Inhibition of SARS-CoV-2 Entry into Host Cells Using Small Molecules

Abstract
1. Introduction
1.1. Classification of Coronaviruses
1.2. General Structure of Coronaviruses
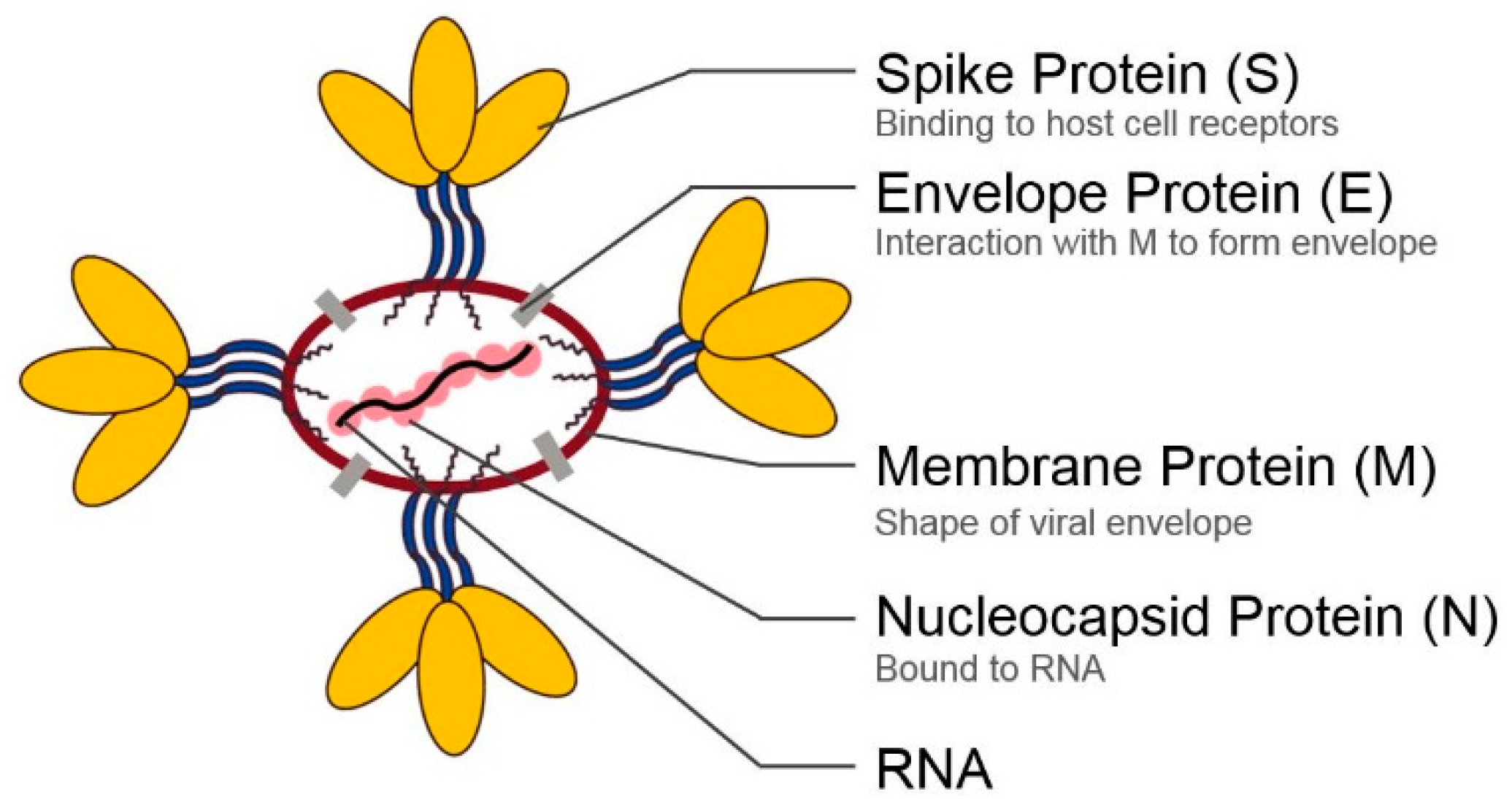
2. Structural Proteins of Coronaviruses
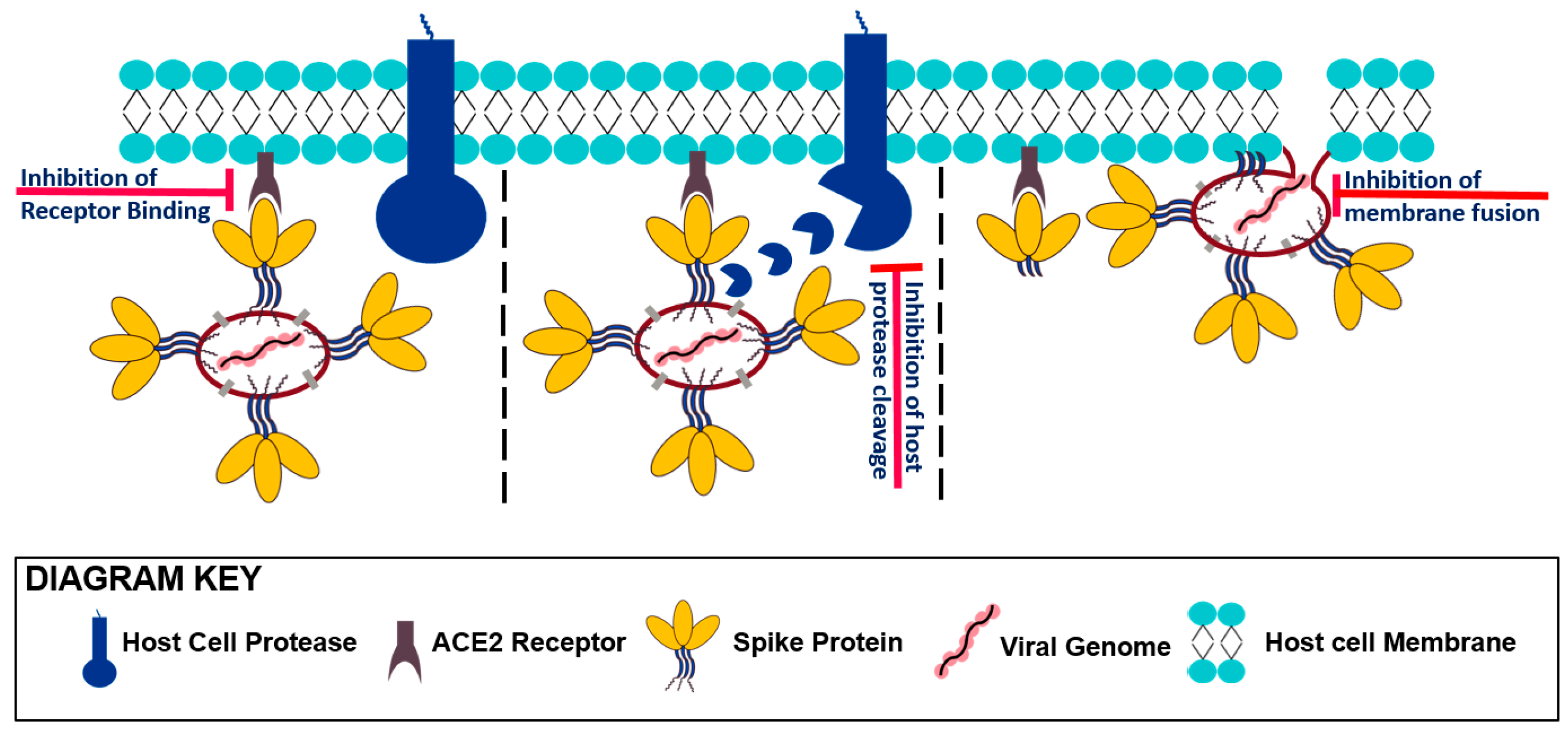
3. Entering Mechanism of SARS-CoV-2 and the Role of Its Spike Protein
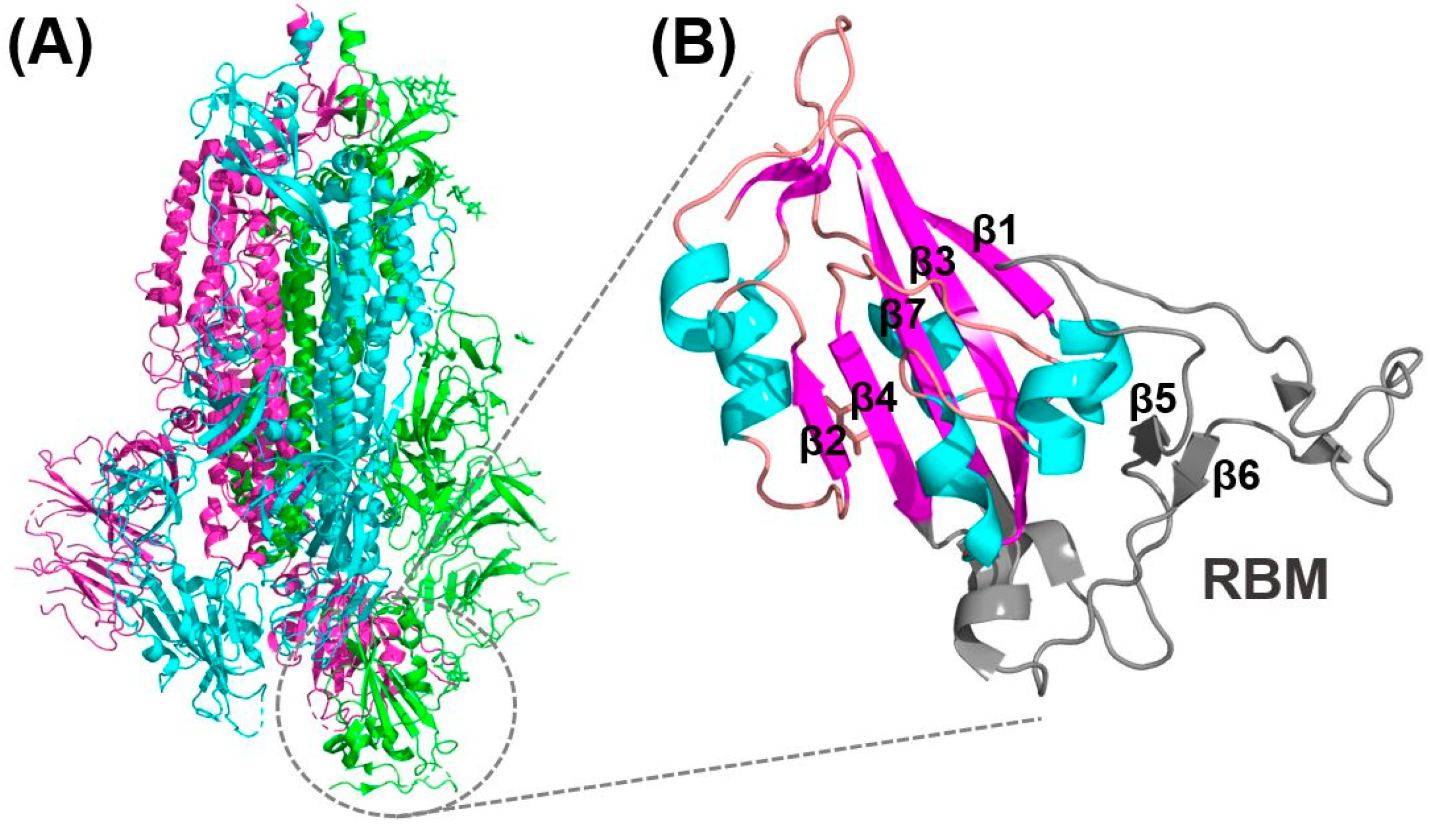
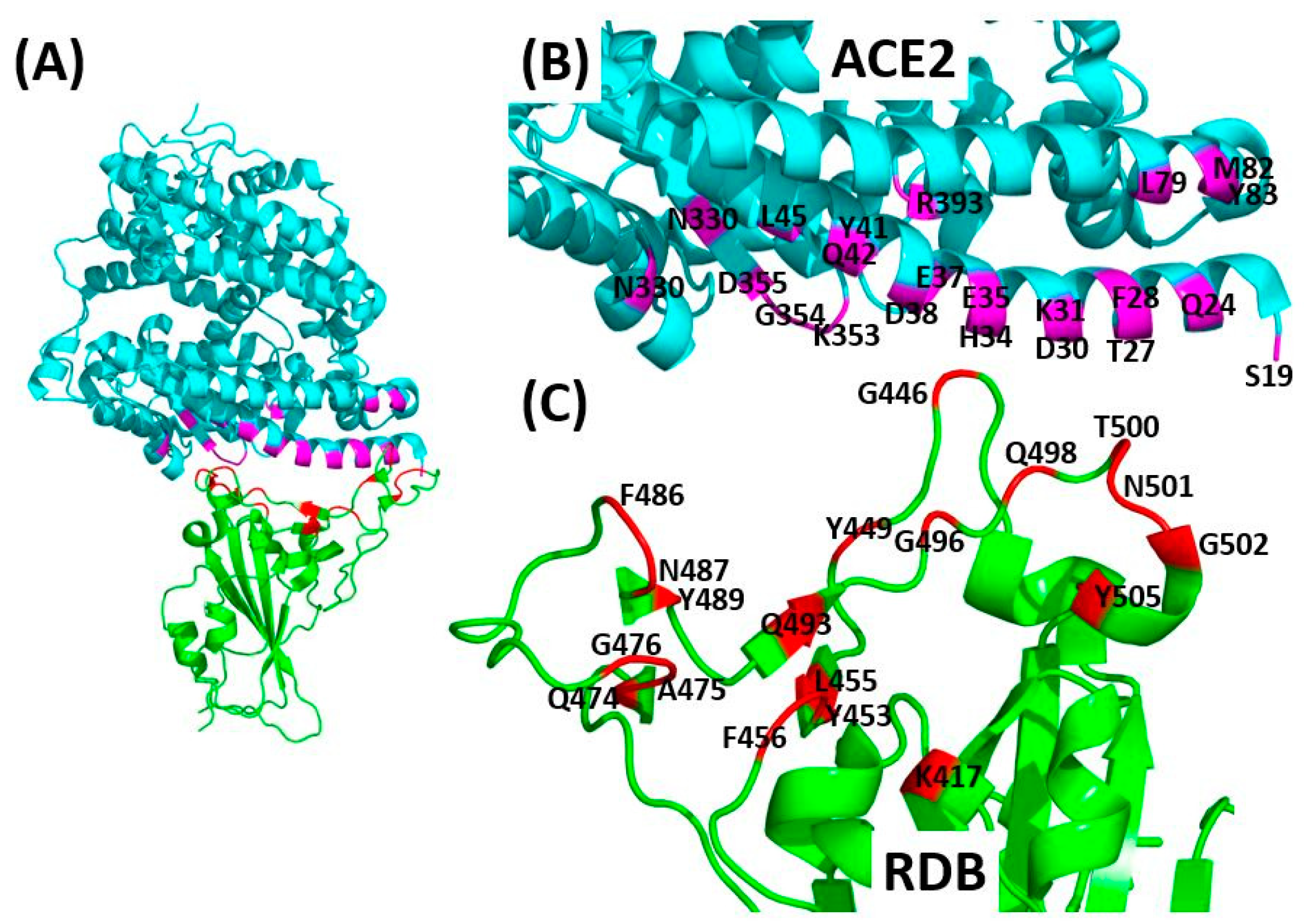
3.1. Receptor Recognition and Binding
3.2. Protease Activation/Proteolytic Processing
3.3. Viral Fusion
4. Small Molecule Inhibitors of SARS-CoV-2 Host Cell Entry
4.1. SARS-CoV-2 Spike Protein as the Drug Target
4.1.1. Small Molecule Inhibitors of Viral Attachment (RBD/ACE2 Interface)
4.1.2. Small Molecule Inhibitors of Viral Fusion (Spike S2 Subdomain)
4.2. Host Cell Receptor and Proteases as the Drug Targets
4.2.1. Protease Small Molecule Inhibitors
4.2.2. ACE2 Small Molecule Inhibitors
5. Conclusions
Funding
Conflicts of Interest
References
- Cascella, M.; Rajnik, M.; Cuomo, A.; Dulebohn, S.C.; Di Napoli, R. Features, Evaluation and Treatment Coronavirus (COVID-19). In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2020. [Google Scholar]
- Fehr, A.R.; Perlman, S. Coronaviruses: An Overview of Their Replication and Pathogenesis. Methods Mol. Biol. 2015, 1282, 1–23. [Google Scholar] [CrossRef]
- Chen, Y.; Liu, Q.; Guo, D. Emerging coronaviruses: Genome structure, replication, and pathogenesis. J. Med. Virol. 2020, 92, 418–423. [Google Scholar] [CrossRef]
- Murray, R.S.; Cai, G.-Y.; Hoel, K.; Zhang, J.-Y.; Soike, K.F.; Cabirac, G.F. Coronavirus infects and causes demyelination in primate central nervous system. Virology 1992, 188, 274–284. [Google Scholar] [CrossRef]
- Bertram, S.; Heurich, A.; Lavender, H.; Gierer, S.; Danisch, S.; Perin, P.; Lucas, J.M.; Nelson, P.S.; Pöhlmann, S.; Soilleux, E.J. Influenza and SARS-Coronavirus Activating Proteases TMPRSS2 and HAT Are Expressed at Multiple Sites in Human Respiratory and Gastrointestinal Tracts. PLoS ONE 2012, 7, e35876. [Google Scholar] [CrossRef]
- Gao, Q.-Y.; Chen, Y.; Fang, J.-Y. 2019 Novel coronavirus infection and gastrointestinal tract. J. Dig. Dis. 2020, 21, 125–126. [Google Scholar] [CrossRef]
- Zhou, J.; Li, C.; Zhao, G.; Chu, H.; Wang, D.; Yan, H.H.-N.; Poon, V.K.-M.; Wen, L.; Wong, B.H.-Y.; Zhao, X.; et al. Human intestinal tract serves as an alternative infection route for Middle East respiratory syndrome coronavirus. Sci. Adv. 2017, 3, eaao4966. [Google Scholar] [CrossRef]
- Bergmann, C.C.; Lane, T.E.; Stohlman, S.A. Coronavirus infection of the central nervous system: Host–virus stand-off. Nat. Rev. Genet. 2006, 4, 121–132. [Google Scholar] [CrossRef]
- Xiao, F.; Tang, M.; Zheng, X.; Liu, Y.; Li, X.; Shan, H. Evidence for Gastrointestinal Infection of SARS-CoV-2. Gastroenterology 2020, 158, 1831–1833. [Google Scholar] [CrossRef]
- Neuman, B.W.; Adair, B.D.; Yoshioka, C.; Quispe, J.D.; Orca, G.; Kuhn, P.; Milligan, R.A.; Yeager, M.; Buchmeier, M.J. Supramolecular Architecture of Severe Acute Respiratory Syndrome Coronavirus Revealed by Electron Cryomicroscopy. J. Virol. 2006, 80, 7918–7928. [Google Scholar] [CrossRef]
- Bárcena, M.; Oostergetel, G.T.; Bartelink, W.; Faas, F.G.A.; Verkleij, A.; Rottier, P.J.M.; Koster, A.J.; Bosch, B.J. Cryo-electron tomography of mouse hepatitis virus: Insights into the structure of the coronavirion. Proc. Natl. Acad. Sci. USA 2009, 106, 582–587. [Google Scholar] [CrossRef]
- Kamitani, W.; Huang, C.; Narayanan, K.R.; Lokugamage, K.G.; Makino, S. A two-pronged strategy to suppress host protein synthesis by SARS coronavirus Nsp1 protein. Nat. Struct. Mol. Biol. 2009, 16, 1134–1140. [Google Scholar] [CrossRef]
- Kamitani, W.; Narayanan, K.; Huang, C.; Lokugamage, K.; Ikegami, T.; Ito, N.; Kubo, H.; Makino, S. Severe acute respiratory syndrome coronavirus nsp1 protein suppresses host gene expression by promoting host mRNA degradation. Proc. Natl. Acad. Sci. USA 2006, 103, 12885–12890. [Google Scholar] [CrossRef]
- Clementz, M.A.; Kanjanahaluethai, A.; O’Brien, T.E.; Baker, S.C. Mutation in murine coronavirus replication protein nsp4 alters assembly of double membrane vesicles. Virology 2008, 375, 118–129. [Google Scholar] [CrossRef]
- Tanaka, T.; Kamitani, W.; DeDiego, M.L.; Enjuanes, L.; Matsuura, Y. Severe Acute Respiratory Syndrome Coronavirus nsp1 Facilitates Efficient Propagation in Cells through a Specific Translational Shutoff of Host mRNA. J. Virol. 2012, 86, 11128–11137. [Google Scholar] [CrossRef]
- Snijder, E.; Decroly, E.; Ziebuhr, J. The Nonstructural Proteins Directing Coronavirus RNA Synthesis and Processing. Adv. Appl. Microbiol. 2016, 96, 59–126. [Google Scholar] [CrossRef]
- Neuman, B.W.; Kiss, G.; Kunding, A.H.; Bhella, D.; Baksh, M.F.; Connelly, S.; Droese, B.; Klaus, J.P.; Makino, S.; Sawicki, S.G.; et al. A structural analysis of M protein in coronavirus assembly and morphology. J. Struct. Biol. 2011, 174, 11–22. [Google Scholar] [CrossRef]
- Nieto-Torres, J.L.; DeDiego, M.L.; Verdiá-Báguena, C.; Jimenez-Guardeño, J.M.; Regla-Nava, J.A.; Fernandez-Delgado, R.; Castaño-Rodriguez, C.; Alcaraz, A.; Torres, J.; Aguilella, V.M.; et al. Severe Acute Respiratory Syndrome Coronavirus Envelope Protein Ion Channel Activity Promotes Virus Fitness and Pathogenesis. PLoS Pathog. 2014, 10, e1004077. [Google Scholar] [CrossRef]
- DeDiego, M.L.; Álvarez, E.; Almazán, F.; Rejas, M.T.; Lamirande, E.; Roberts, A.; Shieh, W.-J.; Zaki, S.R.; Subbarao, K.; Enjuanes, L. A Severe Acute Respiratory Syndrome Coronavirus That Lacks the E Gene Is Attenuated In Vitro and In Vivo. J. Virol. 2006, 81, 1701–1713. [Google Scholar] [CrossRef]
- Beniac, D.R.; Andonov, A.; Grudeski, E.; Booth, T.F. Architecture of the SARS coronavirus prefusion spike. Nat. Struct. Mol. Biol. 2006, 13, 751–752. [Google Scholar] [CrossRef]
- Liu, D.X.; Fung, T.S.; Chong, K.K.-L.; Shukla, A.; Hilgenfeld, R. Accessory proteins of SARS-CoV and other coronaviruses. Antivir. Res. 2014, 109, 97–109. [Google Scholar] [CrossRef]
- Zhao, L.; Jha, B.K.; Wu, A.; Elliott, R.; Ziebuhr, J.; Gorbalenya, A.E.; Silverman, R.H.; Weiss, S.R. Antagonism of the Interferon-Induced OAS-RNase L Pathway by Murine Coronavirus ns2 Protein Is Required for Virus Replication and Liver Pathology. Cell Host Microbe 2012, 11, 607–616. [Google Scholar] [CrossRef] [PubMed]
- Cui, L.; Wang, H.; Ji, Y.; Yang, J.; Xu, S.; Huang, X.; Wang, Z.; Qin, L.; Tien, P.; Zhou, X.; et al. The Nucleocapsid Protein of Coronaviruses Acts as a Viral Suppressor of RNA Silencing in Mammalian Cells. J. Virol. 2015, 89, 9029–9043. [Google Scholar] [CrossRef] [PubMed]
- Molenkamp, R.; Spaan, W.J. Identification of a Specific Interaction between the Coronavirus Mouse Hepatitis Virus A59 Nucleocapsid Protein and Packaging Signal. Virology 1997, 239, 78–86. [Google Scholar] [CrossRef]
- Stohlman, S.A.; Baric, R.S.; Nelson, G.N.; Soe, L.H.; Welter, L.M.; Deans, R.J. Specific interaction between coronavirus leader RNA and nucleocapsid protein. J. Virol. 1988, 62, 4288–4295. [Google Scholar] [CrossRef] [PubMed]
- Stohlman, S.A.; Lai, M.M. Phosphoproteins of murine hepatitis viruses. J. Virol. 1979, 32, 672–675. [Google Scholar] [CrossRef]
- McBride, R.; Van Zyl, M.; Fielding, B. The Coronavirus Nucleocapsid Is a Multifunctional Protein. Viruses 2014, 6, 2991–3018. [Google Scholar] [CrossRef]
- Nal, B.; Chan, C.; Kien, F.; Siu, L.; Tse, J.; Chu, K.; Kam, J.; Staropoli, I.; Crescenzo-Chaigne, B.; Escriou, N.; et al. Differential maturation and subcellular localization of severe acute respiratory syndrome coronavirus surface proteins S, M and E. J. Gen. Virol. 2005, 86, 1423–1434. [Google Scholar] [CrossRef]
- Hurst, K.R.; Koetzner, C.A.; Masters, P.S. Identification of In Vivo-Interacting Domains of the Murine Coronavirus Nucleocapsid Protein. J. Virol. 2009, 83, 7221–7234. [Google Scholar] [CrossRef]
- Arndt, A.L.; Larson, B.J.; Hogue, B.G. A Conserved Domain in the Coronavirus Membrane Protein Tail Is Important for Virus Assembly. J. Virol. 2010, 84, 11418–11428. [Google Scholar] [CrossRef]
- Chen, Y.; Guo, Y.; Pan, Y.; Zhao, Z.J. Structure analysis of the receptor binding of 2019-nCoV. Biochem. Biophys. Res. Commun. 2020, 525, 135–140. [Google Scholar] [CrossRef]
- Ortega, J.T.; Serrano, M.L.; Pujol, F.H.; Rangel, H.R. Role of changes in SARS-CoV-2 spike protein in the interaction with the human ACE2 receptor: An in silico analysis. EXCLI J. 2020, 19, 410–417. [Google Scholar] [PubMed]
- Qamar, M.T.U.; Alqahtani, S.M.; Alamri, M.A.; Chen, L.-L. Structural basis of SARS-CoV-2 3CLpro and anti-COVID-19 drug discovery from medicinal plants. J. Pharm. Anal. 2020, 10, 313–319. [Google Scholar] [CrossRef] [PubMed]
- Zhou, P.; Yang, X.-L.; Wang, X.-G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.-R.; Zhu, Y.; Li, B.; Huang, C.-L.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Krüger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.-H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280. [Google Scholar] [CrossRef] [PubMed]
- Shang, J.; Wan, Y.; Luo, C.; Ye, G.; Geng, Q.; Auerbach, A.; Li, F. Cell entry mechanisms of SARS-CoV-2. Proc. Natl. Acad. Sci. USA 2020, 117, 11727–11734. [Google Scholar] [CrossRef] [PubMed]
- Yan, R.; Zhang, Y.; Li, Y.; Xia, L.; Guo, Y.; Zhou, Q. Structural basis for the recognition of SARS-CoV-2 by full-length human ACE2. Science 2020, 367, 1444–1448. [Google Scholar] [CrossRef]
- Wan, Y.; Shang, J.; Graham, R.; Baric, R.S.; Li, F. Receptor Recognition by the Novel Coronavirus from Wuhan: An Analysis Based on Decade-Long Structural Studies of SARS Coronavirus. J. Virol. 2020, 94, 127. [Google Scholar] [CrossRef]
- Walls, A.C.; Park, Y.-J.; Tortorici, M.A.; Wall, A.; McGuire, A.T.; Veesler, D. Structure, Function, and Antigenicity of the SARS-CoV-2 Spike Glycoprotein. Cell 2020, 181, 281–292. [Google Scholar] [CrossRef]
- Letko, M.; Marzi, A.; Munster, V.J. Functional assessment of cell entry and receptor usage for SARS-CoV-2 and other lineage B betacoronaviruses. Nat. Microbiol. 2020, 5, 562–569. [Google Scholar] [CrossRef]
- Wang, Q.; Zhang, Y.; Wu, L.; Niu, S.; Song, C.; Zhang, Z.; Lu, G.; Qiao, C.; Hu, Y.; Yuen, K.-Y.; et al. Structural and Functional Basis of SARS-CoV-2 Entry by Using Human ACE2. Cell 2020, 181, 894–904. [Google Scholar] [CrossRef]
- Shang, J.; Ye, G.; Shi, K.; Wan, Y.; Luo, C.; Aihara, H.; Geng, Q.; Auerbach, A.; Li, F. Structural basis of receptor recognition by SARS-CoV-2. Nat. Cell Biol. 2020, 581, 221–224. [Google Scholar] [CrossRef]
- Lan, J.; Ge, J.; Yu, J.; Shan, S.; Zhou, H.; Fan, S.; Zhang, Q.; Shi, X.; Wang, Q.; Zhang, L.; et al. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nat. Cell Biol. 2020, 581, 215–220. [Google Scholar] [CrossRef]
- Madeira, F.; Park, Y.M.; Lee, J.; Buso, N.; Gur, T.; Madhusoodanan, N.; Basutkar, P.; Tivey, A.R.N.; Potter, S.C.; Finn, R.D.; et al. The EMBL-EBI search and sequence analysis tools APIs in 2019. Nucleic Acids Res. 2019, 47, W636–W641. [Google Scholar] [CrossRef]
- Wrapp, D.; Wang, N.; Corbett, K.S.; Goldsmith, J.A.; Hsieh, C.-L.; Abiona, O.; Graham, B.S.; McLellan, J.S. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science 2020, 367, 1260–1263. [Google Scholar] [CrossRef]
- Coutard, B.; Valle, C.; De Lamballerie, X.; Canard, B.; Seidah, N.; Decroly, E. The spike glycoprotein of the new coronavirus 2019-nCoV contains a furin-like cleavage site absent in CoV of the same clade. Antivir. Res. 2020, 176, 104742. [Google Scholar] [CrossRef]
- Zang, R.; Castro, M.F.G.; McCune, B.T.; Zeng, Q.; Rothlauf, P.W.; Sonnek, N.M.; Liu, Z.; Brulois, K.F.; Wang, X.; Greenberg, H.B.; et al. TMPRSS2 and TMPRSS4 promote SARS-CoV-2 infection of human small intestinal enterocytes. Sci. Immunol. 2020, 5, eabc3582. [Google Scholar] [CrossRef]
- Jaimes, J.A.; Millet, J.K.; Whittaker, G.R. Proteolytic Cleavage of the SARS-CoV-2 Spike Protein and the Role of the Novel S1/S2 Site. Iscience 2020, 23, 101212. [Google Scholar] [CrossRef]
- Xiu, S.; Dick, A.; Ju, H.; Mirzaie, S.; Abdi, F.; Cocklin, S.; Zhan, P.; Liu, X. Inhibitors of SARS-CoV-2 Entry: Current and Future Opportunities. J. Med. Chem. 2020. [Google Scholar] [CrossRef]
- Romeo, A.; Iacovelli, F.; Falconi, M. Targeting the SARS-CoV-2 spike glycoprotein prefusion conformation: Virtual screening and molecular dynamics simulations applied to the identification of potential fusion inhibitors. Virus Res. 2020, 286, 198068. [Google Scholar] [CrossRef] [PubMed]
- Young, L.M.; Ashcroft, A.E.; Radford, S.E. Small molecule probes of protein aggregation. Curr. Opin. Chem. Biol. 2017, 39, 90–99. [Google Scholar] [CrossRef]
- Shen, Z.; Lou, K.; Wang, W. New small-molecule drug design strategies for fighting resistant influenza A. Acta Pharm. Sin. B 2015, 5, 419–430. [Google Scholar] [CrossRef]
- Litterman, N.; Lipinski, C.A.; Ekins, S. Small molecules with antiviral activity against the Ebola virus. F1000Research 2015, 4, 38. [Google Scholar] [CrossRef]
- Yang, L.; Lu, M. Small Molecule Inhibitors of Hepatitis B Virus Nucleocapsid Assembly: A New Approach to Treat Chronic HBV Infection. Curr. Med. Chem. 2018, 25, 802–813. [Google Scholar] [CrossRef]
- Wang, L.; Liang, R.; Gao, Y.; Li, Y.; Deng, X.; Xiang, R.; Zhang, Y.; Ying, T.; Jiang, S.; Yu, F. Development of Small-Molecule Inhibitors Against Zika Virus Infection. Front. Microbiol. 2019, 10, 2725. [Google Scholar] [CrossRef]
- Rumlová, M.; Ruml, T. In vitro methods for testing antiviral drugs. Biotechnol. Adv. 2018, 36, 557–576. [Google Scholar] [CrossRef]
- Wang, X.; Cao, R.; Zhang, H.; Liu, J.; Xu, M.; Hu, H.; Li, Y.; Zhao, L.; Li, W.; Sun, X.; et al. The anti-influenza virus drug, arbidol is an efficient inhibitor of SARS-CoV-2 in vitro. Cell Discov. 2020, 6, 1–5. [Google Scholar] [CrossRef]
- Vankadari, N. Arbidol: A potential antiviral drug for the treatment of SARS-CoV-2 by blocking trimerization of the spike glycoprotein. Int. J. Antimicrob. Agents 2020, 56, 105998. [Google Scholar] [CrossRef]
- Fan, H.-H.; Wang, L.-Q.; Liu, W.-L.; An, X.-P.; Liu, Z.-D.; He, X.-Q.; Song, L.-H.; Tong, Y.-G. Repurposing of clinically approved drugs for treatment of coronavirus disease 2019 in a 2019-novel coronavirus-related coronavirus model. Chin. Med. J. 2020, 133, 1051–1056. [Google Scholar] [CrossRef]
- Hu, T.; Frieman, M.; Wolfram, J. Insights from nanomedicine into chloroquine efficacy against COVID-19. Nat. Nanotechnol. 2020, 15, 247–249. [Google Scholar] [CrossRef]
- Wang, M.; Cao, R.; Zhang, L.; Yang, X.; Liu, J.; Xu, M.; Shi, Z.; Hu, Z.; Zhong, W.; Xiao, G. Remdesivir and chloroquine effectively inhibit the recently emerged novel coronavirus (2019-nCoV) in vitro. Cell Res. 2020, 30, 269–271. [Google Scholar] [CrossRef]
- Maisonnasse, P.; Guedj, J.; Contreras, V.; Behillil, S.; Solas, C.; Marlin, R.; Naninck, T.; Pizzorno, A.; Lemaitre, J.; Gonçalves, A.; et al. Hydroxychloroquine use against SARS-CoV-2 infection in non-human primates. Nat. Cell Biol. 2020, 1–8. [Google Scholar] [CrossRef]
- Wu, C.; Liu, Y.; Yang, Y.; Zhang, P.; Zhong, W.; Wang, Y.; Wang, Q.; Xu, Y.; Li, M.; Li, X.; et al. Analysis of therapeutic targets for SARS-CoV-2 and discovery of potential drugs by computational methods. Acta Pharm. Sin. B 2020, 10, 766–788. [Google Scholar] [CrossRef]
- Mahdian, S.; Ebrahim-Habibi, A.; Zarrabi, M. Drug repurposing using computational methods to identify therapeutic options for COVID-19. J. Diabetes Metab. Disord. 2020, 1–9. [Google Scholar] [CrossRef]
- Wahedi, H.M.; Ahmad, S.; Abbasi, S.W. Stilbene-based natural compounds as promising drug candidates against COVID-19. J. Biomol. Struct. Dyn. 2020, 1–10. [Google Scholar] [CrossRef]
- Kalathiya, U.; Padariya, M.; Mayordomo, M.; Lisowska, M.; Nicholson, J.; Singh, A.; Baginski, M.; Fåhraeus, R.; Carragher, N.; Ball, K.L.; et al. Highly Conserved Homotrimer Cavity Formed by the SARS-CoV-2 Spike Glycoprotein: A Novel Binding Site. J. Clin. Med. 2020, 9, 1473. [Google Scholar] [CrossRef]
- Hoffmann, M.; Schroeder, S.; Kleine-Weber, H.; Müller, M.A.; Drosten, C.; Pöhlmann, S. Nafamostat Mesylate Blocks Activation of SARS-CoV-2: New Treatment Option for COVID-19. Antimicrob. Agents Chemother. 2020, 64, 754. [Google Scholar] [CrossRef]
- Asakura, H.; Ogawa, H. Potential of heparin and nafamostat combination therapy for COVID-19. J. Thromb. Haemost. 2020, 18, 1521–1522. [Google Scholar] [CrossRef]
- Cheng, Y.-W.; Chao, T.-L.; Li, C.-L.; Chiu, M.-F.; Kao, H.-C.; Wang, S.-H.; Pang, Y.-H.; Lin, C.-H.; Tsai, Y.-M.; Lee, W.-H.; et al. Furin Inhibitors Block SARS-CoV-2 Spike Protein Cleavage to Suppress Virus Production and Cytopathic Effects. SSRN Electron. J. 2020. [Google Scholar] [CrossRef]
- Liu, T.; Luo, S.; Libby, P.; Shi, G.-P. Cathepsin L-selective inhibitors: A potentially promising treatment for COVID-19 patients. Pharmacol. Ther. 2020, 213, 107587. [Google Scholar] [CrossRef]
- Shah, P.P.; Wang, T.; Kaletsky, R.L.; Myers, M.C.; Purvis, J.E.; Jing, H.; Huryn, D.M.; Greenbaum, D.C.; Smith, A.B.; Bates, P.; et al. A Small-Molecule Oxocarbazate Inhibitor of Human Cathepsin L Blocks Severe Acute Respiratory Syndrome and Ebola Pseudotype Virus Infection into Human Embryonic Kidney 293T cells. Mol. Pharmacol. 2010, 78, 319–324. [Google Scholar] [CrossRef]
- Huentelman, M.J.; Zubcevic, J.; Prada, J.A.H.; Xiao, X.; Dimitrov, D.S.; Raizada, M.K.; Ostrov, D.A. Structure-Based Discovery of a Novel Angiotensin-Converting Enzyme 2 Inhibitor. Hypertension 2004, 44, 903–906. [Google Scholar] [CrossRef] [PubMed]
- Towler, P.; Staker, B.; Prasad, S.G.; Menon, S.; Tang, J.; Parsons, T.; Ryan, D.; Fisher, M.; Williams, D.; Dales, N.A.; et al. ACE2 X-Ray Structures Reveal a Large Hinge-bending Motion Important for Inhibitor Binding and Catalysis. J. Biol. Chem. 2004, 279, 17996–18007. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, S.; Yoshiya, T.; Yoshizawa-Kumagaye, K.; Sugiyama, T. Nicotianamine is a novel angiotensin-converting enzyme 2 inhibitor in soybean. Biomed. Res. 2015, 36, 219–224. [Google Scholar] [CrossRef] [PubMed]
- Teralı, K.; Baddal, B.; Gülcan, H.O. Prioritizing potential ACE2 inhibitors in the COVID-19 pandemic: Insights from a molecular mechanics-assisted structure-based virtual screening experiment. J. Mol. Graph. Model. 2020, 100, 107697. [Google Scholar] [CrossRef] [PubMed]
- Benítez-Cardoza, C.G.; Vique-Sánchez, J.L. Potential inhibitors of the interaction between ACE2 and SARS-CoV-2 (RBD), to develop a drug. Life Sci. 2020, 256, 117970. [Google Scholar] [CrossRef]
- Ho, T.-Y.; Wu, S.-L.; Chen, J.-C.; Li, C.-C.; Hsiang, C.-Y. Emodin blocks the SARS coronavirus spike protein and angiotensin-converting enzyme 2 interaction. Antivir. Res. 2007, 74, 92–101. [Google Scholar] [CrossRef]
- Adedeji, A.O.; Severson, W.; Jonsson, C.; Singh, K.; Weiss, S.R.; Sarafianos, S.G. Novel Inhibitors of Severe Acute Respiratory Syndrome Coronavirus Entry That Act by Three Distinct Mechanisms. J. Virol. 2013, 87, 8017–8028. [Google Scholar] [CrossRef]
- Yang, C.; Ke, C.; Yue, D.; Li, W.; Hu, Z.; Liu, W.; Hu, S.; Wang, S.; Liu, J. Effectiveness of Arbidol for COVID-19 Prevention in Health Professionals. Front. Public Health 2020, 8, 249. [Google Scholar] [CrossRef]
- Yao, X.; Ye, F.; Zhang, M.; Cui, C.; Huang, B.; Niu, P.; Liu, X.; Zhao, L.; Dong, E.; Song, C.; et al. In Vitro Antiviral Activity and Projection of Optimized Dosing Design of Hydroxychloroquine for the Treatment of Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2). Clin. Infect. Dis. 2020, 71, 732–739. [Google Scholar] [CrossRef]
- Yu, B.; Li, C.; Chen, P.; Zhou, N.; Wang, L.; Li, J.; Jiang, H.; Wang, D.W. Low dose of hydroxychloroquine reduces fatality of critically ill patients with COVID-19. Sci. China Life Sci. 2020, 63, 1–7. [Google Scholar] [CrossRef]
- Gautret, P.; Lagier, J.C.; Parola, P.; Hoang, V.T.; Meddeb, L.; Mailhe, M.; Doudier, B.; Courjon, J.; Giordanengo, V.; Vieira, V.E.; et al. Hydroxychloroquine and azithromycin as a treatment of COVID-19: Results of an open-label non-randomized clinical trial. Int. J. Antimicrob. Agents 2020, 56, 105949. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, M.; Mösbauer, K.; Hofmann-Winkler, H.; Kaul, A.; Kleine-Weber, H.; Krüger, N.; Gassen, N.C.; Müller, M.A.; Drosten, C.; Pöhlmann, S. Chloroquine does not inhibit infection of human lung cells with SARS-CoV-2. Nat. Cell Biol. 2020, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Abella, B.S.; Jolkovsky, E.L.; Biney, B.T.; Uspal, J.E.; Hyman, M.C.; Frank, I.; Hensley, S.E.; Gill, S.; Vogl, D.T.; Maillard, I.; et al. Efficacy and Safety of Hydroxychloroquine vs Placebo for Pre-exposure SARS-CoV-2 Prophylaxis Among Health Care Workers. JAMA Intern. Med. 2020. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.; Cao, Z.; Han, M.; Wang, Z.; Chen, J.; Sun, W.; Wu, Y.; Xiao, W.; Liu, S.; Chen, E.; et al. Hydroxychloroquine in patients with mainly mild to moderate coronavirus disease 2019: Open label, randomised controlled trial. BMJ 2020, 369, m1849. [Google Scholar] [CrossRef] [PubMed]
- Boulware, D.R.; Pullen, M.F.; Bangdiwala, A.S.; Pastick, K.A.; Lofgren, S.M.; Okafor, E.C.; Skipper, C.P.; Nascene, A.A.; Nicol, M.R.; Abassi, M.; et al. A Randomized Trial of Hydroxychloroquine as Postexposure Prophylaxis for Covid-19. N. Engl. J. Med. 2020, 383, 517–525. [Google Scholar] [CrossRef] [PubMed]
- Geleris, J.; Sun, Y.; Platt, J.; Zucker, J.; Baldwin, M.; Hripcsak, G.; Labella, A.; Manson, D.K.; Kubin, C.; Barr, R.G.; et al. Observational Study of Hydroxychloroquine in Hospitalized Patients with Covid-19. N. Engl. J. Med. 2020, 382, 2411–2418. [Google Scholar] [CrossRef]
- Magagnoli, J.; Narendran, S.; Pereira, F.; Cummings, T.H.; Hardin, J.W.; Sutton, S.S.; Ambati, J. Outcomes of Hydroxychloroquine Usage in United States Veterans Hospitalized with COVID-19. Med 2020. [Google Scholar] [CrossRef]
- Rosenberg, E.S.; Dufort, E.M.; Udo, T.; Wilberschied, L.A.; Kumar, J.; Tesoriero, J.; Weinberg, P.; Kirkwood, J.; Muse, A.; DeHovitz, J.; et al. Association of Treatment with Hydroxychloroquine or Azithromycin with In-Hospital Mortality in Patients With COVID-19 in New York State. JAMA 2020, 323, 2493. [Google Scholar] [CrossRef]
- Molina, J.; Delaugerre, C.; Le Goff, J.; Mela-Lima, B.; Ponscarme, D.; Goldwirt, L.; De Castro, N. No evidence of rapid antiviral clearance or clinical benefit with the combination of hydroxychloroquine and azithromycin in patients with severe COVID-19 infection. Médecine et Maladies Infectieuses 2020, 50, 384. [Google Scholar] [CrossRef] [PubMed]
- Imran, M.; Saleemi, M.K.; Chen, Z.; Wang, X.; Zhou, D.; Li, Y.; Zhao, Z.; Zheng, B.; Li, Q.; Cao, S.; et al. Decanoyl-Arg-Val-Lys-Arg-Chloromethylketone: An Antiviral Compound That Acts against Flaviviruses through the Inhibition of Furin-Mediated prM Cleavage. Viruses 2019, 11, 1011. [Google Scholar] [CrossRef] [PubMed]
- Kawase, M.; Shirato, K.; Van Der Hoek, L.; Taguchi, F.; Matsuyama, S. Simultaneous Treatment of Human Bronchial Epithelial Cells with Serine and Cysteine Protease Inhibitors Prevents Severe Acute Respiratory Syndrome Coronavirus Entry. J. Virol. 2012, 86, 6537–6545. [Google Scholar] [CrossRef] [PubMed]
- Zisman, L.S.; Keller, R.S.; Weaver, B.; Lin, Q.; Speth, R.; Bristow, M.R.; Canver, C.C. Increased Angiotensin-(1-7)–Forming Activity in Failing Human Heart Ventricles. Circulation 2003, 108, 1707–1712. [Google Scholar] [CrossRef]
- Fang, L.; Karakiulakis, G.; Roth, M. Are patients with hypertension and diabetes mellitus at increased risk for COVID-19 infection? Lancet Respir. Med. 2020, 8, e21. [Google Scholar] [CrossRef]
- Ferrario, C.M.; Jessup, J.; Chappell, M.C.; Averill, D.B.; Brosnihan, K.B.; Tallant, E.A.; Diz, D.I.; Gallagher, P.E. Effect of Angiotensin-Converting Enzyme Inhibition and Angiotensin II Receptor Blockers on Cardiac Angiotensin-Converting Enzyme 2. Circulation 2005, 111, 2605–2610. [Google Scholar] [CrossRef]
- Sarzani, R.; Giulietti, F.; Di Pentima, C.; Giordano, P.; Spannella, F. Severe acute respiratory syndrome coronavirus 2 infection, angiotensin-converting enzyme 2 and treatment with angiotensin-converting enzyme inhibitors or angiotensin II type 1 receptor blockers. Eur. J. Prev. Cardiol. 2020. [Google Scholar] [CrossRef]
- Verdecchia, P.; Cavallini, C.; Spanevello, A.; Angeli, F. The pivotal link between ACE2 deficiency and SARS-CoV-2 infection. Eur. J. Intern. Med. 2020, 76, 14–20. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| SARS CoV-2-RBD Residues | K417 | G446 | Y449 | Y453 | L455 | F456 | Q474 | A475 | G476 | F486 | N487 | Y489 | Q493 | G496 | Q498 | T500 | N501 | G502 | Y505 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| PDB: 6M17 [37] | |||||||||||||||||||
| ACE2 residues | D30 | - | - | H34 | - | - | Q24 | - | - | - | - | - | - | - | Q42 | R357 | Y41 K353 R357 | - | - |
| PDB: 6VW1 [42] | |||||||||||||||||||
| ACE2 residues | - | - | D38 | H34 | H34 | T27 K31 | - | S19 T27 | Q24 | L79 M82 Y83 | Q24 Y83 | T27 F28 | K31 E35 | K353 | Y41 Q42 L45 | Y41 N330 D355 R357 | Y41 K353 | K353 G354 | E37 K353 G354 |
| PDB: 6M0J [43] | |||||||||||||||||||
| ACE2 residues | D30 | Q42 | D38 Q42 | - | - | - | - | - | - | - | Q24 Y83 | Y83 | E35 | - | - | Y41 | Y41 | K353 | E37 R393 |
| PDB: 6LZG [41] | |||||||||||||||||||
| ACE2 residues | D30 | Q42 | D38 Q42 | H34 | - | - | - | S19 | - | - | Q24 Y83 | - | - | K353 | Q42 | Y41 D355 | Y41 | K353 | - |
| Small Molecule Name | Structure | Type | Target | IC50/EC50 | Ref. |
|---|---|---|---|---|---|
| Arbidol |  | Anti-influenza virus drug | Viral attachment and entry and targeting the trimerization of SARS-CoV-2 spike | 4.11 μM | [57,58] |
| Cepharanthine (CEP) |  | A naturally occurring anti-inflammatory and anti-neoplastic alkaloid drug | Inhibits entry and post-entry of a SARS-CoV-2 homologous viral model (92% homology) | 0.98 μM | [59] |
| Chloroquine |  | Approved malaria drug | Viral fusion through endocytosis | 1.13 μM | [60,61] |
| Hydroxychloroquine |  | Approved malaria drug | Viral fusion through endocytosis | 0.72 μM | [62] |
| Small Molecule Name | Structure | Type | Target | Docking Score | Ref. |
|---|---|---|---|---|---|
| Hesperidin |  | Bioflavonoid | Spike RBD and ACE2 | −20.63 kcal/mol (RBD) −3.12 kcal/mol (ACE2) | [63,64] |
| Rescinnamine |  | Anti-hypertensive drug | Spike protein (not RBD) | −35.31 kcal/mol | [63] |
| Iloprost |  | Anti-hypertensive drug | Spike protein (not RBD) | −26.19 kcal/mol | [63] |
| Prazosin |  | Anti-hypertensive drug | Spike protein (not RBD) | −19.78 kcal/mol | [63] |
| Posaconazole |  | Antifungal drug | Spike protein (not RBD) | −33.04 kcal/mol | [63] |
| Itraconazole |  | Antifungal drug | Spike protein (not RBD) | −31.03 kcal/mol | [63] |
| Sulfasalazine |  | Antibacterial drug | Spike protein (not RBD) | −22.21 kcal/mol | [63] |
| Azlocillin |  | Antibacterial drug | Spike protein (not RBD) | −36.64 kcal/mol | [63] |
| Penicillin |  | Antibacterial drug | Spike protein (not RBD) | −35.85 kcal/mol | [63] |
| Cefsulodin |  | Antibacterial drug | Spike protein (not RBD) | −35.25 kcal/mol | [63] |
| Dabigatran etexilate |  | Anticoagulant drug | Spike protein (not RBD) | −27.41 kcal/mol | [63] |
| Licoflavonol |  | Natural flavonoid | Spike protein (not RBD) | −12.67 kcal/mol | [63] |
| Cosmosiin |  | Natural flavonoid | Spike protein (not RBD) | −17.54 kcal/mol | [63] |
| Neohesperidin |  | Natural flavonoid | Spike protein (not RBD) | −24.75 kcal/mol | [63] |
| Mangostin |  | Natural flavonoid | Spike protein (not RBD) | −22.77 kcal/mol | [63] |
| Kouitchenside D |  | Natural flavonoid | Spike protein (not RBD) | −9.75 kcal/mol | [63] |
| Excoecariatoxin |  | Natural flavonoid | Spike protein (not RBD) | −17.8 kcal/mol | [63] |
| Phyllaemblicin G7 |  | Natural flavonoid | Spike protein (not RBD) | −12.07 kcal/mol (Spike) | [63] |
| Piceatannol |  | Natural flavonoid | Spike protein (both RBD and non-RBD] | −27.58 kcal/mol | [63,65] |
| Resveratrol |  | Natural polyphenol | ACE2-RBD complex (PDB: 6VW1) | −8.0 kcal/mol | [65] |
| 31h-phthalocyanine |  | Investigational anticancer and antiviral drug | HR1 central cavity of Spike protein | −16.3 kcal/mol | [50] |
| Hypericin |  | Investigational anticancer and antiviral drug | HR1 central cavity of Spike protein | −15.1 kcal/mol | [50] |
| Ergotamine |  | Approved drug for migraine | HR1 central cavity of Spike protein | −13.2 kcal/mol | [50] |
| TMC-647055 |  | Investigational antiviral drug | HR1 central cavity of Spike protein | −12.5 kcal/mol | [50] |
| Quarfloxin |  | Investigational anticancer drug | HR1 central cavity of Spike protein | −12.6 kcal/mol | [50] |
| Tepotinib |  | Investigational anticancer drug | HR1 central cavity of Spike protein | −12.0 kcal/mol | [50] |
| Laniquidar |  | Investigational anticancer drug | HR1 central cavity of Spike protein | −12.8 kcal/mol | [50] |
| Tadalafil |  | Approved drug for erectile dysfunction and pulmonary arterial hypertension | HR1 central cavity of Spike protein | −12.4 kcal/mol | [50] |
| JNJ-10311795 |  | Experimental drug | HR1 central cavity of Spike protein | −13.1 kcal/mol | [50] |
| TZ2PA6 |  | Experimental drug | HR1 central cavity of Spike protein | −12.7 kcal/mol | [50] |
| Chitosan |  | Antibacterial, nontoxic biopolymer | Cavity formed by the three spike S2 subunits | −67.49 kcal/mol | [66] |
| Rapamycin |  | Approved mTOR inhibitor | Cavity formed by the three spike S2 subunits | −49.28 kcal/mol | [66] |
| Paclitaxel |  | Approved anticancer | Cavity formed by the three spike S2 subunits | −45.84 kcal/mol | [66] |
| SelaMeerin (Selamectin) |  | Vet approved antiparasitic | Cavity formed by the three spike S2 subunits | −44.24 kcal/mol | [66] |
| Everolimus |  | Approved mTOR inhibitor | Cavity formed by the three spike S2 subunits | −41.80 kcal/mol | [66] |
| Ritonavir |  | Approved antiviral (HIV protease inhibitor) | Cavity formed by the three spike S2 subunits | −37.92 kcal/mol | [66] |
| Danoprevir |  | Investigational antiviral drug | Cavity formed by the three spike S2 subunits | −35.09 kcal/mol | [66] |
| Small Molecule Name | Structure | Type | Target | IC50/EC50 | Ref. |
|---|---|---|---|---|---|
| Camostat mesylate |  | Chronic pancreatitis drug | Host cell protease: TMPRSS2 | 87 nM | [35,67] |
| Nafamostat mesylate |  | Anticoagulant drug | Host cell protease: TMPRSS2 | 5 nM | [67,68] |
| Naphthofluorescein |  | - | Host cell protease: furin | - | [69,70] |
| EST(23,25)trans-epoxysuccinyl-l-leucylamindo-3-methylbutane ethyl ester |  | - | Host cell protease: Cathepsin-L and Cathepsin-B | - | [69,70] |
| Compound K11777 |  | - | Host cell protease: Cathepsin-L | 0.68 nM | [69,70] |
| Compound 5705213 |  | - | Host cell protease: Cathepsin-L | 9 uM | [69,70] |
| Tetrahydroquinoline oxocarbazate |  | - | Host cell protease: Cathepsin-L | Time-dependent inhibition at IC50 from 6.9 ± 1.0 nM (immediately) to 2.3 ± 0.1 nM (1 h) to 1.2 ± 0.1 nM (2 h) to 0.4 ± 0.1 nM (4 h); | [69,70,71] |
| Compound SSAA09E1 |  | - | Host cell protease: Cathepsin-L | 5.22 uM | [69,70] |
| NAAE |  | Experimental ACE2 inhibitor | ACE2 | −23.7 kcal/mol IC50 of 57 ± 7 μmol/L | [72] |
| MLN-4760 |  | Investigational ACE2 inhibitor | ACE2 | 0.44 nM | [73] |
| Nicotianamine |  | Natural metal chelator | ACE2 | 84 nM | [74] |
| Small Molecule Name | Structure | Type | Target | Docking Score | Ref. |
|---|---|---|---|---|---|
| Lividomycin |  | - | ACE2 | −2145.79 kcal/mol | [75] |
| Burixafor |  | Investigational anticancer drug | ACE2 | −2108.82 kcal/mol | [75] |
| Quisinostat |  | Investigational anticancer drug | ACE2 | −1998.77 kcal/mol | [75] |
| Fluprofylline |  | - | ACE2 | −1785.00 kcal/mol | [75] |
| Pemetrexed |  | Anticancer drug | ACE2 | −1602.58 kcal/mol | [75] |
| Spirofylline |  | - | ACE2 | −1541.73 kcal/mol | [75] |
| Edotecarin |  | Investigational drug for inhibiting DNA topoisomerase-1 | ACE2 | −1312.19 kcal/mol | [75] |
| Diniprofylline |  | - | ACE2 | −1292.42 kcal/mol | [75] |
| Troglitazone |  | Anti-diabetes drug | ACE2 | −21.10 kcal/mol | [63] |
| Losartan |  | Anti-hypertensive drug | ACE2 | −21.49 kcal/mol | [63] |
| Ergotamine |  | Analgesia drug | ACE2 | −14.74 kcal/mol | [63] |
| Cefmenoxime |  | Antibacterial drug | ACE2 | −13.49 kcal/mol | [63] |
| Silybin |  | Hepatoprotective drug | ACE2 | −25.93 kcal/mol | [63] |
| Xanthones |  | Antiviral and anti-inflammatory effect | ACE2 | −29.40 kcal/mol | [63] |
| N′,N″-[oxybis(4,1-phenylenecarbonyl)]bis(3-methoxybenzohydrazide) |  | - | ACE2 | −5.87 kcal/mol | [76] |
| 2,2′-{1,4-butanediylbis[(4-ethyl-4H-1,2,4-triazole-5,3-diyl)thio]}bis(1-phenylethanone) |  | - | ACE2 | −5.84 kcal/mol | [76] |
| N,N′-bis{4-[(benzylamino)carbonyl]phenyl}malonamide |  | - | ACE2 | −5.83 kcal/mol | [76] |
| N,N′-[methylenebis(2-hydroxy-4,1-phenylene)]bis[2-(3,4-dimethoxyphenyl)acetamide] |  | - | ACE2 | −5.77 kcal/mol | [76] |
| ethyl 4-({[(4-allyl-5-{2-[(2,4-dimethylphenyl)amino]-2-oxoethyl}-4H-1,2,4-triazol-3-yl)thio]acetyl}amino)benzoate |  | - | ACE2 | −5.69 kcal/mol | [76] |
| 2-(4-methoxyphenyl)-N-{[4-methyl-5-({2-[(5-methyl-4-phenyl-1,3-thiazol-2-yl)amino]-2-oxoethyl}thio)-4H-1,2,4-triazol-3-yl]methyl}acetamide |  | - | ACE2 | −5.69 kcal/mol | [76] |
| N,N′-1,6-hexanediylbis[2 -(4-isopropylphenoxy)acetamide] |  | - | ACE2 | −5.65 kcal/mol | [76] |
| N′,N″-[oxybis(4,1-phenylenecarbonyl)]bis(2-chlorobenzohydrazide) |  | - | ACE2 | −5.62 kcal/mol | [76] |
| N-(5-{[2-(dibenzo[b,d]furan-3-ylamino)-2-oxoethyl]thio}-1,3,4-thiadiazol-2-yl)-2-methoxybenzamide |  | - | ACE2 | −5.56 kcal/mol | [76] |
| N,N′-1,2-phenylenebis[2-(4-ethylphenoxy)acetamide] |  | - | ACE2 | −5.54 kcal/mol | [76] |
| ethyl 3-({[(5-{[(3,4-dimethoxybenzoyl)amino]methyl}-4-methyl-4H-1,2,4-triazol-3-yl)thio]acetyl}amino)benzoate |  | - | ACE2 | −5.54 kcal/mol | [76] |
| N,N′-(oxydi-4,1-phenylene)bis[2-(2-methoxyphenoxy)acetamide] |  | - | ACE2 | −5.53 kcal/mol | [76] |
| N,N′-4,4′-biphenyldiylbis[2-(2-methoxyphenoxy)acetamide] |  | - | ACE2 | −5.53 kcal/mol | [76] |
| 1-[(3,4-dimethoxyphenyl)acetyl]-N,N′-bis(2-thienylmethyl)-1H-1,2,4-triazole-3,5-diamine |  | - | ACE2 | −5.53 kcal/mol | [76] |
| N,N′-1,2-propanediylbis[2-(4-tert-butylphenoxy)acetamide] |  | - | ACE2 | −5.52 kcal/mol | [76] |
| 4,4′-oxybis[N-(2-ethoxyphenyl)benzamide] |  | - | ACE2 | −5.51 kcal/mol | [76] |
| 2-chloro-N-{2-[5-({2-[(3-cyano-4,5,6,7-tetrahydro-1-benzothien-2-yl)amino]-2-oxoethyl}thio)-4-ethyl-4H-1,2,4-triazol-3-yl]ethyl}benzamide |  | - | ACE2 | −5.51 kcal/mol | [76] |
| N-{2-[4-allyl-5-({2-oxo-2-[(4-phenyl-1,3-thiazol-2-yl)amino]ethyl}thio)-4H-1,2,4-triazol-3-yl]ethyl}-4-methylbenzamide |  | - | ACE2 | −5.51 kcal/mol | [76] |
| N-[2-(3,4-dimethoxyphenyl)ethyl]-2-{4-[(isobutylamino)sulfonyl]-2-methylphenoxy}acetamide |  | - | ACE2 | −5.51 kcal/mol | [76] |
| N~2~-[4-(benzyloxy)phenyl]-N~1~-(4-{[(2,6-dimethylphenyl)amino]sulfonyl}phenyl)-N~2~-(methylsulfonyl)glycinamide |  | - | ACE2 | −5.50 kcal/mol | [76] |
| Phyllaemblicin G7 |  | Natural flavonoid | ACE2 | −4.33 kcal/mol | [63] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Al Adem, K.; Shanti, A.; Stefanini, C.; Lee, S. Inhibition of SARS-CoV-2 Entry into Host Cells Using Small Molecules. Pharmaceuticals 2020, 13, 447. https://doi.org/10.3390/ph13120447
Al Adem K, Shanti A, Stefanini C, Lee S. Inhibition of SARS-CoV-2 Entry into Host Cells Using Small Molecules. Pharmaceuticals. 2020; 13(12):447. https://doi.org/10.3390/ph13120447
Chicago/Turabian StyleAl Adem, Kenana, Aya Shanti, Cesare Stefanini, and Sungmun Lee. 2020. "Inhibition of SARS-CoV-2 Entry into Host Cells Using Small Molecules" Pharmaceuticals 13, no. 12: 447. https://doi.org/10.3390/ph13120447
APA StyleAl Adem, K., Shanti, A., Stefanini, C., & Lee, S. (2020). Inhibition of SARS-CoV-2 Entry into Host Cells Using Small Molecules. Pharmaceuticals, 13(12), 447. https://doi.org/10.3390/ph13120447