Formulation and Evaluation of Loperamide HCl Oro Dispersible Tablets



Abstract
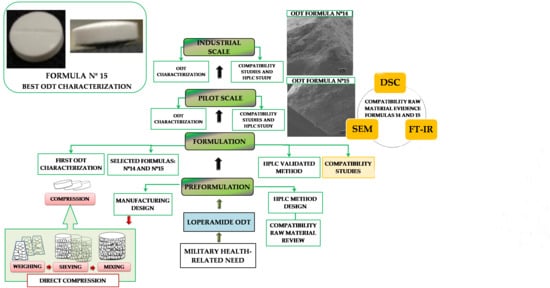
1. Introduction
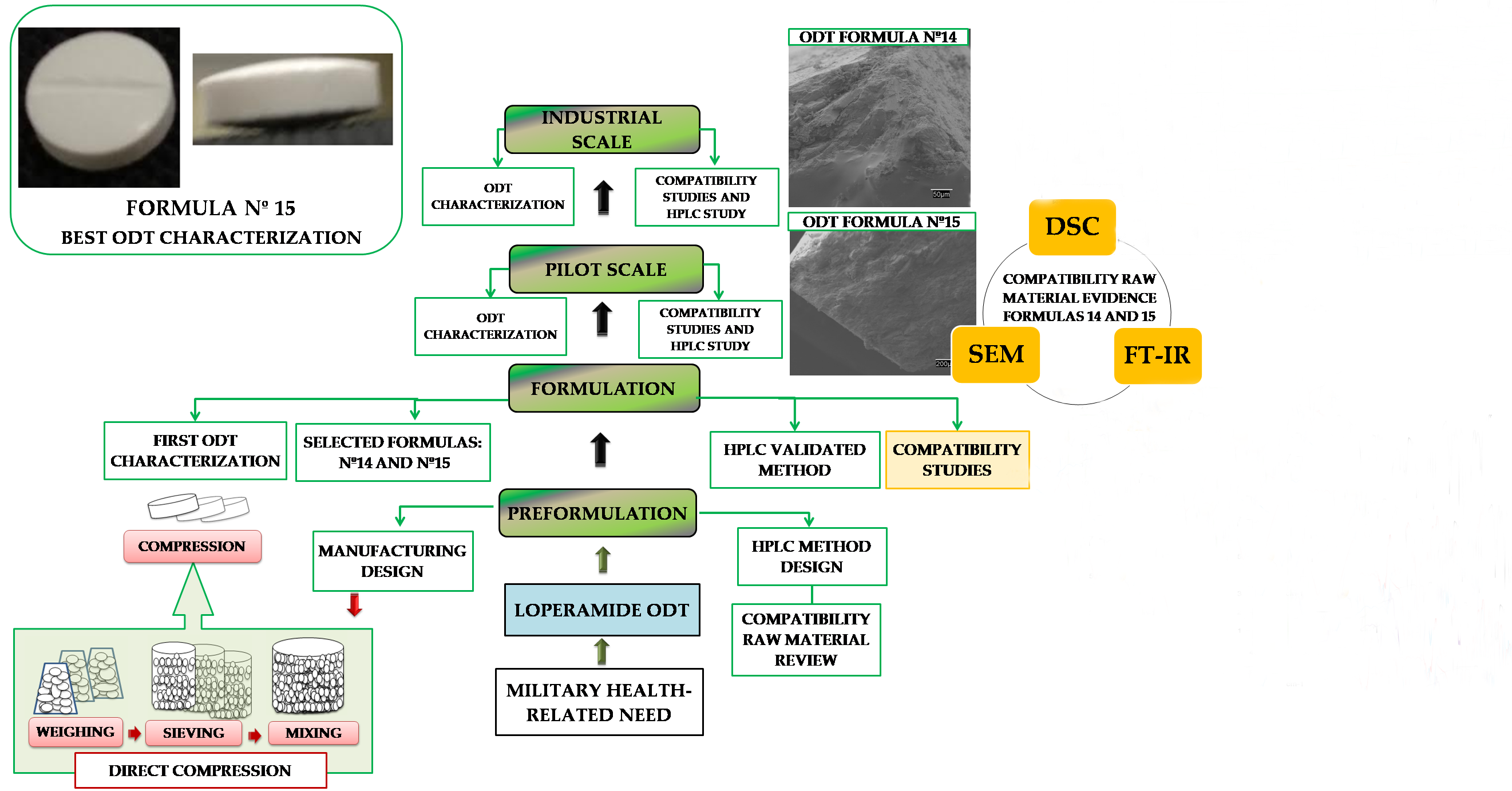
2. Results and Discussion
2.1. Preformulation
2.2. Formulation
2.3. Solid-State Characterization
2.3.1. Thermal Analysis
2.3.2. FT-IR
2.3.3. SEM Studies
2.4. Pilot Scale
2.5. Industrial Scale
2.6. Critical Quality Attributes of Loperamide ODT
3. Experimental
3.1. Materials
3.1.1. Preparation of Physical Mixtures
3.1.2. Preparation of Artificial Saliva
3.2. High-Performance Liquid Chromatography (HPLC) Analysis
3.3. Differential Scanning Calorimetry (DSC) Analysis
3.4. Fourier Transforms Infrared Spectroscopy (FT-IR) Analysis
3.5. Scanning Electron Microscopy (SEM) Studies
3.6. Water Absorption Ratio
3.7. Preparation of Orally Disintegrating Tablets (ODTs)
3.8. Tablets Characterization
3.8.1. Weight Variation
3.8.2. Thickness
3.8.3. Hardness
3.8.4. Diameter
3.8.5. Friability Test
3.8.6. Disintegration Time
3.8.7. Dissolution Studies
3.8.8. Content Uniformity
3.8.9. Divisibility Study
3.8.10. Water Content
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Boletín Oficial de Defensa N° 059. Article N°6; Centro Militar de Farmacia de la Defensa, Ministerio de Defensa: Madrid, Spain, 2012.
- Boletín Oficial del Estado. Resolución de 22 de agosto de 2019, de la Subsecretaría, por la que se publica el Convenio entre el Ministerio de Defensa y la Agencia Española de Medicamentos y Productos Sanitarios, Para la Custodia y Gestión del Depósito Estatal Estratégico de Medicamentos y Productos Sanitarios Para Emergencias y Catástrofes, el Depósito Estatal de Antivirales, el Depósito Contra la Viruela, el Depósito de Antitoxinas y Medicamentos de Urgencia, y la Fabricación de Medicamentos Para Situaciones Especiales. Available online: https://www.boe.es/boe/dias/2019/08/29/pdfs/BOE-A-2019-12545.pdf (accessed on 1 September 2019).
- Spanish Ministry of Defence. Foreign Missions. Available online: https://www.defensa.gob.es/misiones/en_exterior/ (accessed on 1 September 2019).
- Boletín Oficial de Defensa. Orden Ministerial 8/2014, de 30 de Enero, Por La Que Se Establece El Petitorio de Farmacia del Ministerio de Defensa; Ministerio de Defensa: Madrid, Spain, 2014.
- World Health Organization. World Health Organization Model List of Essential Medicines. 21st List. Available online: https://apps.who.int/iris/bitstream/handle/10665/325771/WHO-MVP-EMP-IAU-2019.06-eng.pdf?ua=1 (accessed on 1 September 2019).
- Connor, B.A. The prevalent consultation: Travellers´ diarrhea. In Centers for Disease Control and Prevention. CDC Yellow Book: Health Information for International Travel; Oxford University Press: New York, NY, USA, 2020. [Google Scholar]
- Riddle, M.S.; Connor, B.A.; Beeching, N.J.; DuPont, H.L.; Hamer, D.H.; Kozarsky, P.; Libman, M.; Steffen, R.; Taylor, D.; Tribble, D.R.; et al. Guidelines for the prevention and treatment of travelers’ diarrhea: A graded expert panel report. J. Travel Med. 2017, 24, 57–74. [Google Scholar] [CrossRef] [PubMed]
- Baker, D.E. Loperamide: A pharmacological review. Rev. Gastroenterol. Disord. 2007, 7, 11–18. [Google Scholar]
- Regnard, C.; Twycross, R.; Mihalyo, M.; Wilcock, A. Therapeutic reviews loperamide. J. Pain Symptom Manag. 2011, 42, 319–323. [Google Scholar] [CrossRef] [PubMed]
- Vogler, S.; Zimmermann, N.; Leopold, C.; Joncheere, K.D. Pharmaceutical policies in European countries in response to the global financial crisis. South. Med. Rev. 2011, 482, 69–79. [Google Scholar] [CrossRef] [PubMed]
- Nenni, M.E.; Giustiniano, L.; Pirolo, L. Improvement of manufacturing operations through a lean management approach: A case study in the pharmaceutical industry. Int. J. Eng. Bus. Manag. 2014, 6, 1–6. [Google Scholar] [CrossRef]
- Haleem, R.M.; Salem, M.Y.; Fatahallah, F.A.; Abdelfattah, L.E. Quality in the pharmaceutical industry. A literature review. Saudi Pharm. J. 2015, 23, 463–469. [Google Scholar] [CrossRef]
- Agiba, A.M.; Eldin, A.B. Insights into formulation technologies and novel strategies for the design of orally disintegrating dosage forms: A comprehensive industrial review. Int. J. Pharm. Pharm. Sci. 2019, 11, 8–20. [Google Scholar] [CrossRef][Green Version]
- Khan, A.B.; Tripuraneni, A. Fast dissolving tablets–a novel approach in drug delivery. Rguhs J. Pharm. Sci. 2014, 4, 7–16. [Google Scholar] [CrossRef]
- Niazi, S.K. Guidance on formulating compressed solids. In Handbook of Pharmaceutical Manufacturing Formulations Compressed Solid Products, 2nd ed.; Informa Helthcare: New York, NY, USA, 2009. [Google Scholar]
- Davies, P. Oral solid dosage forms. In Pharmaceutical Preformulation and Formulation, 2nd ed.; Gibson, M., Ed.; Informa Helthcare: New York, NY, USA, 2009. [Google Scholar]
- Medicine Online Information Center of AEMPS-CIMA. Ficha Técnica Salva colina Flas 2 mg comprimidos Bucodispersables. Available online: https://cima.aemps.es/cima/pdfs/es/ft/82401/FT_82401.pdf (accessed on 9 January 2020).
- Kotkoskie, L.A.; Weiner, M. Excipient Toxicity and Safety, 2nd ed.; Marcel Dekker Inc.: New York, NY, USA, 2000. [Google Scholar]
- Schwartz, J.B. Scale up of the compaction and tableting process. InPharmaceutical Process Scale Up; Marcel Dekker Inc.: New York, NY, USA, 2002. [Google Scholar]
- Tannebaum, E.J. Oral solid dosage facilities. In Good Design Practices for GMP Pharmaceutical Facilities; CRC Press Taylor&Francis Group: New York, NY, USA, 2005. [Google Scholar]
- Lee, B.J. Pharmaceutical preformulation: Physicochemical properties of excipients and powders and tablet characterization. In Pharmaceutical Manufacturing Handbook: Production and Processes; John Wiley & Sons Inc.: Hoboken, NJ, USA, 2008. [Google Scholar]
- Narang, A.S.; Mantri, R.V.; Ragahavan, K.S. Excipient compatibility and functionality. In Developing Solid Oral Dosage Forms. Pharmaceutical Theory and Practice, 2nd ed.; Elsevier Inc.: Amsterdam, The Netherlands, 2017. [Google Scholar]
- Moretom, R.C. Calcium phosphate dibasic dihydrate. In Handbook of Pharmaceutical Excipients, 6th ed.; Pharmaceutical Press and American Pharmacists Association: London, UK, 2009. [Google Scholar]
- Debord, B.; Lefebvre, C.; Guyot-Hermann, A.M.; Hubert, J.; Bouché, R.; Cuyot, J.C. Study of different crystalline forms of mannitol: Comparative behaviour under compression. Drug Dev. Ind. Pharm. 1987, 13, 1533–1546. [Google Scholar] [CrossRef]
- Molokhia, A.M.; Al-Shora, H.I.; Hammad, A.A. Aging of tablets prepared by direct compression of bases with different moisture content. Drug Dev. Ind. Pharm. 1987, 13, 1933–1946. [Google Scholar] [CrossRef]
- Armstrong, N.A. Mannitol. In Handbook of Pharmaceutical Excipients, 6th ed.; Pharmaceutical Press and American Pharmacists Association: London, UK, 2009. [Google Scholar]
- Yoshinari, T.; Forbes, R.T.; York, P.; Kawashima, Y. The improved compaction properties of mannitol after a moisture-induced polymorphic transition. Int. J. Pharm. 2003, 258, 121–131. [Google Scholar] [CrossRef]
- Rogers, T.L. Hypromellose. In Handbook of Pharmaceutical Excipients, 6th ed.; Pharmaceutical Press and American Pharmacists Association: London, UK, 2009. [Google Scholar]
- Young, P.M. Sodium starch glycolate. In Handbook of Pharmaceutical Excipients, 6th ed.; Pharmaceutical Press and American Pharmacists Association: London, UK, 2009. [Google Scholar]
- Hernández-Torres, J.E.; Melgoza-Contreras, L.M. Principales superdisgregantes sintéticos, mecanismos y factores que influyenensuactividad. Rev. Colomb. Cienc. Quím. Farm. 2014, 43, 234–247. [Google Scholar] [CrossRef]
- Goggin, P.L. Sodium cyclamate. In Handbook of Pharmaceutical Excipients, 6th ed.; Pharmaceutical Press and American Pharmacists Association: London, UK, 2009. [Google Scholar]
- Langdon, B.A.; Mullarney, P.M. Menthol. In Handbook of Pharmaceutical Excipients, 6th ed.; Pharmaceutical Press and American Pharmacists Association: London, UK, 2009. [Google Scholar]
- Chauhan, R. Taste masking: A unique approach for bitter drugs. J. Stem.Cell. Bio. Transplant. 2017, 1, 2–12. [Google Scholar]
- Inoue, Y.; Funato, S.; Suzuki, R.; Morita, Y.; Murata, I.; Kanamoto, I. Human sensory testing of loperamide hydrochloride preparations for children to improve their palatability. World J. Pharm. Sci. 2015, 3, 570–579. [Google Scholar]
- Shende, M.; Chavan, K.D. Formulation of furosemide oral disintegrating tablets using natural and synthetic superdisintegrants by SeDeM expert design system. JDDT 2019, 9, 55–63. [Google Scholar] [CrossRef]
- European Pharmacopoeia (Ph. Eur.) European Department for Quality of Medicines, 10th ed.; EDQM: Strasboug, France, 2020.
- Chadha, R.; Swati, B.S. Drug–Excipient compatibility screening. Role of thermoanalytical and spectroscopic techniques. J. Pharm. Biomed. Anal. 2014, 87, 82–97. [Google Scholar] [CrossRef]
- Caira, M.R.; Gerber, J.J.; Lotter, A.P. Hydration of drug molecules: Cavity inclusion of water in crystals of loperamide hydrochloride tetrahydrate. Supramol. Chem. 1995, 5, 225–230. [Google Scholar] [CrossRef]
- Newman, A.W.; Andres, M.C.; Stahly, G.P.; Ohannessian, L.; Adair, A. Unsolvatedenantiotropic polymorphs of loperamide hydrochloride. AAPS PharmSci. 2002, 4, 1816. [Google Scholar]
- Woertz, C.; Kleinebudde, P. Development of orodispersible polymer films with focus on the solid-state characterization of crystalline loperamide. Eur. J. Pharm. Biopharm. 2015, 94, 52–63. [Google Scholar] [CrossRef]
- Rompay, J.; Carter, J.E. Clorhidrato de loperamida. In Perfiles Analíticos de Sustancias y Excipientes de Drogas; Florey, K., Ed.; Academic Press: Orlando, FL, USA, 1990. [Google Scholar]
- Carlton, R.A. Experimental methods in the study of thermodynamic polymorph stability relationships. Am. Pharm. Rev. 2006, 9, 88–93. [Google Scholar]
- Landin, M.; Rowe, R.C.; York, P. Structural changes during the dehydration of dicalcium phosphate dehydrate. Eur. J. Pharm. Sci. 1994, 2, 245–252. [Google Scholar] [CrossRef]
- Medina, D.; Ferreira, A.; Cavalheiro, E.; Vargas, D. Polymorphism and thermal behavior of sodium cyclamate. J. Therm. Anal. Calorim. 2019, 137, 1307–1313. [Google Scholar] [CrossRef]
- Ali, F.; Kumar, R.; Sahu, P.L.; Singh, G.N. Physicochemical characterization and compatibility study of roflumilast with various pharmaceutical excipients. J. Therm. Anal. Calorim. 2017, 130, 1627–1641. [Google Scholar] [CrossRef]
- Chavan, R.B.; Shastri, N.R. Polymorphic transformation as a result of atovaquone incompatibility with selected excipients. J. Therm. Anal. Calorim. 2018, 131, 2129–2139. [Google Scholar] [CrossRef]
- Georgy, K.R.; Farid, R.M.; Latif, R.; Benda, E.R. A new design for a chronological release profile of etodolac from coated bilayer tablets: In-vitro and in-vivo assessment. J. Adv. Res. 2018, 15, 37–47. [Google Scholar] [CrossRef]
- Indrayanto, G.; Mugihardjo, H.R. Compatibility study between famotidine and some excipients using differential scanning calorimetry. Drug Dev. Ind. Pharm. 1994, 20, 911–920. [Google Scholar] [CrossRef]
- Thomas, V.H.; Naath, M. Design and utilization of the drug–excipient chemical compatibility automated system. Int. J. Pharm. 2008, 359, 150–157. [Google Scholar] [CrossRef]
- Kiss, D.; Zelkó, R.; Novák, C.; Éhen, Z. Application of DSC and NIRS to study the compatibility of metronidazole with different pharmaceutical excipients. J. Therm. Anal. Cal. 2006, 84, 447–451. [Google Scholar] [CrossRef]
- Sachin, T.V.; Deodhar, M.N.; Prakya, V. Advances in analytical techniques used in predicting drug-excipient interactions. Int. J. Pharm. Tech. 2014, 6, 6388–6417. [Google Scholar]
- Venkateswarlu, K.; Preethi, J.K.; Chandrasekhar, K.B. Enhancement of loperamide dissolution rate by liquid-solid compact technique. Adv. Pharm. Bull. 2016, 6, 385–390. [Google Scholar] [CrossRef]
- Aigner, Z.; Heinrich, R.; Sipos, E.; Farkas, G.; Ciurba, A.; Berkesi, O.; Szabó-Révész, P. Compatibility studies of aceclofenac with retard tablet excipients by means of thermal and FT-IR spectroscopic methods. J. Therm. Anal. Calorim. 2011, 104, 265–271. [Google Scholar] [CrossRef]
- Stulzer, H.K.; Tagliari, M.P.; Cruz, A.P.; Silva, M.A.; Laranjeira, M. Compatibility studies between piroxicam and pharmaceutical excipients used in solid dosage forms. Pharm. Chem. J. 2008, 42, 215–219. [Google Scholar] [CrossRef]
- Ríos, M. Developments in scanning electron microscopy for tablet and granule characterization. Pharm. Tech. 2008, 2, 36–38. [Google Scholar]
- Dadhich, T.; Kumar, M.; Pathak, K. Capsulated surface solid dispersion of loperamide for targeted delivery. Pharm. Chem. 2016, 3, 78–90. [Google Scholar]
- USP 42 - NF 37. In The United States Pharmacopeia and National Formulary; United States Pharmacopeial Convention: Brasília, Brasil, 2019.
- Torrado, G.; Aberturas, M.R.; Molpeceres, J.; Peña, M.A.; Villarrubia, A. Estudio comparativo de disgregación de diferentes formulaciones de olanzapina. An. R. Acad. Farm. 2011, 77, 58–75. [Google Scholar]
- Halder, A.; Behera, A.; Si, S.; Biswal, I.; Dinda, A. Preparation of loperamide hydrochloride chewable tablet: Method validation by HPLC. Int. J. Pharm. Pharmaceut. Sci. 2012, 4, 372–381. [Google Scholar]
- Sujatha, T.; Balmuralikrishna, K.; Raju, R. A validated RP-HPLC method for the estimation of loperamide hydrochloride in tablet dosage forms. Int. J. Chem Tech Res. 2014, 6, 1097–1102. [Google Scholar]
- Sonawane, A.M.; Dudhe, P.B.; Chalke, N.H.; Bhagat, K.B. Development and validation RP-HPLC method for the simultaneous determination of loperamide hydrochloride and norfloxacin in pharmaceutical formulation. Int. J. Pharm. Sci. Res. 2016, 7, 3441–3445. [Google Scholar]
- Kabir, H.; Paul, R.K.; Rahaman, M.S.; Ahmad, M.F.; Bhattacharjya, D.K.; Rahaman, M.S. Method validation for assay of loperamide hydrochloride by HPLC in loperamide hydrochloride tablets. Int. J. Adv. Res. Chem. Sci. 2017, 4, 11–27. [Google Scholar]
- Suneetha, A.; Sharmila, N.; Purnima, M. Stability indicating RP-HPLC method for the determination &validation of loperamide hydrochloride & simethicone in pharmaceutical dosage form. World J. Pharm. Pharm. Sci. 2017, 6, 955–971. [Google Scholar]
- Draksiene, G.; Kopustinskiene, D.M.; Lazauskas, R.; Bernatoniene, J. Psyllium (Plantago Ovata Forsk) Husk Powder as a natural superdisintegrant for orodispersible formulations: A study on meloxicam tablets. Molecules 2019, 6, 3255. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| FORMULA | LOPERAMIDE | CHPD | STARCH | TALC | MAGNESSIUM STEARATUM | HC | CS | SACCHARIN SODIUM | MENTHOL | ANISE EXTRACT | HPMC | XYLITOL | CROSPOVIDONE | MANNITOL | SODIUM STARCH GLYCOLATE TYPE A | SODIUM CYCLAMATE | OBJECTIVE | FAILURES | PHOTOS |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| N°1 | 1.33% | 89.67% | 5.00% | 3.00% | 1.00% | - | - | - | - | - | - | - | - | - | - | - | T | CAPPING + CHIPPING |  |
| 150 mg | |||||||||||||||||||
| N°2 | 1.33% | 79.67% | 5.00% | 3.00% | 1.00% | 10.00% | - | - | - | - | - | - | - | - | - | - | T | CAPPING |  |
| 150 mg | |||||||||||||||||||
| N°3 | 1.33% | 89.67% | - | 3.00% | 1.00% | - | 5.00% | - | - | - | - | - | - | - | - | - | T + ES | DISGREGATION CORE´S ODT ZONE |  |
| 150 mg | |||||||||||||||||||
| N°4 | 1.33% | 86.67% | - | 3.00% | 1.00% | - | 5.00% | 1.00% | 1.00% | 1.00% | - | - | - | - | - | - | T + P | CAP PING + P FAILURES |  |
| 150 mg | |||||||||||||||||||
| N°5 | 1.33% | - | - | 3.00% | 1.00% | - | 7.50% | 1.00% | 0.33% | 1.00% | 10.00% | 74.84% | - | - | - | - | T + P | FRIABILITY TEST |  |
| 150 mg | |||||||||||||||||||
| N°6 | 1.33% | - | - | 3.00% | 1.00% | - | 7.50% | 1.00% | 0.33% | 1.00% | 24.92% | 49.92% | 10.00% | - | - | - | T | FRIABILITY TEST |  |
| 150 mg | |||||||||||||||||||
| N°7 | 1.33% | - | - | 3.00% | 1.00% | - | 7.50% | 1.00% | 0.33% | 1.00% | 25.00% | 30.00% | 29.84% | - | - | - | T | DISGREGATION TEST |  |
| 150 mg | |||||||||||||||||||
| N°8 | 1.33% | - | - | 3.00% | 1.00% | - | 15.00% | 1.00% | 0.33% | 1.00% | 25.00% | 22.50% | 29.84% | - | - | - | T | DISGREGATION TEST |  |
| 150 mg | |||||||||||||||||||
| N°9 | 1.33% | - | 15.00% | 3.00% | 1.00% | - | - | 1.00% | 0.33% | 1.00% | 15.00% | 33.34% | 29.00% | - | - | - | T | CAPPING + CHIPPING |  |
| 150 mg | |||||||||||||||||||
| N°10 | 1.33% | 44.42% | - | 3.00% | 3.00% | - | - | 1.00% | 0.33% | 1.00% | - | - | - | 44.42% | 1.50% | - | T | DISGREGATION TEST |  |
| 150 mg | |||||||||||||||||||
| N°11 | 1.33% | 71.835% | - | - | 10.00% | - | - | 1.00% | 0.33% | 1.00% | - | - | - | 71.835% | 3.00% | - | % SSGTA | INTERFERENCE HPLC LECTURE OF SACCHARIN SODIUM |  |
| 150 mg | |||||||||||||||||||
| N°12 | 1.33% | 45.835% | - | - | 1.00% | - | - | 1.00% | 0.33% | 1.00% | - | - | - | 45.835% | 5.00% | - | % SSGTA | INTERFERENCE HPLC LECTURE OF SACCHARIN SODIUM |  |
| 150 mg | |||||||||||||||||||
| N°13 | 1.33% | 45.17% | - | - | 1.00% | - | - | - | 0.33% | 1.00% | - | - | - | 45.17% | 5.00% | 1.00% | T | CAPPING |  |
| 150 mg | |||||||||||||||||||
| N°14 | 1.33% | 42.67% | - | - | 1.00% | - | - | - | 0.33% | 1.00% | 5.00% | - | - | 42.67% | 5.00% | 1.00% | T | - |  |
| 150 mg | |||||||||||||||||||
| N°15 | 1.00% | 42.835% | - | - | 1.00% | - | - | - | 0.33% | 1.00% | 5.00% | - | - | 42.835% | 5.00% | 1.00% | T | - |  |
| 200 mg |
| EXCIPIENTS | OH | CH | CO | ALKYL CHAIN | CARBOXYLATE ANION | NH | SO | (H2PO4)- | COO- | AROMATIC GROUP | C-O-C |
|---|---|---|---|---|---|---|---|---|---|---|---|
| MANNITOL | 3200 | 2947 | 1520 | ||||||||
| (Figure 2A) | |||||||||||
| SODIUM CYCLAMATE | 3400 | 1220 | |||||||||
| (Figure 2B) | |||||||||||
 | |||||||||||
| CALCIUM HYDROGEN PHOSPHATE DYHIDRATE | 1040–1100 | ||||||||||
| (Figure 2C) | |||||||||||
 | |||||||||||
| SODIUM STARCH GLYCOLATE | 3270 | 1002 | |||||||||
| (Figure 2D) | |||||||||||
 | |||||||||||
| MAGNESIUM STEARATE | 2916 and 2849 | 1446 and 1570 | 1567–1464 | ||||||||
| (Figure 2E) | |||||||||||
 | |||||||||||
| HYPROMELLOSE | 1900 | 1400 | |||||||||
| (Figure 2F) | |||||||||||
 | |||||||||||
| MENTHOL | 3256.24 | 2872.22 | |||||||||
| (Figure 2G) | |||||||||||
 | |||||||||||
| ANISE EXTRACT | 2346.36 | 2930.29 | |||||||||
| (Figure 2H) | |||||||||||
 |
| Loperamide | Mannitol | Sodium Cyclamate | Emcompress® |
|---|---|---|---|
| - | 3402.8 | 3419.72 | 3736.90 |
| 3235.93 | 3286.08 | 3278.31 | 3275.76 |
| 2959.11 | 3059.19 | 2935.42 | - |
| 2635.49 | 2637.2 | 2854.09 | - |
| 2497.04 | - | 2366.01 | 2368.92 |
| SAMPLES | ORGANOLEPTIC AND PHYSICAL ATTRIBUTES, DIMENSION, THICKNESS | WEIGHT VARIATION | HARDNESS | FRIABILITY | DISSOLUTION | DISGREGATION | DISGREGATION IN VITRO | WATER CONTENT | DIVISIBILITY TEST | CONTENT UNIFORMITY |
|---|---|---|---|---|---|---|---|---|---|---|
| ODT 150 mg | Bright white Grooved Palatable Ø = 8.026 mm T = 2.431 mm (1) | = 150.98 mg | 23.68 Nw | W0 = 6.9047 g Wf = 6.8503 g D = 0.7879% (2) | nearly 100% | Between 9.00–9.57 s | = 14.21 s (3) | W→ = 1.568 g H→ = 1.41% (5) | = 79.94 mg One fractional mass out of 85–115% (94.2 mg) (6) | = 2.05 mg/ODT (7) |
| = 13.92 s (4) | ||||||||||
| ODT 200 mg | Bright white Grooved Palatable Ø = 8.027mm T = 3.029 mm (1) | = 194.93 mg | 24.74 Nw | W0 = 6.8135 g Wf = 6.78 g D = 0.4975% (2) | nearly 100% | Between 9.30–9.63 s | = 13.99 s (3) | W→ = 1.519 g H→ = 1.28% (5) | = 103.43 mg All fractional mass between 85–115% (6) | = 2.07 mg/ODT (7) |
| = 13.83 s (4) |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alejandro, B.; Guillermo, T.; Ángeles, P.M. Formulation and Evaluation of Loperamide HCl Oro Dispersible Tablets. Pharmaceuticals 2020, 13, 100. https://doi.org/10.3390/ph13050100
Alejandro B, Guillermo T, Ángeles PM. Formulation and Evaluation of Loperamide HCl Oro Dispersible Tablets. Pharmaceuticals. 2020; 13(5):100. https://doi.org/10.3390/ph13050100
Chicago/Turabian StyleAlejandro, Blasco, Torrado Guillermo, and Peña M Ángeles. 2020. "Formulation and Evaluation of Loperamide HCl Oro Dispersible Tablets" Pharmaceuticals 13, no. 5: 100. https://doi.org/10.3390/ph13050100
APA StyleAlejandro, B., Guillermo, T., & Ángeles, P. M. (2020). Formulation and Evaluation of Loperamide HCl Oro Dispersible Tablets. Pharmaceuticals, 13(5), 100. https://doi.org/10.3390/ph13050100