Small-Dose Sunitinib Modulates p53, Bcl-2, STAT3, and ERK1/2 Pathways and Protects against Adenine-Induced Nephrotoxicity



Abstract
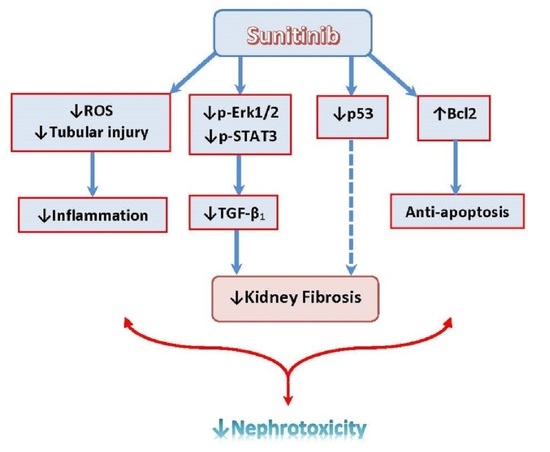
1. Introduction
2. Results
3. Discussion
4. Materials and Methods
4.1. Study Design
4.2. Kidney Function Biomarkers
4.3. Oxidative Stress and Antioxidant Biomarkers
4.4. ELISA
4.5. Tubular Injury and Interstitial Fibrosis
4.6. Immunohistochemistry
4.7. Statistical Analyses
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Meran, S.; Steadman, R. Fibroblasts and myofibroblasts in renal fibrosis. Int. J. Exp. Pathol. 2011, 92, 158–167. [Google Scholar] [CrossRef]
- Liu, Y. Cellular and molecular mechanisms of renal fibrosis. Nat. Rev. Nephrol. 2011, 7, 684–696. [Google Scholar] [CrossRef] [PubMed]
- Woolf, A.S.; Yuan, H.T. Angiopoietin growth factors and Tie receptor tyrosine kinases in renal vascular development. Pediatr. Nephrol. 2001, 16, 177–184. [Google Scholar] [CrossRef] [PubMed]
- Beyer, C.; Distler, J.H. Tyrosine kinase signaling in fibrotic disorders: Translation of basic research to human disease. Biochim. Biophys. Acta 2013, 1832, 897–904. [Google Scholar] [CrossRef] [PubMed]
- He, F.F.; Zhang, D.; Chen, Q.; Zhao, Y.; Wu, L.; Li, Z.Q.; Zhang, C.; Jiang, Z.H.; Wang, Y.M. Angiopoietin-Tie signaling in kidney diseases: An updated review. FEBS Lett. 2019, 593, 2706–2715. [Google Scholar] [CrossRef] [PubMed]
- Tang, J.; Liu, N.; Zhuang, S. Role of epidermal growth factor receptor in acute and chronic kidney injury. Kidney Int. 2013, 83, 804–810. [Google Scholar] [CrossRef]
- de Vriese, A.S.; Tilton, R.G.; Elger, M.; Stephan, C.C.; Kriz, W.; Lameire, N.H. Antibodies against vascular endothelial growth factor improve early renal dysfunction in experimental diabetes. J. Am. Soc. Nephrol. 2001, 12, 993–1000. [Google Scholar]
- Chen, Y.T.; Chang, F.C.; Wu, C.F.; Chou, Y.H.; Hsu, H.L.; Chiang, W.C.; Shen, J.; Chen, Y.M.; Wu, K.D.; Tsai, T.J.; et al. Platelet-derived growth factor receptor signaling activates pericyte-myofibroblast transition in obstructive and post-ischemic kidney fibrosis. Kidney Int. 2011, 80, 1170–1181. [Google Scholar] [CrossRef]
- Bigaeva, E.; Stribos, E.G.D.; Mutsaers, H.A.M.; Piersma, B.; Leliveld, A.M.; de Jong, I.J.; Bank, R.A.; Seelen, M.A.; van Goor, H.; Wollin, L.; et al. Inhibition of tyrosine kinase receptor signaling attenuates fibrogenesis in an ex vivo model of human renal fibrosis. Am. J. Physiol. Renal. Physiol. 2020, 318, F117–F134. [Google Scholar] [CrossRef]
- Motzer, R.J.; Escudier, B.; Gannon, A.; Figlin, R.A. Sunitinib: Ten Years of Successful Clinical Use and Study in Advanced Renal Cell Carcinoma. Oncologist 2017, 22, 41–52. [Google Scholar] [CrossRef]
- Eitner, F.; Ostendorf, T.; Van Roeyen, C.; Kitahara, M.; Li, X.; Aase, K.; Grone, H.J.; Eriksson, U.; Floege, J. Expression of a novel PDGF isoform, PDGF-C, in normal and diseased rat kidney. J. Am. Soc. Nephrol. 2002, 13, 910–917. [Google Scholar] [PubMed]
- Eitner, F.; Ostendorf, T.; Kretzler, M.; Cohen, C.D.; Eriksson, U.; Grone, H.J.; Floege, J.; Consortium, E. PDGF-C expression in the developing and normal adult human kidney and in glomerular diseases. J. Am. Soc. Nephrol. 2003, 14, 1145–1153. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Ziyadeh, F.N. Vascular endothelial growth factor and diabetic nephropathy. Curr. Diab. Rep. 2008, 8, 470–476. [Google Scholar] [CrossRef] [PubMed]
- Liu, E.; Morimoto, M.; Kitajima, S.; Koike, T.; Yu, Y.; Shiiki, H.; Nagata, M.; Watanabe, T.; Fan, J. Increased expression of vascular endothelial growth factor in kidney leads to progressive impairment of glomerular functions. J. Am. Soc. Nephrol. 2007, 18, 2094–2104. [Google Scholar] [CrossRef] [PubMed]
- Hawthorne, T.; Giot, L.; Blake, L.; Kuang, B.; Gerwien, R.; Smithson, G.; Hahne, W.; Mansfield, T.; Starling, G.C.; Pochart, P.; et al. A phase I study of CR002, a fully-human monoclonal antibody against platelet-derived growth factor-D. Int. J. Clin. Pharmacol. Ther. 2008, 46, 236–244. [Google Scholar] [CrossRef]
- Lieberthal, W.; Levine, J.S. The role of the mammalian target of rapamycin (mTOR) in renal disease. J. Am. Soc. Nephrol. 2009, 20, 2493–2502. [Google Scholar] [CrossRef]
- Kasinath, B.S.; Mariappan, M.M.; Sataranatarajan, K.; Lee, M.J.; Ghosh Choudhury, G.; Feliers, D. Novel mechanisms of protein synthesis in diabetic nephropathy—Role of mRNA translation. Rev. Endocr. Metab. Disord. 2008, 9, 255–266. [Google Scholar] [CrossRef]
- Papaioannou, I.; Xu, S.; Denton, C.P.; Abraham, D.J.; Ponticos, M. STAT3 controls COL1A2 enhancer activation cooperatively with JunB, regulates type I collagen synthesis posttranscriptionally, and is essential for lung myofibroblast differentiation. Mol. Biol. Cell. 2018, 29, 84–95. [Google Scholar] [CrossRef]
- Bowman, T.; Garcia, R.; Turkson, J.; Jove, R. STATs in oncogenesis. Oncogene 2000, 19, 2474–2488. [Google Scholar] [CrossRef]
- Saleh, M.A.; Antar, S.A.; Hazem, R.M.; El-Azab, M.F. Pirfenidone and Vitamin D Ameliorate Cardiac Fibrosis Induced by Doxorubicin in Ehrlich Ascites Carcinoma Bearing Mice: Modulation of Monocyte Chemoattractant Protein-1 and Jun N-terminal Kinase-1 Pathways. Pharmaceuticals 2020, 13, 348. [Google Scholar] [CrossRef]
- Li, C.; Iness, A.; Yoon, J.; Grider, J.R.; Murthy, K.S.; Kellum, J.M.; Kuemmerle, J.F. Noncanonical STAT3 activation regulates excess TGF-beta1 and collagen I expression in muscle of stricturing Crohn’s disease. J. Immunol. Baltim. Md. 1950 2015, 194, 3422–3431. [Google Scholar] [CrossRef]
- Pang, M.; Ma, L.; Gong, R.; Tolbert, E.; Mao, H.; Ponnusamy, M.; Chin, Y.E.; Yan, H.; Dworkin, L.D.; Zhuang, S. A novel STAT3 inhibitor, S3I-201, attenuates renal interstitial fibroblast activation and interstitial fibrosis in obstructive nephropathy. Kidney Int. 2010, 78, 257–268. [Google Scholar] [CrossRef] [PubMed]
- Zheng, C.; Huang, L.; Luo, W.; Yu, W.; Hu, X.; Guan, X.; Cai, Y.; Zou, C.; Yin, H.; Xu, Z.; et al. Inhibition of STAT3 in tubular epithelial cells prevents kidney fibrosis and nephropathy in STZ-induced diabetic mice. Cell Death Dis. 2019, 10, 848. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Feng, H.; Kan, T.; Huang, B.; Zhang, M.; Li, Y.; Shi, C.; Wu, M.; Luo, Y.; Yang, J.; et al. Bevacizumab attenuates hepatic fibrosis in rats by inhibiting activation of hepatic stellate cells. PLoS ONE 2013, 8, e73492. [Google Scholar] [CrossRef]
- Nam, E.H.; Park, S.R.; Kim, P.H. TGF-beta1 induces mouse dendritic cells to express VEGF and its receptor (Flt-1) under hypoxic conditions. Exp. Mol. Med. 2010, 42, 606–613. [Google Scholar] [CrossRef]
- Aubrey, B.J.; Kelly, G.L.; Janic, A.; Herold, M.J.; Strasser, A. How does p53 induce apoptosis and how does this relate to p53-mediated tumour suppression? Cell Death Differ. 2018, 25, 104–113. [Google Scholar] [CrossRef]
- Zhou, L.; Fu, P.; Huang, X.R.; Liu, F.; Lai, K.N.; Lan, H.Y. Activation of p53 promotes renal injury in acute aristolochic acid nephropathy. J. Am. Soc. Nephrol. 2010, 21, 31–41. [Google Scholar] [CrossRef]
- Cordenonsi, M.; Montagner, M.; Adorno, M.; Zacchigna, L.; Martello, G.; Mamidi, A.; Soligo, S.; Dupont, S.; Piccolo, S. Integration of TGF-beta and Ras/MAPK signaling through p53 phosphorylation. Science 2007, 315, 840–843. [Google Scholar] [CrossRef]
- Yang, R.; Xu, X.; Li, H.; Chen, J.; Xiang, X.; Dong, Z.; Zhang, D. p53 induces miR199a-3p to suppress SOCS7 for STAT3 activation and renal fibrosis in UUO. Sci. Rep. 2017, 7, 43409. [Google Scholar] [CrossRef]
- Zhang, G.; Oldroyd, S.D.; Huang, L.H.; Yang, B.; Li, Y.; Ye, R.; El Nahas, A.M. Role of apoptosis and Bcl-2/Bax in the development of tubulointerstitial fibrosis during experimental obstructive nephropathy. Exp. Nephrol. 2001, 9, 71–80. [Google Scholar] [CrossRef]
- Jin, J.; Xiong, Y.; Cen, B. Bcl-2 and Bcl-xL mediate resistance to receptor tyrosine kinase-targeted therapy in lung and gastric cancer. Anticancer Drugs 2017, 28, 1141–1149. [Google Scholar] [CrossRef] [PubMed]
- Stoyanoff, T.R.; Todaro, J.S.; Aguirre, M.V.; Zimmermann, M.C.; Brandan, N.C. Amelioration of lipopolysaccharide-induced acute kidney injury by erythropoietin: Involvement of mitochondria-regulated apoptosis. Toxicology 2014, 318, 13–21. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.T.; Wang, T.E.; Hsu, Y.T.; Chou, C.C.; Huang, K.H.; Hsu, C.C.; Liang, H.J.; Chang, H.W.; Lee, T.H.; Tsai, P.S. Nanoparticulated Honokiol Mitigates Cisplatin-Induced Chronic Kidney Injury by Maintaining Mitochondria Antioxidant Capacity and Reducing Caspase 3-Associated Cellular Apoptosis. Antioxidants 2019, 8, 466. [Google Scholar] [CrossRef]
- Nemmar, A.; Karaca, T.; Beegam, S.; Yuvaraju, P.; Yasin, J.; Hamadi, N.K.; Ali, B.H. Prolonged Pulmonary Exposure to Diesel Exhaust Particles Exacerbates Renal Oxidative Stress, Inflammation and DNA Damage in Mice with Adenine-Induced Chronic Renal Failure. Cell Physiol. Biochem. 2016, 38, 1703–1713. [Google Scholar] [CrossRef] [PubMed]
- Awad, A.M.; Saleh, M.A.; Abu-Elsaad, N.M.; Ibrahim, T.M. Erlotinib can halt adenine induced nephrotoxicity in mice through modulating ERK1/2, STAT3, p53 and apoptotic pathways. Sci. Rep. 2020, 10, 11524. [Google Scholar] [CrossRef] [PubMed]
- Tan, J.R.; Chakravarthi, S.; Judson, J.P.; Haleagrahara, N.; Segarra, I. Potential protective effect of sunitinib after administration of diclofenac: Biochemical and histopathological drug-drug interaction assessment in a mouse model. Naunyn. Schmiedebergs Arch Pharmacol. 2013, 386, 619–633. [Google Scholar] [CrossRef]
- Ohkawa, H.; Ohishi, N.; Yagi, K. Assay for lipid peroxides in animal tissues by thiobarbituric acid reaction. Anal. Biochem. 1979, 95, 351–358. [Google Scholar] [CrossRef]
- Aebi, H.E. Catalase. In Methods of Enzymatic Analysis; Bergmeyer, H.U., Bergmeyer, J., Grabi, M., Eds.; Verlag Chimie: Weinheim, Germany, 1983; pp. 273–286. [Google Scholar]
- Kinomura, M.; Kitamura, S.; Tanabe, K.; Ichinose, K.; Hirokoshi, K.; Takazawa, Y.; Kitayama, H.; Nasu, T.; Sugiyama, H.; Yamasaki, Y.; et al. Amelioration of cisplatin-induced acute renal injury by renal progenitor-like cells derived from the adult rat kidney. Cell Transpl. 2008, 17, 143–158. [Google Scholar] [CrossRef]
- Yamate, J.; Tatsumi, M.; Nakatsuji, S.; Kuwamura, M.; Kotani, T.; Sakuma, S. Immunohistochemical observations on the kinetics of macrophages and myofibroblasts in rat renal interstitial fibrosis induced by cis-diamminedichloroplatinum. J. Comp. Pathol. 1995, 112, 27–39. [Google Scholar] [CrossRef]
- Percicote, A.P. Immunohistochemical expression of p53, BCL-2, BAX and VEGFR1 proteins in nephroblastomas. J. Bras. Patol. Med. Lab. 2013, 49, 50–56. [Google Scholar] [CrossRef]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Treatment Groups (n = 10 Per Group) | Tubular Injury | Fibrosis Score | Caspase-3 |
|---|---|---|---|
| normal | 0.00 ± 0.000 | 1.14 ± 0.034 | 3 ± 0.611 |
| adenine | 2.35 ± 0.150 | 3.32 ± 0.094 *** | 58.14 ± 4.444 *** |
| sunitinib | 1.45 ± 0.114 ††† | 1.57 ± 0.110 **††† | 3 ± 0.583 ††† |
| adenine+sunitinib | 1.65 ± 0.131 †† | 1.52 ± 0.068 **††† | 13.14 ± 1.334 **††† |
| Treatment Groups (n = 10 Per Group) | Week | ||||
|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | ||
| normal | □ | □ | □ | □ | Sacrifice |
| adenine | ☼□ | ☼□ | ☼□ | ☼□ | |
| sunitinib | ■ | ■ | ■ | ■ | |
| adenine+sunitinib | ☼■ | ☼■ | ☼■ | ☼■ | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Saleh, M.A.; Awad, A.M.; Ibrahim, T.M.; Abu-Elsaad, N.M. Small-Dose Sunitinib Modulates p53, Bcl-2, STAT3, and ERK1/2 Pathways and Protects against Adenine-Induced Nephrotoxicity. Pharmaceuticals 2020, 13, 397. https://doi.org/10.3390/ph13110397
Saleh MA, Awad AM, Ibrahim TM, Abu-Elsaad NM. Small-Dose Sunitinib Modulates p53, Bcl-2, STAT3, and ERK1/2 Pathways and Protects against Adenine-Induced Nephrotoxicity. Pharmaceuticals. 2020; 13(11):397. https://doi.org/10.3390/ph13110397
Chicago/Turabian StyleSaleh, Mohamed A., Ahmed M. Awad, Tarek M. Ibrahim, and Nashwa M. Abu-Elsaad. 2020. "Small-Dose Sunitinib Modulates p53, Bcl-2, STAT3, and ERK1/2 Pathways and Protects against Adenine-Induced Nephrotoxicity" Pharmaceuticals 13, no. 11: 397. https://doi.org/10.3390/ph13110397
APA StyleSaleh, M. A., Awad, A. M., Ibrahim, T. M., & Abu-Elsaad, N. M. (2020). Small-Dose Sunitinib Modulates p53, Bcl-2, STAT3, and ERK1/2 Pathways and Protects against Adenine-Induced Nephrotoxicity. Pharmaceuticals, 13(11), 397. https://doi.org/10.3390/ph13110397