Anticancer Efficiency and Acute Toxicity of Anthrafuran for Oral Administration †

 ,
,
Abstract
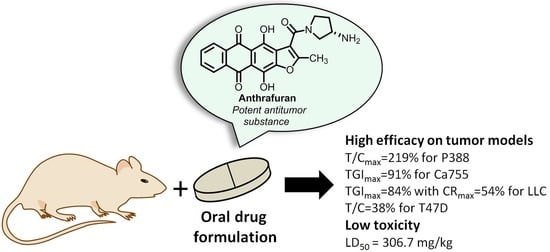
1. Introduction
2. Results and Discussions
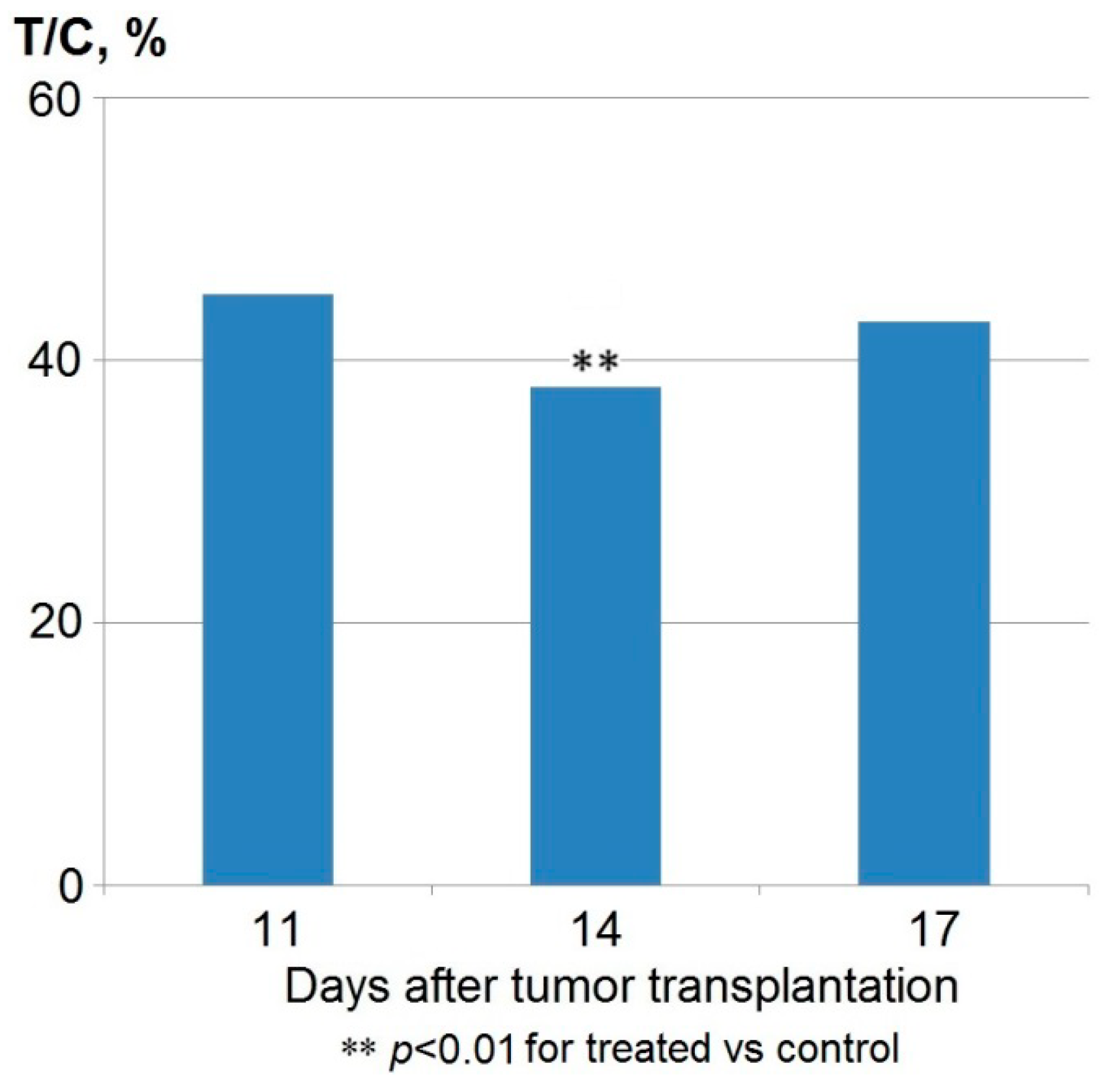
2.1. Antitumor Efficacy
2.2. Acute Toxicity
3. Materials and Methods
3.1. Materials
3.2. In Vivo Tumor Models and Mice
3.3. Evaluation of Antitumor Activity
3.4. Acute Toxicity
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Jonkman-de Vries, J.D.; Flora, K.P.; Bult, A.; Beijnen, J.H. Pharmaceutical development of (investigational) anticancer agents for parenteral use—A review. J. Drug Develop. Ind. Pharm. 1996, 22, 475–494. [Google Scholar] [CrossRef]
- Olusanya, T.O.B.; Ahmad, R.R.H.; Ibegbu, D.M.; Smith, J.R.; Elkordy, A.A. Liposomal drug delivery systems and anticancer drugs. Molecules 2018, 23, 907. [Google Scholar] [CrossRef] [PubMed]
- Ruddy, K.; Mayer, E.; Partridge, A. Patient adherence and persistence with oral anticancer treatment. CA Cancer J. Clin. 2009, 59, 56–66. [Google Scholar] [CrossRef] [PubMed]
- Tran, P.; Pyo, Y.C.; Kim, D.H.; Lee, S.E.; Kim, J.K.; Park, J.S. Overview of the manufacturing methods of solid dispersion technology for improving the solubility of poorly water-soluble drugs and application to anticancer drugs. Pharmaceutics 2019, 11, 132. [Google Scholar] [CrossRef]
- Banna, G.L.; Collova, E.; Gebbia, V.; Lipari, H.; Giuffrida, P.; Cavallaro, S.; Condorelli, R.; Buscarino, C.; Tralongo, P.; Ferrau, F. Anticancer oral therapy: Emerging related issues. Cancer Treat. Rev. 2010, 36, 595–605. [Google Scholar] [CrossRef] [PubMed]
- Thanki, K.; Gangwal, R.P.; Sangamwar, A.T.; Jain, S. Oral delivery of anticancer drugs: Challenges and opportunities. J. Controlled Release 2013, 170, 15–40. [Google Scholar] [CrossRef] [PubMed]
- Alam, M.A.; Al-Jenoobi, F.I.; Al-Mohizea, A.M.; Ali, R. Understanding and managing oral bioavailability: Physiological concepts and patents. Rec. Pat. Anticancer Drug Discov. 2015, 10, 87–96. [Google Scholar] [CrossRef]
- Lennernäs, H.; Aarons, L.; Augustijns, P.; Beato, S.; Bolger, M.; Box, K.; Brewster, M.; Butler, J.; Dressman, J.; Holm, R.; et al. Oral biopharmaceutics tools—Time for a new initiative—An introduction to the IMI project OrBiTo. Eur. J. Pharm. Sci. 2014, 57, 292–299. [Google Scholar] [CrossRef]
- Sawicki, E.; Schellens, J.H.; Beijnen, J.H.; Nuijen, B. Inventory of oral anticancer agents: Pharmaceutical formulation aspects with focus on the solid dispersion technique. Cancer Treat. Rev. 2016, 50, 247–263. [Google Scholar] [CrossRef]
- Stuurman, F.E.; Nuijen, B.; Beijnen, J.H.; Schellens, J.H. Oral anticancer drugs: Mechanisms of low bioavailability and strategies for improvement. Clin. Pharmacokinet. 2013, 52, 399–414. [Google Scholar] [CrossRef]
- Volodina, Y.L.; Dezhenkova, L.G.; Tikhomirov, A.S.; Tatarskiy, V.V.; Kaluzhny, D.N.; Moisenovich, A.M.; Moisenovich, M.M.; Isagulieva, A.K.; Shtil, A.A.; Tsvetkov, V.B.; et al. New anthra[2,3-b]furancarboxamides: A role of positioning of the carboxamide moiety in antitumor properties. Eur. J. Med. Chem. 2019, 165, 31–45. [Google Scholar] [CrossRef] [PubMed]
- Tikhomirov, A.S.; Shtil, A.A.; Shchekotikhin, A.E. Advances in the discovery of anthraquinone-based anticancer agents. Rec. Pat. Anticancer Drug Discov. 2018, 13, 159–183. [Google Scholar] [CrossRef] [PubMed]
- Shchekotikhin, A.E.; Dezhenkova, L.G.; Tsvetkov, V.B.; Luzikov, Y.N.; Volodina, Y.L.; Tatarskiy, V.V.; Kalinina, A.A.; Treshalin, M.I.; Treshalina, H.M.; Romanenko, V.I.; et al. Discovery of antitumor anthra[2,3-b]furan-3-carboxamides: Optimization of synthesis and evaluation of antitumor properties. Eur. J. Med. Chem. 2016, 112, 114–129. [Google Scholar] [CrossRef] [PubMed]
- Tikhomirov, A.S.; Lin, C.-Y.; Volodina, Y.L.; Dezhenkova, L.G.; Tatarskiy, V.V.; Schols, D.; Shtil, A.A.; Kaur, P.; Chueh, P.J.; Shchekotikhin, A.E. New antitumor anthra[2,3-b]furan-3-carboxamides: Synthesis and structure-activity relationship. Eur. J. Med. Chem. 2018, 148, 128–139. [Google Scholar] [CrossRef] [PubMed]
- Treshalina, H.M.; Romanenko, V.I.; Kaluzhny, D.N.; Treshalin, M.I.; Nikitin, A.A.; Tikhomirov, A.S.; Shchekotikhin, A.E. Development and pharmaceutical evaluation of the anticancer Anthrafuran/Cavitron complex, a prototypic parenteral drug formulation. Eur. J. Pharm. Sci. 2017, 109, 631–637. [Google Scholar] [CrossRef] [PubMed]
- Schellens, J.H.M.; Malingre, M.M.; Kruijtzer, C.M.F.; Bardelmeijer, H.A.; van Tellingen, O.; Schinkel, A.H.; Beijnen, J.H. Modulation of oral bioavailability of anticancer drugs: From mouse to man. Eur. J. Pharm. Sci. 2000, 12, 103–110. [Google Scholar] [CrossRef]
- Fields, S.M.; Koeller, J.M. Idarubicin: A second-generation anthracycline. Ann. Pharmcotherapy 1991, 25, 505–517. [Google Scholar] [CrossRef]
- Portoy, Y.A.; Dovzhenko, S.A.; Cobrin, M.B.; Pereverzeva, E.R.; Treshchalin, M.I.; Golibrodo, V.A.; Shchekotikhin, A.E.; Firsov, A.A. Pharmacokinetics and acute toxicity of Anthrafuran, a novel antitumor agent (pre-clinical study). Pharm. Chem. J. 2020. [Google Scholar] [CrossRef]
- Treschalin, M.I.; Treschalin, I.D.; Golibrodo, V.A.; Shchekotikhin, A.E.; Pereverzeva, E.R. Experimental evaluation of toxic properties of LCTA-2034 by the oral route of administration. Rus. J. Biother. 2018, 17, 81–88. [Google Scholar] [CrossRef]
- Golibrodo, V.A.; Treshchalin, I.D.; Shchekotikhin, A.E.; Pereverzeva, E.R. Neurotoxic properties of new antitumor agent Anthrafuran. Rus. J. Biother. 2019, 18, 75–79. [Google Scholar] [CrossRef]
- Globally Harmonized System of Classification and Labelling of Chemicals (GHS), United Nations, New York and Geneva, 2011, Fourth Revised Edition, p. 109. Available online: https://www.unece.org/fileadmin/DAM/trans/danger/publi/ghs/ghs_rev04/English/ST-SG-AC10-30-Rev4e.pdf (accessed on 17 August 2011).
- Council of Europe European Convention for the Protection of Vertebrate Animals Used for Experimental and Other Purposes. Strasbourg: 1986, 18.III.1986, Council of Europe, ETS No. 123. Available online: https://rm.coe.int/168007a67b (accessed on 28 August 2018).
- Directive 2010/63/EU on the Protection of Animals Used for Scientific Purposes EN. Official Journal of the European Union, L 276/33-276/79 (20.10.2010). Available online: https://eur-lex.europa.eu/LexUriServ/LexUriServ.do?uri=OJ:L:2010:276:0033:0079:EN:PDF (accessed on 27 April 2020).
- National State Standard GOST P 53434-2009 the Russian Federation standard “The Principles of Good Laboratory Practice” (Approved and Put into Effect by the Order of the Federal Agency for Technical Regulation and Metrology of December 2, 2009), No 544. Available online: http://docs.cntd.ru/document/1200075972 (accessed on 3 March 2010). (In Russian).
- Treshalina, H.M. Immunodeficient Balb/c nude mice and modeling of different tumor growth options for preclinical studies. Rus. J. Biother. 2017, 16, 6–13. [Google Scholar] [CrossRef]
- Cobrett, T. In vivo methods for screening and preclinical testing. In Anticancer Drug Development Guide: Preclinical Screening, Clinical Trials, and Approval, 2nd ed.; Teicher, B.A., Andrews, P.A., Eds.; Humana Press: Totowa, NJ, USA, 2004; pp. 99–123. [Google Scholar]
- Treshalina, H.M.; Andronova, N.V.; Garin, A.M. Preclinical investigation of the anticancer drugs. Rational Pharmacotherapy in Oncology. In Rational Pharmacotherapy in Oncology; Litterra: Moscow, Russia, 2016; pp. 75–82. (In Russian) [Google Scholar]
- Andronova, N.V.; Smirnova, G.B.; Borisova, J.A.; Kalish’yan, M.S.; Treshchalina, E.M. Modeling of carcinomatosis in the intraperitoneal implantation of solid tumors of mice and human. Russ. J. Oncol. 2016, 21, 41–45. Available online: https://rucont.ru/efd/427984. (accessed on 10 April 2016). (In Russian).




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group | Single Dose | Total Dose | Parameters | |
|---|---|---|---|---|
| L ± m (days) | T/C (%) | |||
| Control | 0.5 mL * | 2.5 mL | 9.5 ± 0.7 | 100 |
| Anthrafuran | 40 mg/kg | 200 mg/kg | 12.4 ± 0.9 ** | 130 |
| 60 mg/kg | 300 mg/kg | 14.5 ± 1.1 ** | 153 | |
| 80 mg/kg | 400 mg/kg | 20.8 ± 3.2 ** | 219 | |
| 100 mg/kg | 500 mg/kg | 19.9 ± 3.7 ** | 202 | |
| 120 mg/kg | Lethal toxicity *** | |||
| Group | Single Dose | Regimen | Total Dose | Parameters | |
|---|---|---|---|---|---|
| L ± m (days) | T/C (%) | ||||
| Control | 0.5 mL * | 8 × 24 h | 4.0 mL | 9.7 ± 0.9 | 100 |
| Anthrafuran | 80 mg/kg | 5 × 24 h | 400 mg/kg | 18.4 ± 1.7 ** | 190 |
| 5 × 48 h | 400 mg/kg | 18.4 ± 2.1 ** | 190 | ||
| 8 × 24 h | 640 mg/kg | 17.4 ± 2.2 ** | 179 | ||
| Parameter | Days after Tumor Transplantation | ||
|---|---|---|---|
| 9 | 13 | 19 | |
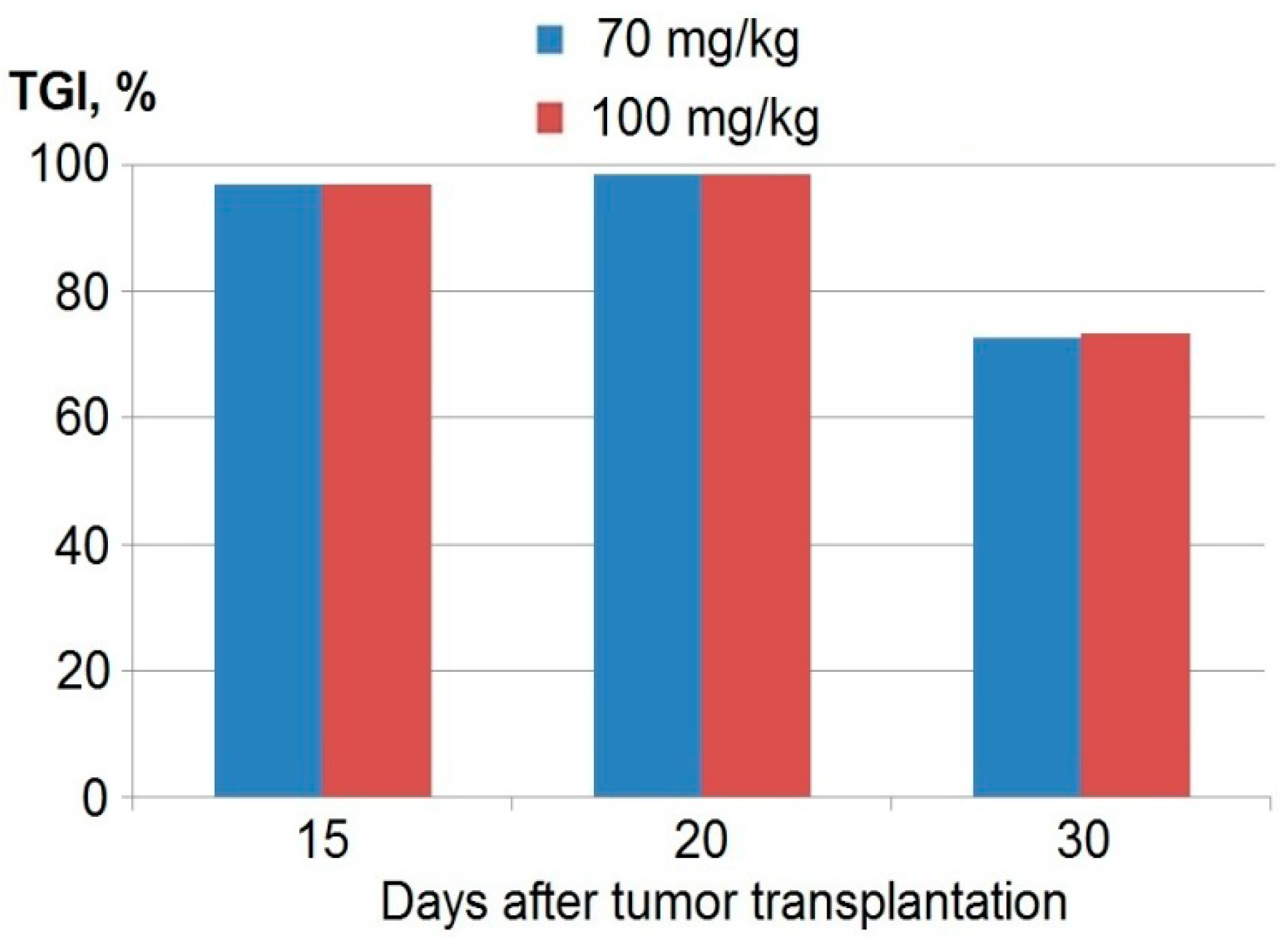
| TGI% * | 84 | 61 | 49 |
| Complete remission (number of mice) | 7 | 3 | 1 |
| Route of Administration | Parameter | Doses, mg/kg | |
|---|---|---|---|
| Males | Females | ||
| Intraperitoneal | LD50 | 52.5 (47.1 ÷ 57.9) * | 53.1 (48.3 ÷ 58.4) * |
| LD10 | 39.4 | 38.4 | |
| Oral | LD50 | 306.7 (209.1 ÷ 404.3) * | 309.2 (237.4 ÷ 387.7) * |
| LD10 | 128.7 | 129.9 | |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shchekotikhin, A.E.; Treshalina, H.M.; Treshchalin, M.I.; Pereverzeva, E.R.; Isakova, H.B.; Tikhomirov, A.S. Experimental Evaluation of Anticancer Efficiency and Acute Toxicity of Anthrafuran for Oral Administration. Pharmaceuticals 2020, 13, 81. https://doi.org/10.3390/ph13050081
Shchekotikhin AE, Treshalina HM, Treshchalin MI, Pereverzeva ER, Isakova HB, Tikhomirov AS. Experimental Evaluation of Anticancer Efficiency and Acute Toxicity of Anthrafuran for Oral Administration. Pharmaceuticals. 2020; 13(5):81. https://doi.org/10.3390/ph13050081
Chicago/Turabian StyleShchekotikhin, Andrey E., Helen M. Treshalina, Michael I. Treshchalin, Eleonora R. Pereverzeva, Helen B. Isakova, and Alexander S. Tikhomirov. 2020. "Experimental Evaluation of Anticancer Efficiency and Acute Toxicity of Anthrafuran for Oral Administration" Pharmaceuticals 13, no. 5: 81. https://doi.org/10.3390/ph13050081
APA StyleShchekotikhin, A. E., Treshalina, H. M., Treshchalin, M. I., Pereverzeva, E. R., Isakova, H. B., & Tikhomirov, A. S. (2020). Experimental Evaluation of Anticancer Efficiency and Acute Toxicity of Anthrafuran for Oral Administration. Pharmaceuticals, 13(5), 81. https://doi.org/10.3390/ph13050081