New Insights into the Binding Features of F508del CFTR Potentiators: A Molecular Docking, Pharmacophore Mapping and QSAR Analysis Approach

 , ,
, ,  ,
,  , ,
, ,  and
and 
Abstract
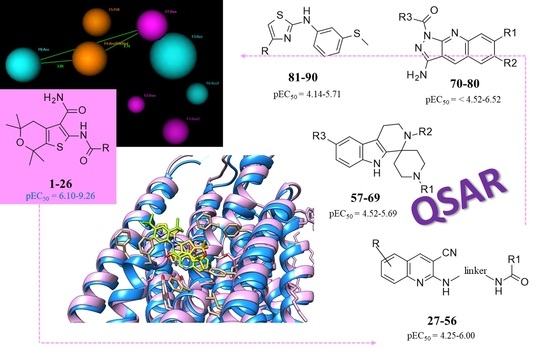
1. Introduction
2. Results
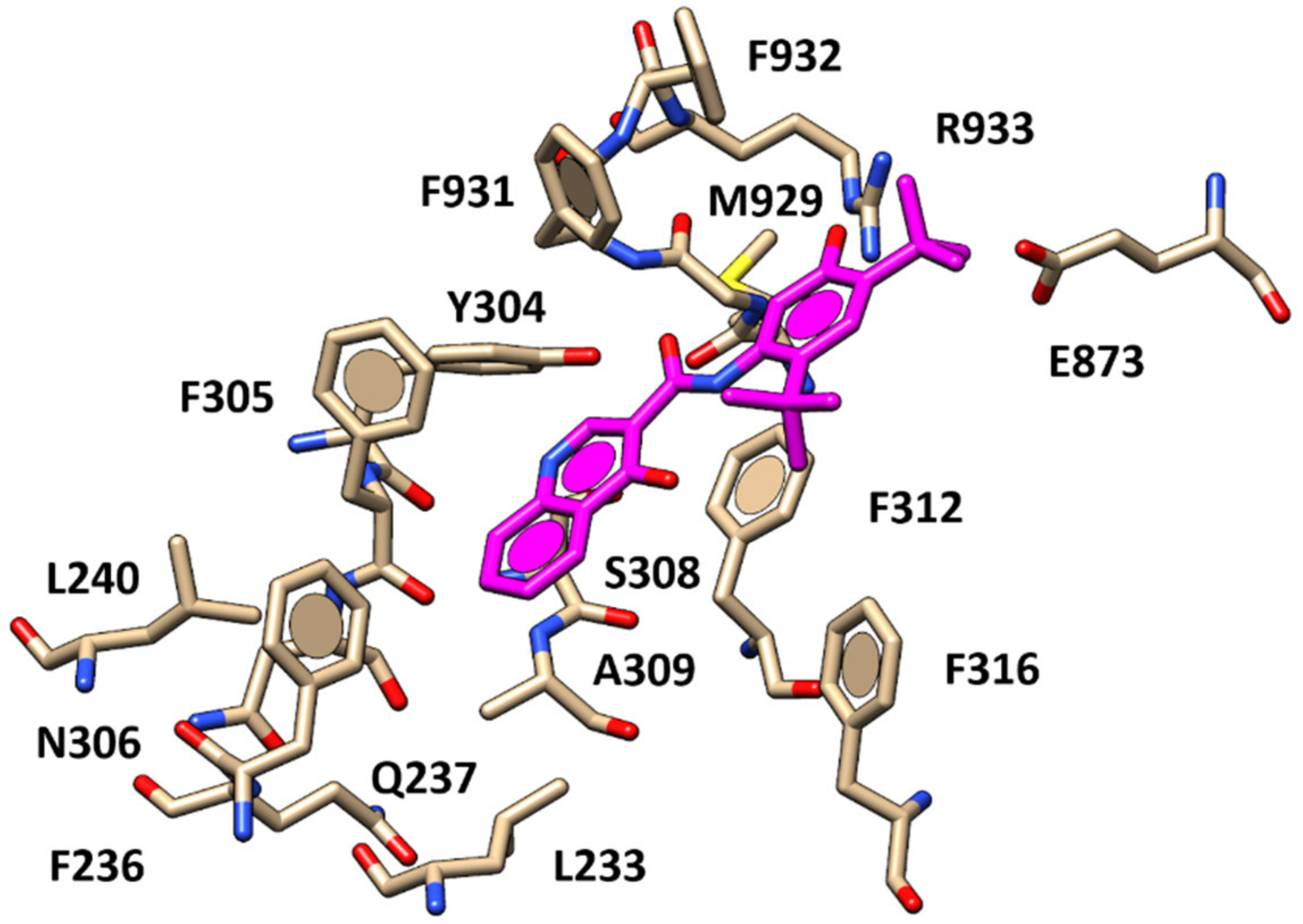
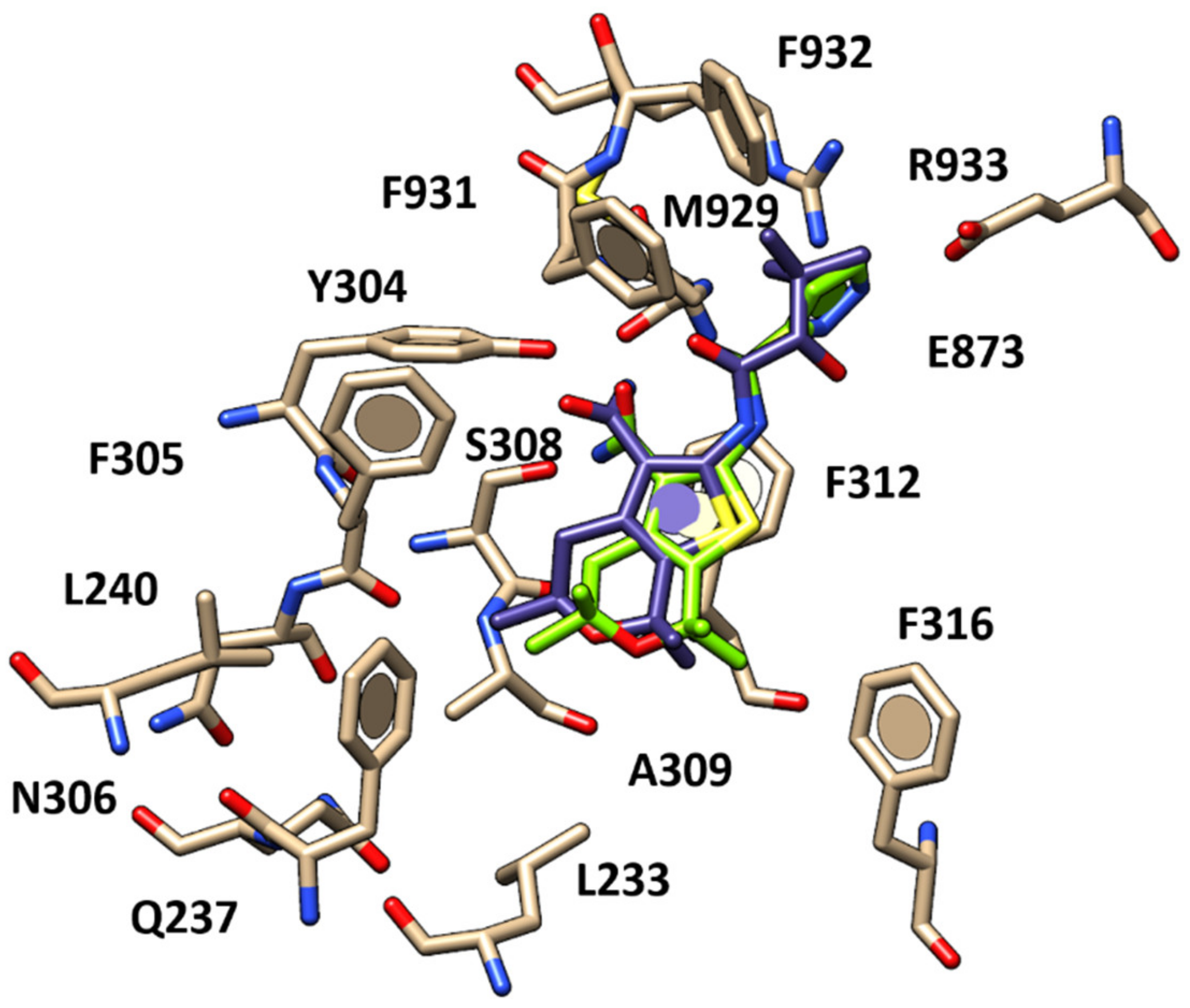
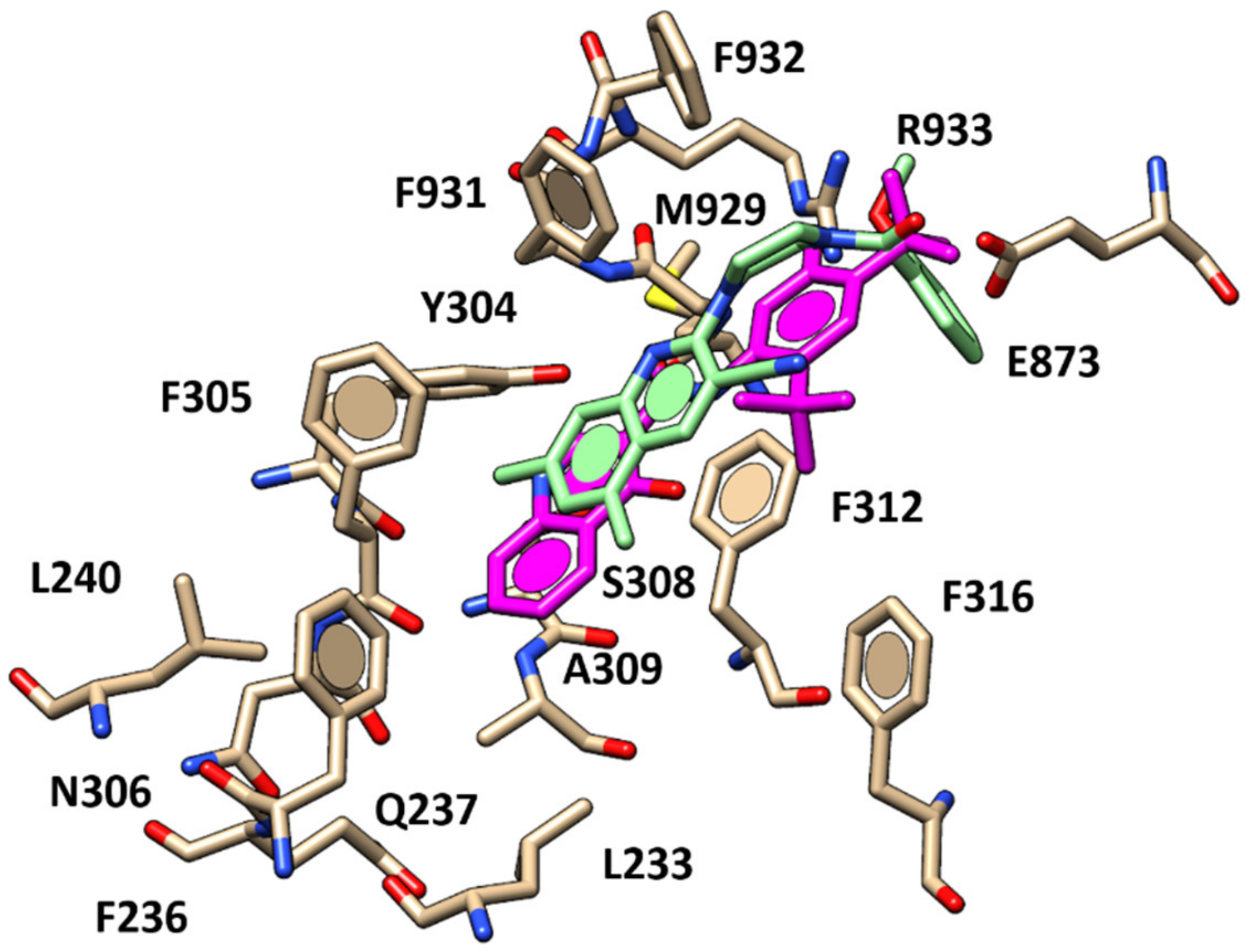
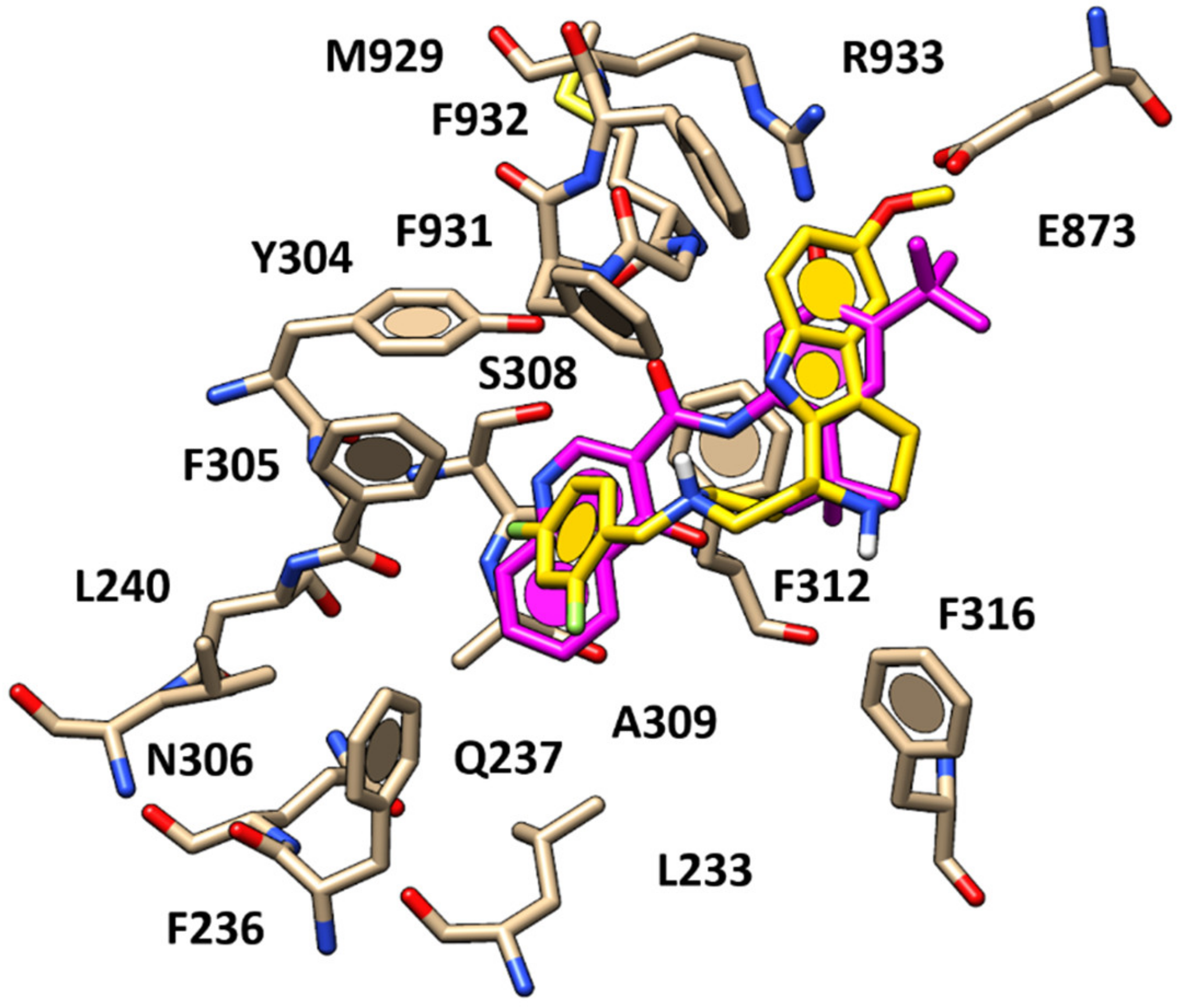
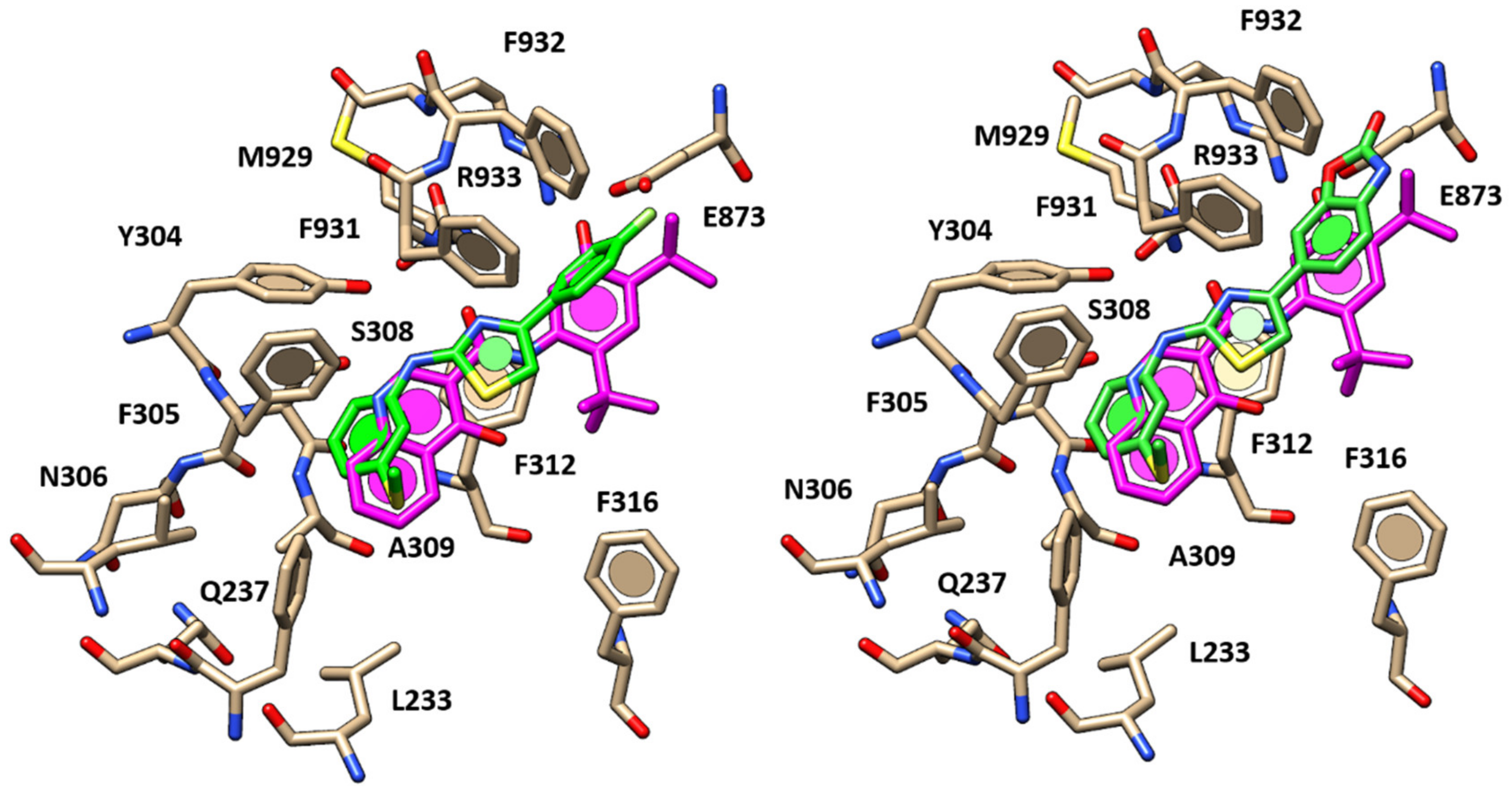
2.1. Molecular Docking Studies
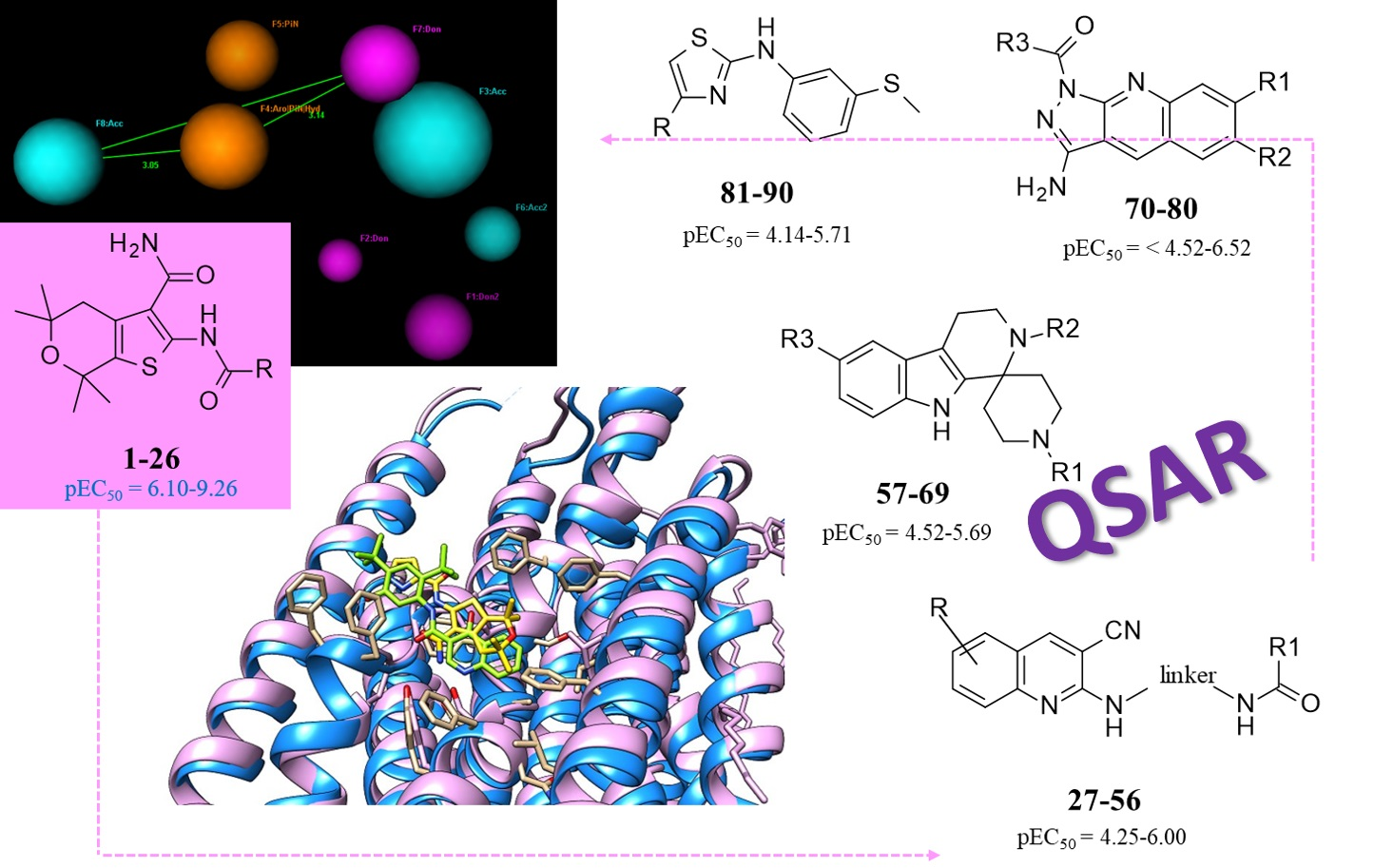
2.2. CFTR Potentiator Pharmacophore Model
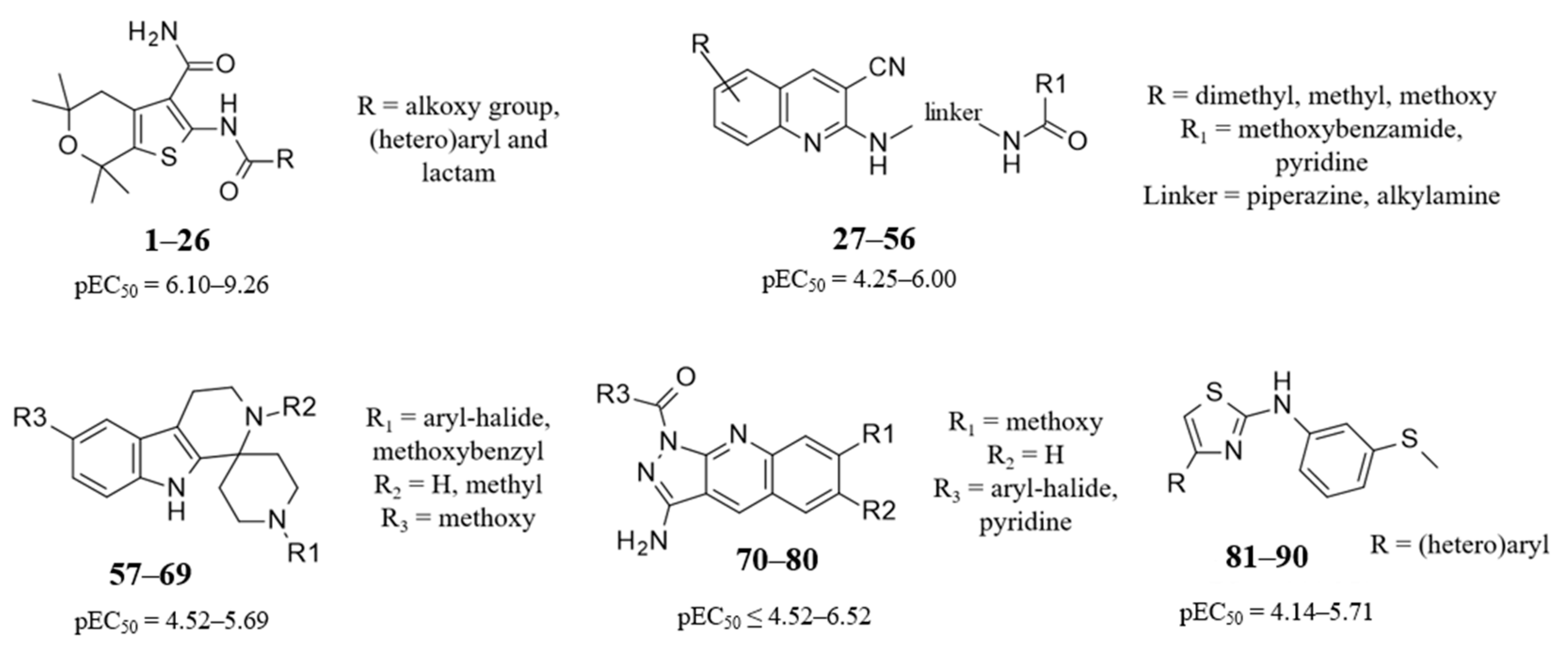
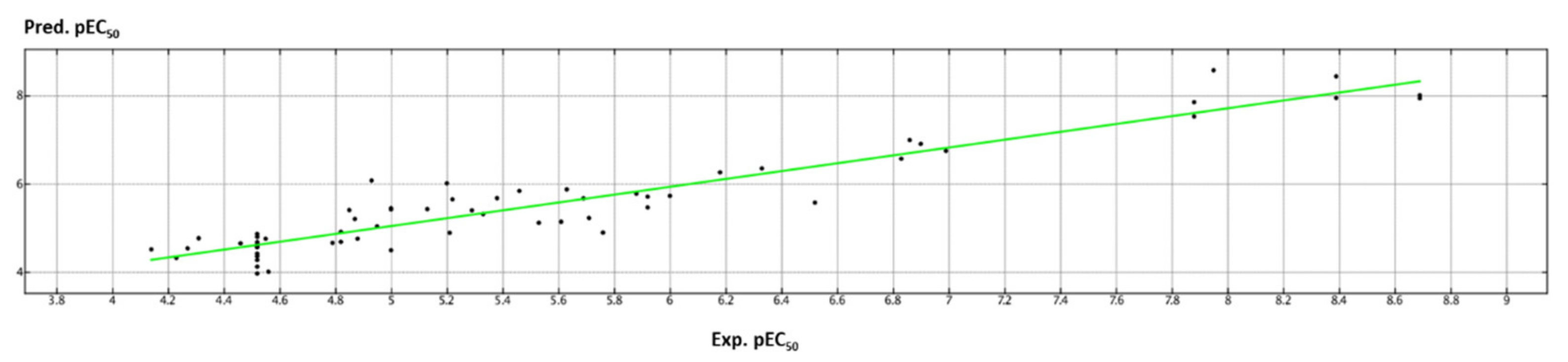
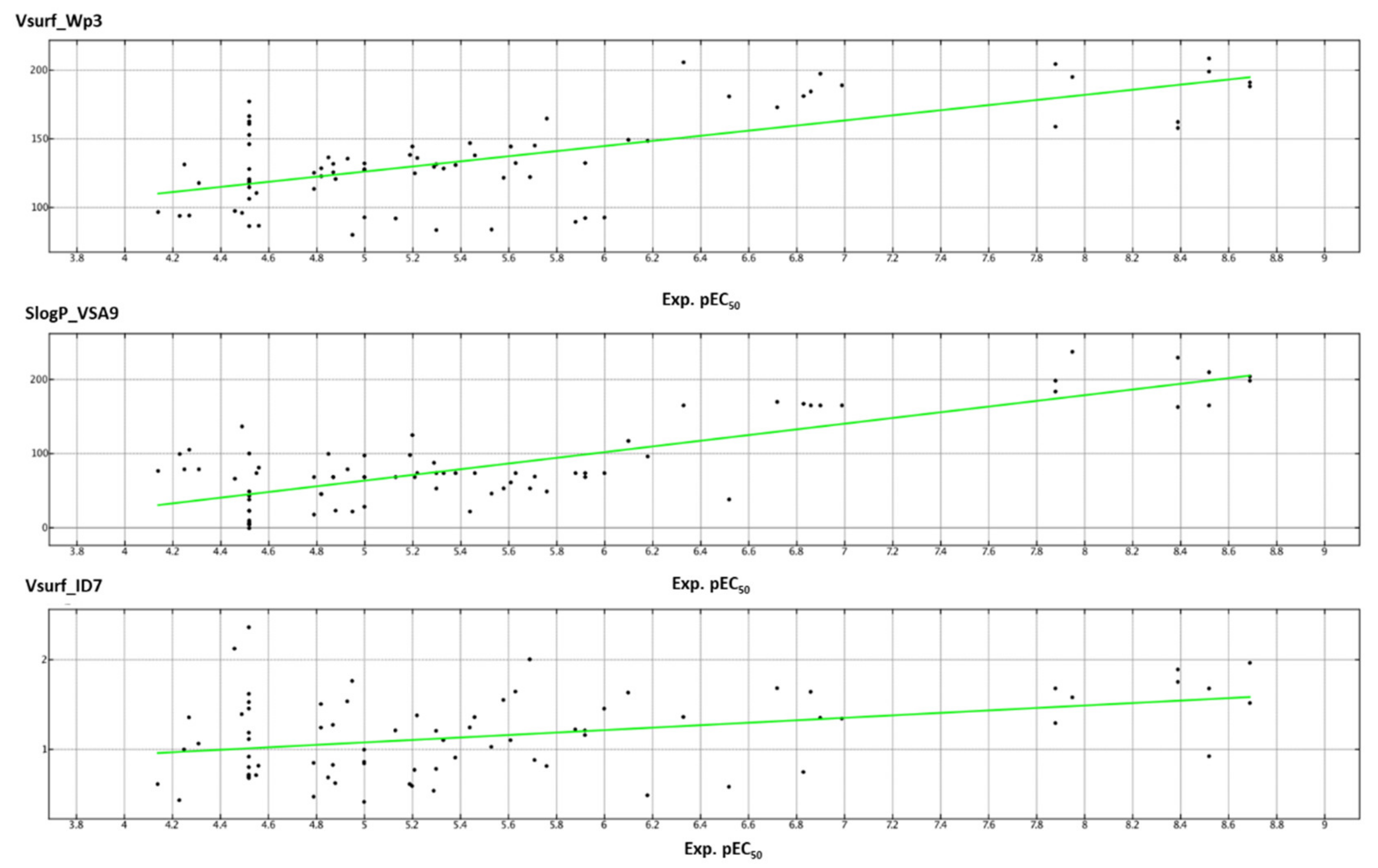
2.3. QSAR Analysis
2.4. Prediction of ADMET Properties
3. Discussion
4. Materials and Methods
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AAT | aminoarylthiazole |
| CF | cystic fibrosis |
| CFTR | cystic fibrosis transmembrane conductance regulator |
| F508del | deletion of phenylalanine at position 508 |
| PDB | protein data bank |
| pEC50 | negative logarithm of the half maximal effective concentration (EC50) |
References
- Bobadilla, J.L.; Macek, M.; Fine, J.P.; Farrell, P.M. Cystic fibrosis: A worldwide analysis of CFTR mutations--correlation with incidence data and application to screening. Hum. Mutat. 2002, 19, 575–606. [Google Scholar] [CrossRef]
- Lubamba, B.; Dhooghe, B.; Noel, S.; Leal, T. Cystic fibrosis: Insight into CFTR pathophysiology and pharmacotherapy. Clin. Biochem. 2012, 15, 1132–1144. [Google Scholar] [CrossRef]
- Hanrahan, J.W.; Sampson, H.M.; Thomas, D.Y. Novel pharmacological strategies to treat cystic fibrosis. Trends Pharmacol. Sci. 2013, 34, 119–125. [Google Scholar] [CrossRef]
- Gadsby, D.C.; Nairn, A.C. Control of CFTR channel gating by phosphorylation and nucleotide hydrolysis. Physiol. Rev. 1999, 79, 77–107. [Google Scholar] [CrossRef]
- Vergani, P.; Lockless, S.W.; Nairn, A.C.; Gadsby, D.C. CFTR channel opening by ATP-driven tight dimerization of its nucleotide-binding domains. Nature 2005, 433, 876–880. [Google Scholar] [CrossRef] [PubMed]
- Mondejar-Lopez, P.; Pastor-Vivero, M.D.; Sanchez-Solis, M.; Escribano, A. Cystic fibrosis treatment: Targeting the basic defect. Expert Opin. Orphan Drugs 2017, 5, 181–192. [Google Scholar] [CrossRef]
- Bell, C.; De Boeck, K.; Amaral, M.D. New pharmacological approaches for cystic fibrosis: Promises, progress, pitfalls. Pharmacol. Ther. 2015, 145, 19–34. [Google Scholar] [CrossRef] [PubMed]
- Sermet-Gaudelus, I. Ivacaftor treatment in patients with cystic fibrosis and the G551D-CFTR mutation. Eur. Respir. Rev. 2013, 22, 66–71. [Google Scholar] [CrossRef] [PubMed]
- Veit, G.; Avramescu, R.G.; Chiang, A.N.; Houck, S.A.; Cai, Z.; Peters, K.W.; Hong, J.S.; Pollard, H.B.; Guggino, W.B.; Balch, W.E.; et al. From CFTR biology toward combinatorial pharmacotherapy: Expanded classification of cystic fibrosis mutations. Mol. Biol. Cell. 2016, 27, 424–433. [Google Scholar] [CrossRef]
- Lopes-Pacheco, M. CFTR Modulators: Shedding Light on Precision Medicine for Cystic Fibrosis. Front. Pharmacol. 2016, 7, 275. [Google Scholar] [CrossRef]
- Pedemonte, N.; Sonawane, N.D.; Taddei, A.; Hu, J.; Zegarra-Moran, O.; Suen, Y.F.; Robins, L.I.; Dicus, C.W.; Willenbring, D.; Nantz, M.H.; et al. Phenylglycine and Sulfonamide Correctors of Defective Delta F508 and G551D Cystic Fibrosis Transmembrane Conductance Regulator Chloride-Channel Gating. Mol. Pharmacol. 2005, 67, 1797–1807. [Google Scholar] [CrossRef] [PubMed]
- Van Goor, F.; Hadida, S.; Grootenhuis, P.D.J.; Burton, B.; Stack, J.H.; Straley, K.S.; Decker, C.J.; Miller, M.; McCartney, J.; Olson, E.R.; et al. Correction of the F508del-CFTR protein processing defect in vitro by the investigational drug VX-809. Proc. Natl. Acad. Sci. USA 2011, 108, 18843–18848. [Google Scholar] [CrossRef] [PubMed]
- Moran, O. Model of the cAMP activation of chloride transport by CFTR channel and the mechanism of potentiators. J. Theor. Biol. 2010, 262, 73–79. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Martiniano, S.L.; Sagel, S.D.; Zemanick, E.T. Cystic fibrosis: A model system for precision medicine. Curr. Opin. Pediatr. 2016, 28, 312–317. [Google Scholar] [CrossRef]
- Van Goor, F.; Hadida, S.; Grootenhuis, P.D.J.; Burton, B.; Stack, J.H.; Straley, K.S.; Decker, C.J.; Miller, M.; McCartney, J.; Olson, E.R.; et al. Rescue of CF airway epithelial cell function in vitro by a CFTR potentiator, VX-770. Proc. Natl. Acad. Sci. USA 2009, 106, 18825–18830. [Google Scholar] [CrossRef]
- Clancy, J.P.; Rowe, S.M.; Accurso, F.J.; Aitken, M.L.; Amin, R.S.; Ashlock, M.A.; Ballmann, M.; Boyle, M.P.; Bronsveld, I.; Campbell, P.W.; et al. Results of a phase IIa study of VX-809, an investigational CFTR corrector compound, in subjects with cystic fibrosis homozygous for the F508del-CFTR mutation. Thorax 2012, 67, 12–18. [Google Scholar] [CrossRef]
- Flume, P.A.; Liou, T.G.; Borowitz, D.S.; Li, H.; Yen, K.; Ordoñez, C.L.; Geller, D. Ivacaftor in subjects with cystic fibrosis who are homozygous for the F508del-CFTR mutation. Chest 2012, 142, 718–724. [Google Scholar] [CrossRef]
- Wainwright, C.E.; Elborn, J.S.; Ramsey, B.W.; Marigowda, G.; Huang, X.; Cipolli, M.; Colombo, C.; Davies, J.C.; De Boeck, K.; Flume, P.A.; et al. Lumacaftor-ivacaftor in patients with cystic fibrosis homozygous for Phe508del CFTR. N. Engl. J. Med. 2015, 373, 220–231. [Google Scholar] [CrossRef]
- Cuyx, S.; De Boeck, K. Treating the Underlying Cystic Fibrosis Transmembrane Conductance Regulator Defect in Patients with Cystic Fibrosis. Semin. Respir. Crit. Care Med. 2019, 40, 762–774. [Google Scholar] [CrossRef]
- Ridley, K.; Condren, M. Elexacaftor-Tezacaftor-Ivacaftor: The First Triple-Combination Cystic Fibrosis Transmembrane Conductance Regulator Modulating Therapy. Pediatr. Pharmacol. Ther. 2020, 25, 192–197. [Google Scholar] [CrossRef]
- Liu, F.; Zhang, Z.; Levit, A.; Levring, J.; Touhara, K.K.; Shoichet, B.K.; Chen, J. Structural identification of a hotspot on CFTR for potentiation. Science 2019, 364, 1184–1188. [Google Scholar] [CrossRef] [PubMed]
- Liessi, N.; Pesce, E.; Salis, A.; Damonte, G.; Tasso, B.; Cichero, E.; Pedemonte, N.; Millo, E. Synthesis and structure-activity relationship of aminoarylthiazole derivatives as potential potentiators of the chloride transport defect in cystic fibrosis. Med. Chem. 2020. [Google Scholar] [CrossRef] [PubMed]
- Pesce, E.; Bellotti, M.; Liessi, N.; Guariento, S.; Damonte, G.; Cichero, E.; Galatini, A.; Salis, A.; Gianotti, A.; Pedemonte, N.; et al. Synthesis and structure-activity relationship of aminoarylthiazole derivatives as correctors of the chloride transport defect in cystic fibrosis. Eur. J. Med. Chem. 2015, 99, 14–35. [Google Scholar] [CrossRef] [PubMed]
- Pedemonte, N.; Tomati, V.; Sondo, E.; Caci, E.; Millo, E.; Armirotti, A.; Damonte, G.; Moran, O.; Galietta, L.J.V. Dual activity of aminoarylthiazoles on the trafficking and gating defects of the cystic fibrosis transmembrane conductance regulator chloride channel caused by cystic fibrosis mutations. J. Biol. Chem. 2011, 17, 15215–15226. [Google Scholar] [CrossRef]
- Liessi, N.; Cichero, E.; Pesce, E.; Arkel, M.; Salis, A.; Tomati, V.; Paccagnella, M.; Damonte, G.; Tasso, B.; Galietta, L.J.V.; et al. Synthesis and biological evaluation of novel thiazole- VX-809 hybrid derivatives as F508del correctors by QSAR-based filtering tools. Eur. J. Med. Chem. 2018, 144, 179–200. [Google Scholar] [CrossRef]
- Phuan, P.; Tan, J.A.; Rivera, A.A.; Zlock, L.; Nielson, D.W.; Finkbeiner, W.E.; Haggie, P.M.; Verkmann, A.S. Nanomolar-potency ‘co-potentiator’ therapy for cystic fibrosis caused by a defined subset of minimal function CFTR mutants. Sci. Rep. 2019, 9, 17640. [Google Scholar] [CrossRef]
- Van der Plas, S.E.; Kelgtermans, H.; De Munck, T.; Martina, S.; Dropsit, S.; Quinton, E.; De Blieck, A.; Joannesse, C.; Tomaskovic, L.; Jans, M.; et al. Discovery of N-(3-Carbamoyl-5,5,7,7-tetramethyl-5,7-dihydro-4H-thieno[2,3-c]pyran-2-yl)-lH-pyrazole-5carboxamide (GLPG1837), a Novel Potentiator which can open class III mutant cystic fibrosis transmembrane conductance to a high extent. J. Med. Chem. 2018, 61, 1425–1435. [Google Scholar] [CrossRef]
- Phuan, P.W.; Yang, B.; Knapp, J.M.; Wood, A.B.; Lukacs, G.L.; Kurth, M.J.; Verkman, A.S. Cyanoquinolines with independent corrector and potentiator activities restore ∆Phe508-Cystic fibrosis transmembrane conductance regulator chloride channel function in cystic fibrosis. Mol. Pharmacol. 2011, 80, 683–693. [Google Scholar] [CrossRef]
- Knapp, J.M.; Wood, A.B.; Phuan, P.W.; Lodewyk, M.W.; Tantillo, D.J.; Verkman, A.S.; Kurth, M.J. Structure-activity relationships of cyanoquinolines with corrector-potentiator activity in delta-F508-cystic fibrosis transmembrane conductance regulator protein. J. Med. Chem. 2012, 55, 1242–1251. [Google Scholar] [CrossRef]
- Chemical Computing Group Inc. MOE. Montreal. H3A2R7 Canada. Available online: http://www.chemcomp.com (accessed on 29 October 2019).
- Vandeginste, B. PARVUS: An Extendable Package of Programs for Data Exploration, Classification and Correlation; Forina, M., Leardi, R., Armanino, C., Lanteri, S., Eds.; Elsevier: Amsterdam, The Netherlands, 1988. [Google Scholar]
- Wildman, S.A.; Crippen, G.M. Prediction of Physiochemical Parameters by Atomic Contributions. J. Chem. Inf. Comput. Sci. 1999, 5, 868–873. [Google Scholar] [CrossRef]
- Hou, T.J.; Xia, K.; Zhang, W.; Xu, X.J. ADME Evaluation in Drug Discovery. 4. Prediction of Aqueous Solubility Based on Atom Contribution Approach. J. Chem. Inf. Comput. Sci. 2004, 44, 266–275. [Google Scholar] [CrossRef] [PubMed]
- Pearlman, R.S.; Smith, K.M. Novel software tools for chemical diversity. Persp. Drug. Discov. Des. 2002, 9–11, 339–353. [Google Scholar] [CrossRef]
- Kennard, R.W.; Stone, L.A. Computer aided design of experiments. Technometrics 1969, 11, 137–148. [Google Scholar] [CrossRef]
- Waterbeemd, H.; Gifford, E. ADMET in silico modelling: Towards prediction paradise? Nat. Rev. Drug Discov. 2003, 2, 192–204. [Google Scholar] [CrossRef]
- Franchini, S.; Manasieva, L.I.; Sorbi, C.; Battisti, U.M.; Fossa, P.; Cichero, E.; Denora, N.; Iacobazzi, R.M.; Cilia, A.; Pirona, L.; et al. Synthesis, biological evaluation and molecular modeling of 1-oxa-4-thiaspiro- and 1,4-dithiaspiro[4.5]decane derivatives as potent and selective 5-HT1A receptor agonists. Eur. J. Med. Chem. 2017, 125, 435–452. [Google Scholar] [CrossRef]
- Tonelli, M.; Espinoza, S.; Gainetdinov, R.R.; Cichero, E. Novel biguanide-based derivatives scouted as TAAR1 agonists: Synthesis, biological evaluation, ADME prediction and molecular docking studies. Eur. J. Med. Chem. 2017, 127, 781–792. [Google Scholar] [CrossRef]
- Francesconi, V.; Cichero, E.; Kanov, E.V.; Laurini, E.; Pricl, S.; Gainetdinov, R.R.; Tonelli, M. Novel 1-Amidino-4-Phenylpiperazines as Potent Agonists at Human TAAR1 Receptor: Rational Design, Synthesis, Biological Evaluation and Molecular Docking Studies. Pharmaceuticals 2020, 13, E391. [Google Scholar] [CrossRef]
- Pesce, E.; Pedemonte, N.; Leoni, A.; Locatelli, A.; Morigi, R. Synthesis and biological evaluation of thiazole derivatives on basic defects underlying cystic fibrosis. Bioorg. Med. Chem. Lett. 2020, 30, 127473. [Google Scholar] [CrossRef]
- Böhm, H.J. The computer program LUDI: A new method for the de novo design of enzyme inhibitors. J. Comput. Aided Mol. Des. 1992, 6, 61–78. [Google Scholar] [CrossRef]
- Böhm, H.J. The development of a simple empirical scoring function to estimate the binding constant for a protein–ligand complex of known three-dimensional structure. J. Comput. Aided Mol. Des. 1994, 8, 243–256. [Google Scholar] [CrossRef]
- Rarey, M.; Kramer, B.; Lengauer, T.; Klebe, G. A fast flexible docking method using an incremental construction algorithm. J. Mol. Biol. 1996, 261, 470–489. [Google Scholar] [CrossRef] [PubMed]
- Bichmann, L.; Wang, Y.T.; Fischer, W.B. Docking assay of small molecule antivirals to p7 of HCV. Comput. Biol. Chem. 2014, 53, 308–317. [Google Scholar] [CrossRef] [PubMed]
- Reulecke, I.; Lange, G.; Albrecht, J.; Klein, R.; Rarey, M. Towards an integrated description of hydrogen bonding and dehydration: Decreasing false positives in virtual screening with the HYDE scoring function. ChemMedChem 2008, 3, 885–897. [Google Scholar] [CrossRef]
- Schneider, N.; Hindle, S.; Lange, G.; Klein, R.; Albrecht, J.; Briem, H.; Beyer, K.; Claußen, H.; Gastreich, M.; Lemmen, C.; et al. Substantial improvements in large-scale redocking and screening using the novel HYDE scoring function. J. Comput. Aided Mol. Des. 2012, 26, 701–723. [Google Scholar] [CrossRef] [PubMed]
- Wolber, G.; Seidel, T.; Bendix, F.; Langer, T. Molecule-pharmacophore superpositioning and pattern matchingin computational drug design. Drug Discov. Today 2008, 13, 23–29. [Google Scholar] [CrossRef]
- Khalid, S.; Hanif, R.; Jabeen, I.; Mansoor, Q.; Ismail, M. Pharmacophore modeling for identification of anti-IGF-1R drugs and in-vitro validation of fulvestrant as a potential inhibitor. PLoS ONE 2018, 5, e0196312. [Google Scholar] [CrossRef]
- Mining, H.S.; Bouaziz, Z.; Marminon, C.; Laitinen, T.; Poso, A.; Le Borgne, M.; Jose, J. Development of Pharmacophore Model for Indeno[1,2-b] indoles as Human Protein Kinase CK2 Inhibitors and Database Mining. Pharmaceuticals 2017, 10, 8. [Google Scholar] [CrossRef]
- Cichero, E.; Menozzi, G.; Spallarossa, A.; Mosti, L.; Fossa, P. Exploring the binding features of rimonabant analogues and acyclic CB1 antagonists: Docking studies and QSAR analysis. J. Mol. Model. 2008, 12, 1131–1145. [Google Scholar] [CrossRef]
- Boggia, R.; Forina, M.; Fossa, P.; Mosti, L. Zupan’s descriptors in QSAR applied to the study of a new class of cardiotonic agents. Farmaco 1997, 52, 411–419. [Google Scholar]

















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Descriptor | Type | Series | RI |
|---|---|---|---|
| SlogP_VSA4 | Sum of vi such that Li is in (0.1, 0.15) | 2D-II | 0.293739 |
| SlogP_VSA5 | Sum of vi such that Li is in (0.15, 0.20) | 2D-II | 0.501133 |
| SlogP_VSA9 | Sum of vi such that Li > 0.40 | 2D-II | 0.686395 |
| SMR_VSA2 | Sum of vi such that Ri is in (0.26, 0.35) | 2D-II | 0.429419 |
| SMR_VSA4 | Sum of vi such that Ri is in (0.39, 0.44) | 2D-II | 0.234752 |
| PEOE_VSA+5 | Sum of vi where qi is in the range (0.25, 0.30) | 2D-V | 0.053586 |
| PEOE_VSA-6 | Sum of vi where qi is less than −0.30 | 2D-V | 0.461873 |
| Descriptor | Type | Series | RI |
|---|---|---|---|
| BCUT_SLOGP_1 | The BCUT descriptors using atomic contribution to logP | 2D-VII | 0.578303 |
| BCUT_SMR_1 | The BCUT descriptors using atomic contribution to molar refractivity | 2D-VII | 0.500605 |
| BCUT_SMR_2 | The BCUT descriptors using atomic contribution to molar refractivity | 2D-VII | 0.029230 |
| BCUT_SMR_3 | The BCUT descriptors using atomic contribution to molar refractivity | 2D-VII | 0.720350 |
| a_hyd | Number of hydrophobic atoms | 2D-VI | 0.101682 |
| LogS | Log of the aqueous solubility (mol/L). This property is calculated from an atom contribution linear atom type model with r2 = 0.90, ~1200 molecules [33] | 2D-I | 0.950723 |
| b_1rotR | Fraction of rotatable single bonds: number of rotatable bonds (b_1rotN) divided by number of bonds between heavy atoms (b_heavy) | 2D-III | 0.211130 |
| Descriptor | Type | Series | RI |
|---|---|---|---|
| vsurf_ID1 | Hydrophobic integy moment | 3D-III | 0.119071 |
| vsurf_ID7 | Hydrophobic integy moment | 3D-III | 0.582277 |
| vsurf_Wp2 | Polar volume | 3D-III | 0.243649 |
| vsurf_Wp3 | Polar volume | 3D-III | 1.000000 |
| dipoleY | The y component of the dipole moment | 3D-V | 0.492821 |
| ASA+ | Water-accessible surface area of all atoms with positive partial charge | 3D-V | 0.309621 |
| Potentiator Series | b_1rotRmean | a_hydmean | Vsurf_WP3mean | Vsurf_ID7mean | SlogP_VSA9mean | pEC50 |
|---|---|---|---|---|---|---|
| Thienopyrans | 0.11934 | 15.8667 | 183.5333 | 1.4341 | 178.6650 | 6.10–9.26 |
| Cyanoquinolines | 0.1830 | 19.0741 | 118.6481 | 1.1386 | 68.4367 | 4.25–6.00 |
| Indole-based derivatives | 0.0901 | 21.8461 | 117.8750 | 1.09374 | 26.9052 | 4.52–5.69 |
| Pyrazoloquinolines | 0.1313 | 17.1000 | 160.6875 | 0.9737 | 40.7683 | 4.52–6.52 |
| Amino aryl Thiazoles | 0.17156 | 17.5454 | 108.8875 | 0.98697 | 96.0631 | 4.14–5.71 |
| Comp. | cLogP | LogBB a | LogPS b | HIA (%) c | Vd (l/kg) d | %PPB | LogKa HSA | %F (Oral) | Solubility (mg/mL) |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 2.98 | 0.18 | −1.4 | 100 | 2.1 | 93.86 | 3.86 | 95.0 | 0.10 |
| 3 | 2.56 | −0.19 | −1.8 | 100 | 1.3 | 92.00 | 3.72 | 99.2 | 0.25 |
| 4 | 2.93 | 0.02 | −1.6 | 100 | 1.5 | 93.70 | 3.99 | 94.6 | 0.14 |
| 6 | 3.46 | 0.29 | −1.4 | 100 | 1.7 | 95.20 | 4.20 | 83.2 | 0.07 |
| 7 | 2.90 | −0.00 | −2.6 | 100 | 1.5 | 94.37 | 4.18 | 97.9 | 0.41 |
| 8 | 1.55 | −0.54 | −3.7 | 100 | 0.29 | 91.58 | 4.65 | 96.7 | 0.92 |
| 9 | 0.88 | −0.63 | −2.8 | 100 | 0.32 | 84.39 | 3.59 | 98.4 | 0.75 |
| 10 | 2.16 | 0.17 | −2.2 | 100 | 1.3 | 77.92 | 3.36 | 99.4 | 0.57 |
| 11 | 2.16 | 0.18 | −2.2 | 100 | 1.3 | 77.92 | 3.36 | 99.4 | 0.57 |
| 12 | 2.29 | 0.06 | −2.3 | 100 | 1.2 | 77.50 | 3.41 | 99.4 | 0.47 |
| 22 | 2.71 | 0.26 | −1.9 | 100 | 1.2 | 88.45 | 3.69 | 99.0 | 0.26 |
| 23 | 1.78 | 0.25 | −2.2 | 100 | 1.4 | 76.56 | 3.38 | 98.6 | 0.34 |
| 24 | 2.33 | 0.50 | −2.2 | 100 | 1.5 | 85.40 | 3.47 | 86.9 | 0.31 |
| 25 | 1.78 | 0.25 | −2.2 | 100 | 1.4 | 76.56 | 3.38 | 86.9 | 0.34 |
| 26 | 2.54 | 0.41 | −1.8 | 100 | 1.3 | 87.76 | 3.72 | 94.0 | 0.14 |
| VX-770 | 4.84 | 0.29 | −1.3 | 100 | 3.6 | 98.82 | 4.87 | 49.2 | 0.09 |
| Compound | LD50 a (mg/kg) (RI ≥ 0.30) Mouse, Oral | hERG Inhibitor (RI ≥ 0.40) | Endocrine System Disruption b (RI ≥ 0.50) | CYP3A4 and CYP2D6 (RI ≥ 0.40) | ||
|---|---|---|---|---|---|---|
| LogRBA > −3 | LogRBA > 0 | Inhibitor < 10 mM | Substrate | |||
| 1 | 1100 | No inhibitor | No binder | No binder | 0.02 | CYP3A4 |
| 3 | 1000 | No inhibitor | No binder | No binder | 0.01 | CYP3A4 |
| 4 | 840 | No inhibitor | No binder | No binder | 0.01 | CYP3A4 |
| 6 | 820 | No inhibitor | No binder | No binder | 0.01 | CYP3A4 |
| 7 | 790 | No inhibitor | No binder | No binder | 0.01 | CYP3A4 |
| 8 | 1500 | No inhibitor | No binder | No binder | 0.01 | CYP3A4 |
| 9 | 2700 | No inhibitor | No binder | No binder | 0.02 | CYP3A4 |
| 10 | 1200 | No inhibitor | No binder | No binder | 0.02 | CYP3A4 |
| 11 | 1200 | No inhibitor | No binder | No binder | 0.02 | CYP3A4 |
| 12 | 1000 | No inhibitor | No binder | No binder | 0.01 | CYP3A4 |
| 22 | 1000 | No inhibitor | No binder | No binder | 0.01 | CYP3A4 |
| 23 | 1000 | No inhibitor | No binder | No binder | 0.02 | CYP3A4 |
| 24 | 510 | No inhibitor | No binder | No binder | 0.01 | CYP3A4 |
| 25 | 1000 | No inhibitor | No binder | No binder | 0.01 | CYP3A4 |
| 26 | 1000 | No inhibitor | No binder | No binder | 0.01 | CYP3A4 |
| VX-770 | 780 | No inhibitor | No binder | No binder | 0.01 | CYP3A4 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Righetti, G.; Casale, M.; Tonelli, M.; Liessi, N.; Fossa, P.; Pedemonte, N.; Millo, E.; Cichero, E. New Insights into the Binding Features of F508del CFTR Potentiators: A Molecular Docking, Pharmacophore Mapping and QSAR Analysis Approach. Pharmaceuticals 2020, 13, 445. https://doi.org/10.3390/ph13120445
Righetti G, Casale M, Tonelli M, Liessi N, Fossa P, Pedemonte N, Millo E, Cichero E. New Insights into the Binding Features of F508del CFTR Potentiators: A Molecular Docking, Pharmacophore Mapping and QSAR Analysis Approach. Pharmaceuticals. 2020; 13(12):445. https://doi.org/10.3390/ph13120445
Chicago/Turabian StyleRighetti, Giada, Monica Casale, Michele Tonelli, Nara Liessi, Paola Fossa, Nicoletta Pedemonte, Enrico Millo, and Elena Cichero. 2020. "New Insights into the Binding Features of F508del CFTR Potentiators: A Molecular Docking, Pharmacophore Mapping and QSAR Analysis Approach" Pharmaceuticals 13, no. 12: 445. https://doi.org/10.3390/ph13120445
APA StyleRighetti, G., Casale, M., Tonelli, M., Liessi, N., Fossa, P., Pedemonte, N., Millo, E., & Cichero, E. (2020). New Insights into the Binding Features of F508del CFTR Potentiators: A Molecular Docking, Pharmacophore Mapping and QSAR Analysis Approach. Pharmaceuticals, 13(12), 445. https://doi.org/10.3390/ph13120445