New Class of Efficient T2 Magnetic Resonance Imaging Contrast Agent: Carbon-Coated Paramagnetic Dysprosium Oxide Nanoparticles

 ,
,  ,
,  , ,
, , 
Abstract

1. Introduction
2. Results
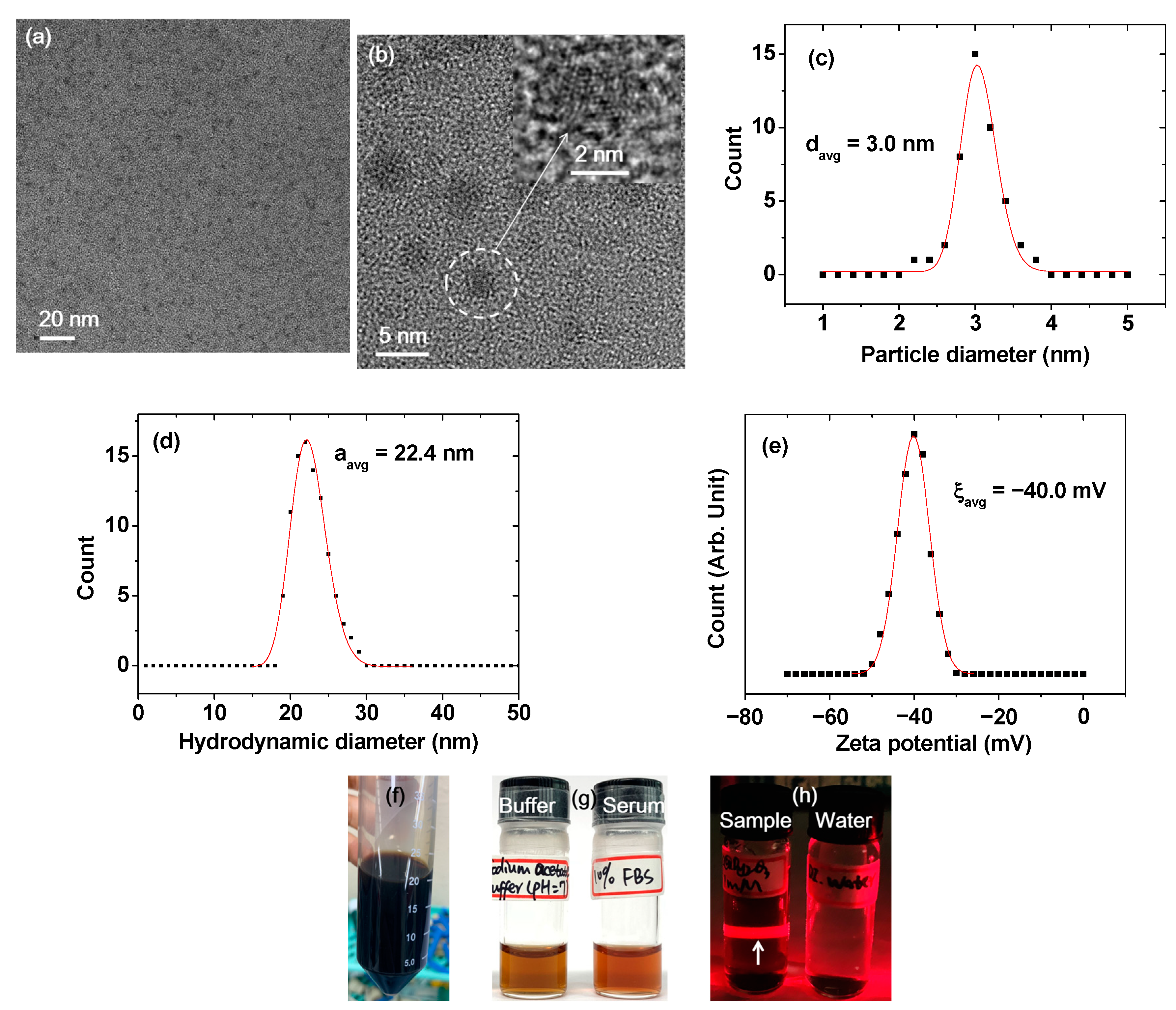
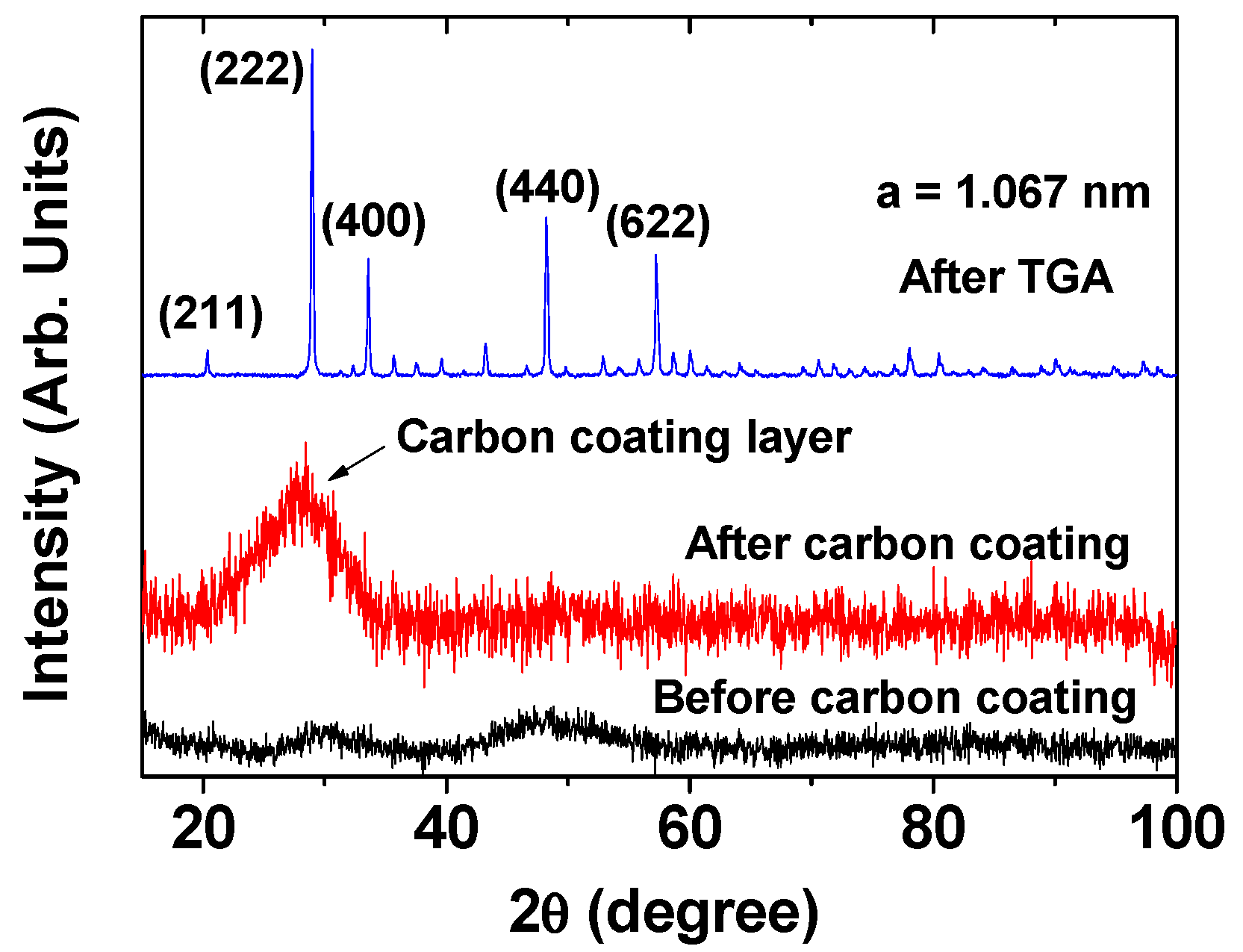
2.1. Size, Colloidal Stability, and Crystallinity of DYO@C Nanoparticles
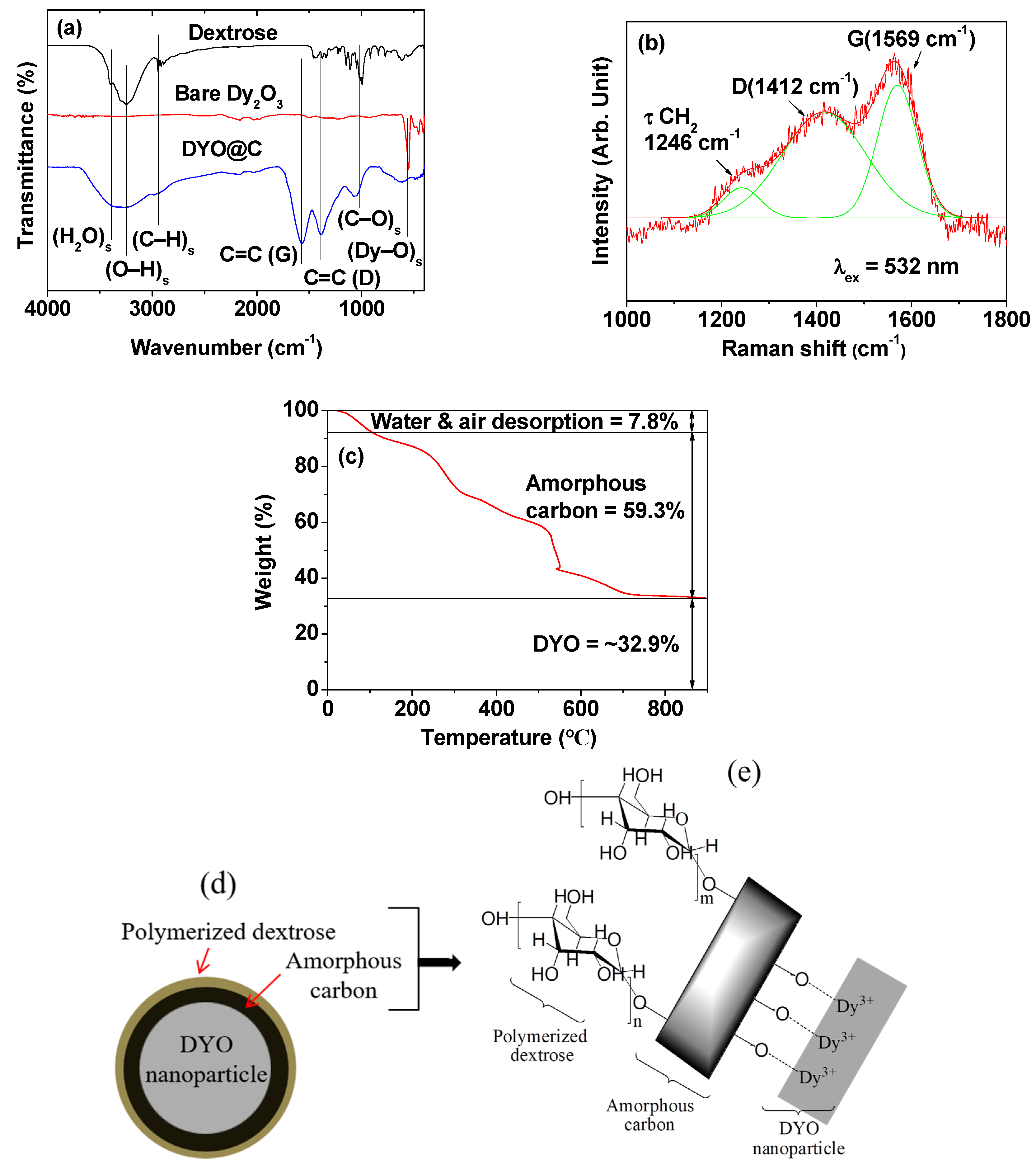
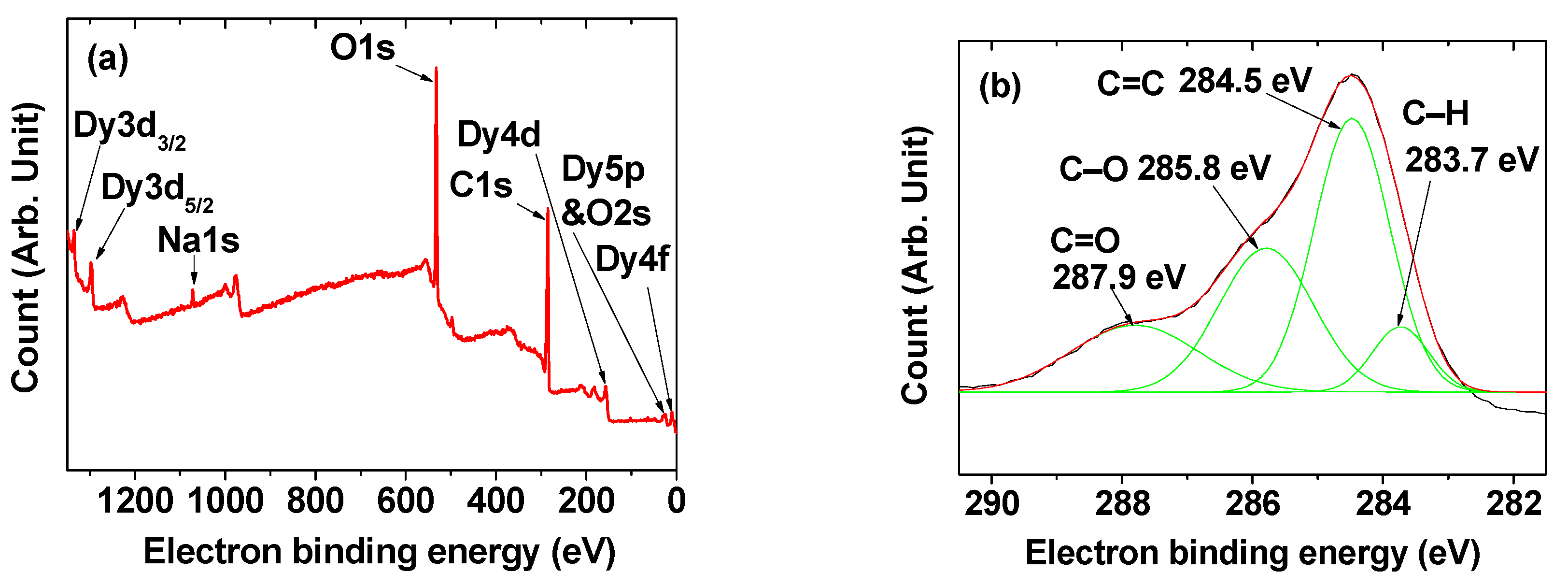
2.2. Surface-Coating Amount and Surface-Coating Structure
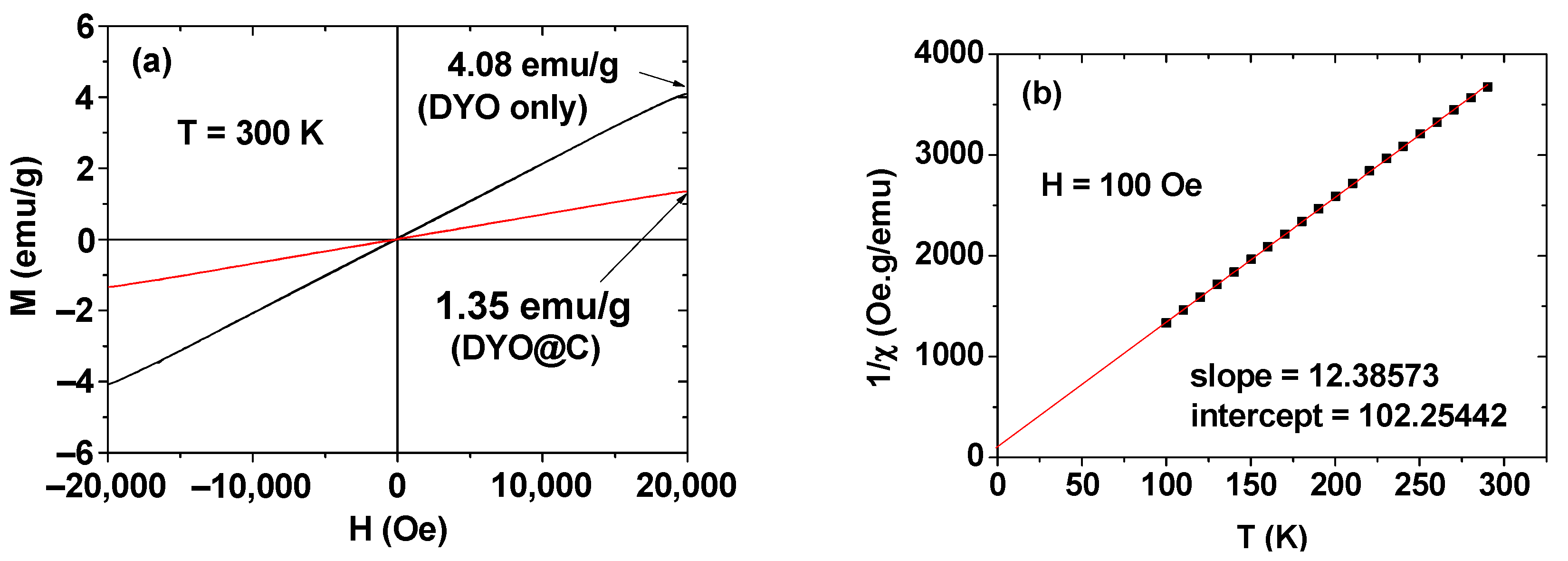
2.3. Magnetic Properties
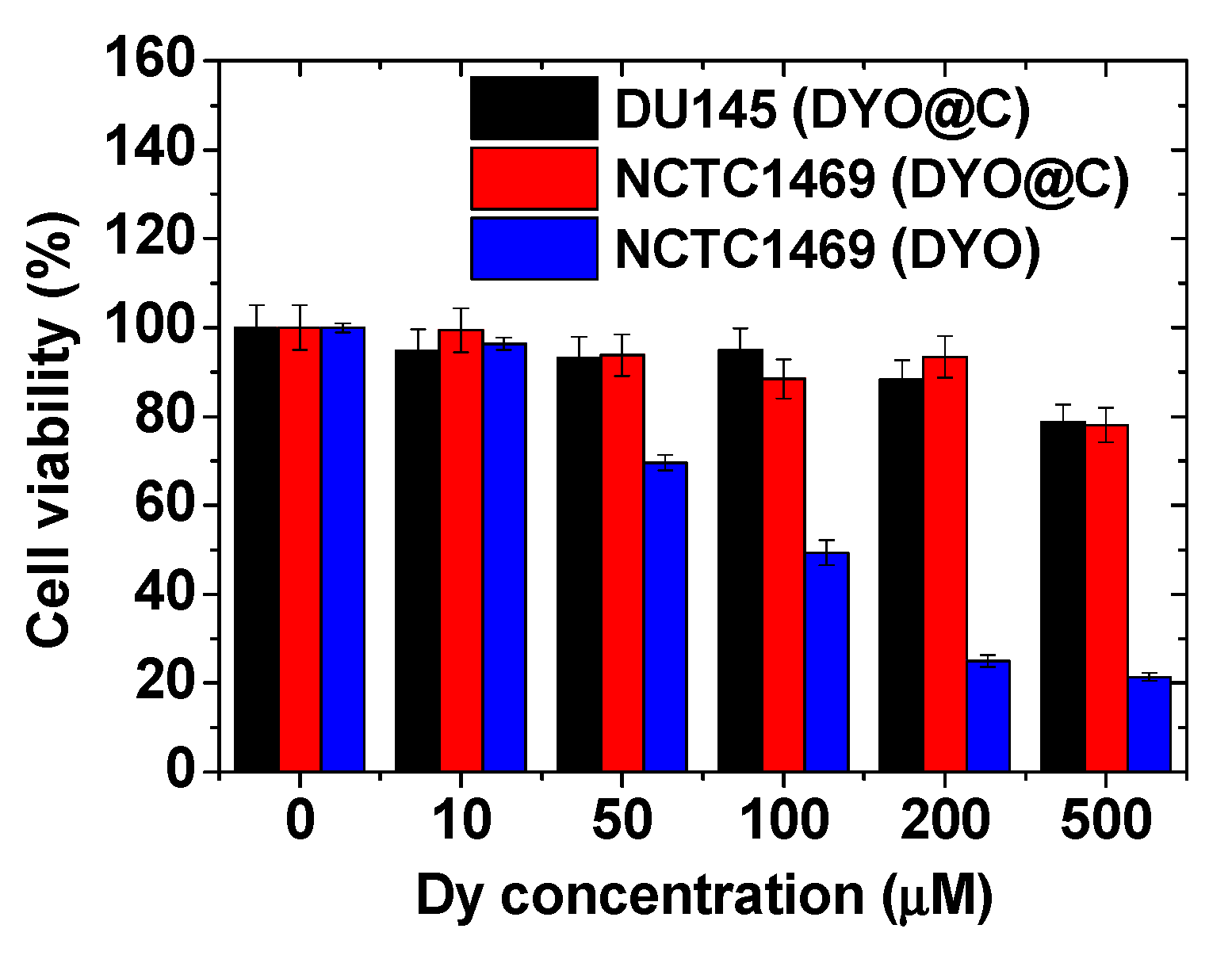
2.4. In Vitro Cytotoxicity Results
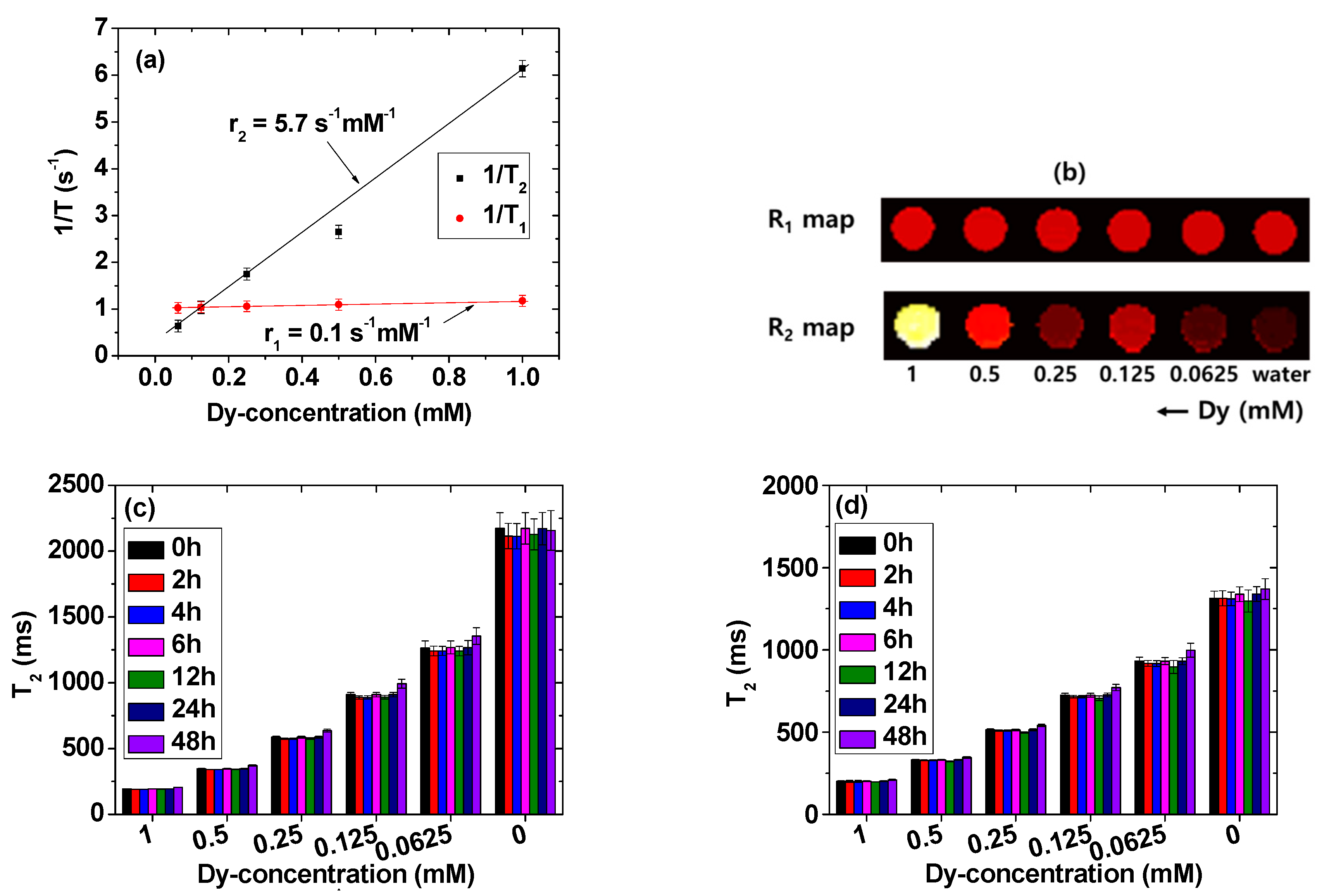
2.5. Longitudinal (r1) and Transverse (r2) Water Proton Spin Relaxivities
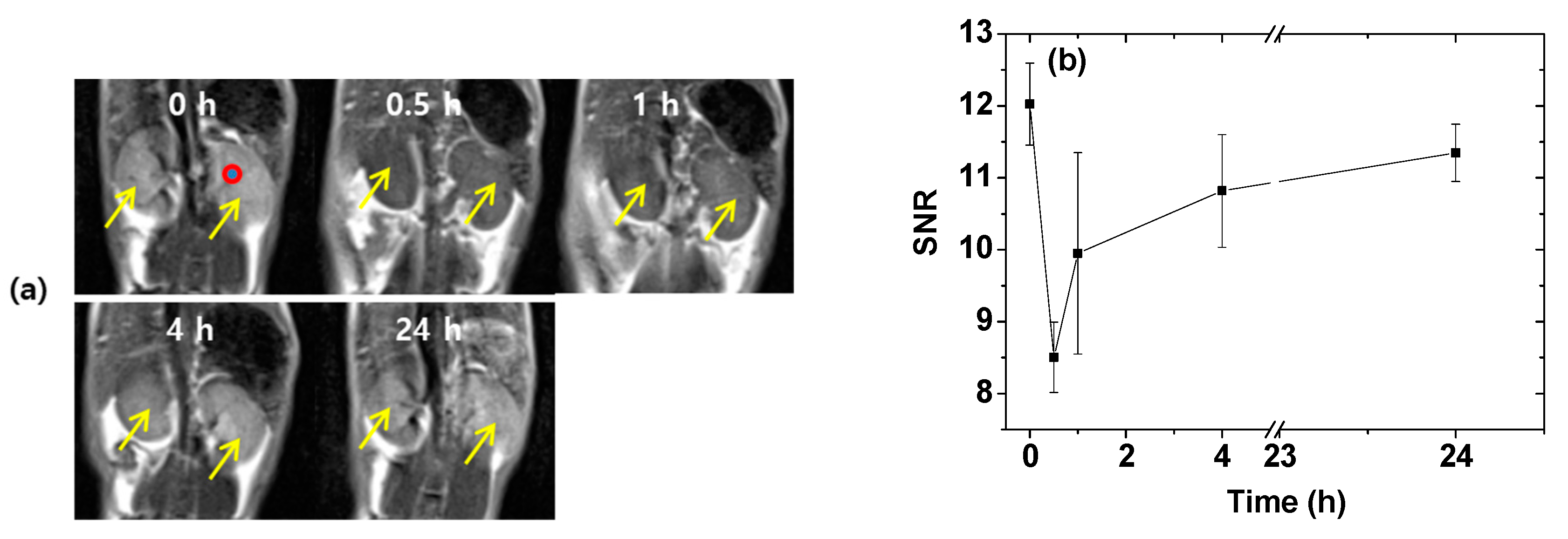
2.6. In Vivo T2 MR Images
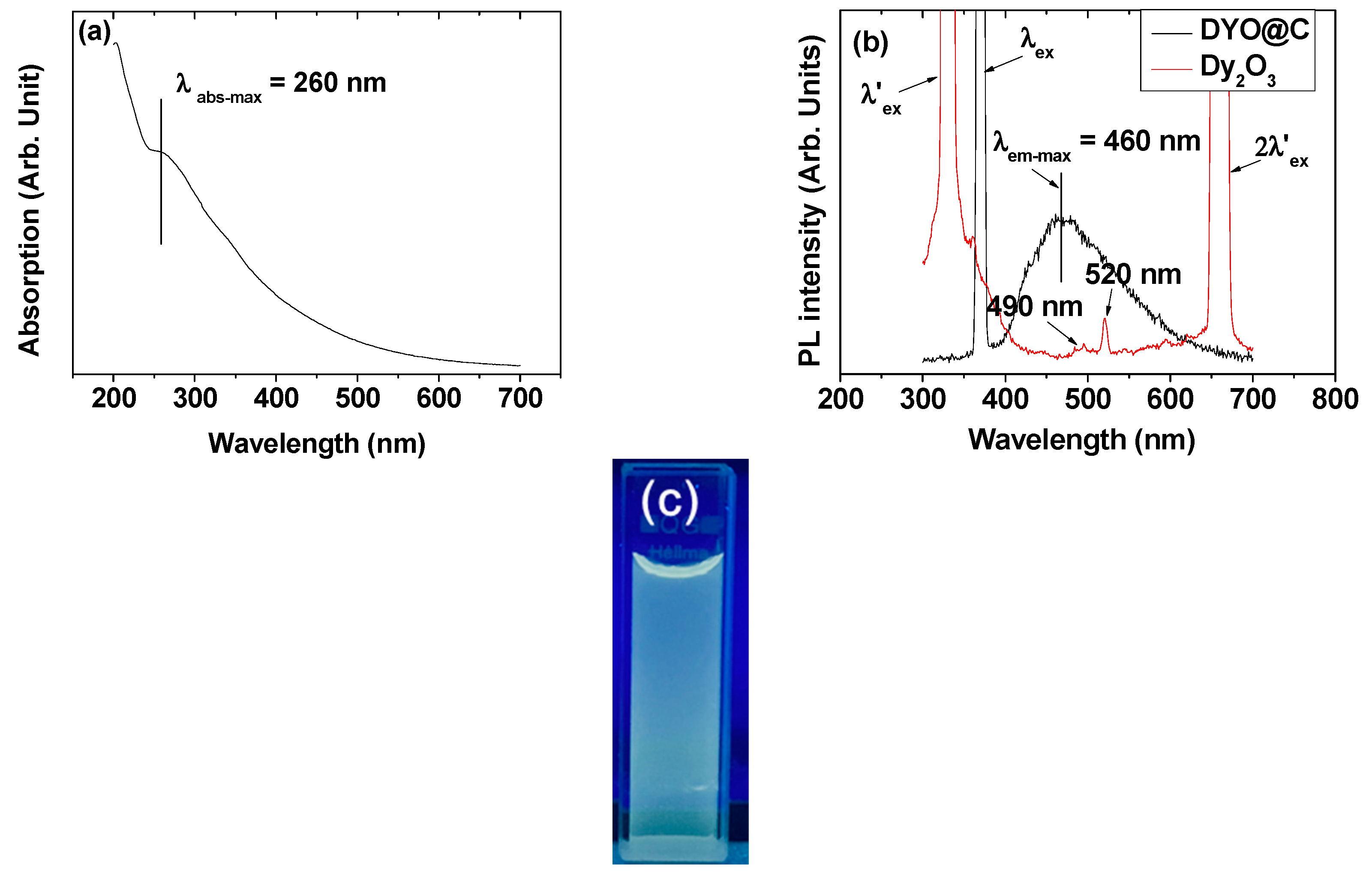
2.7. Optical Properties: Ultraviolet (UV)-Visible Absorption and Photoluminescent (PL) Spectra
3. Discussion
4. Materials and Methods
4.1. Materials
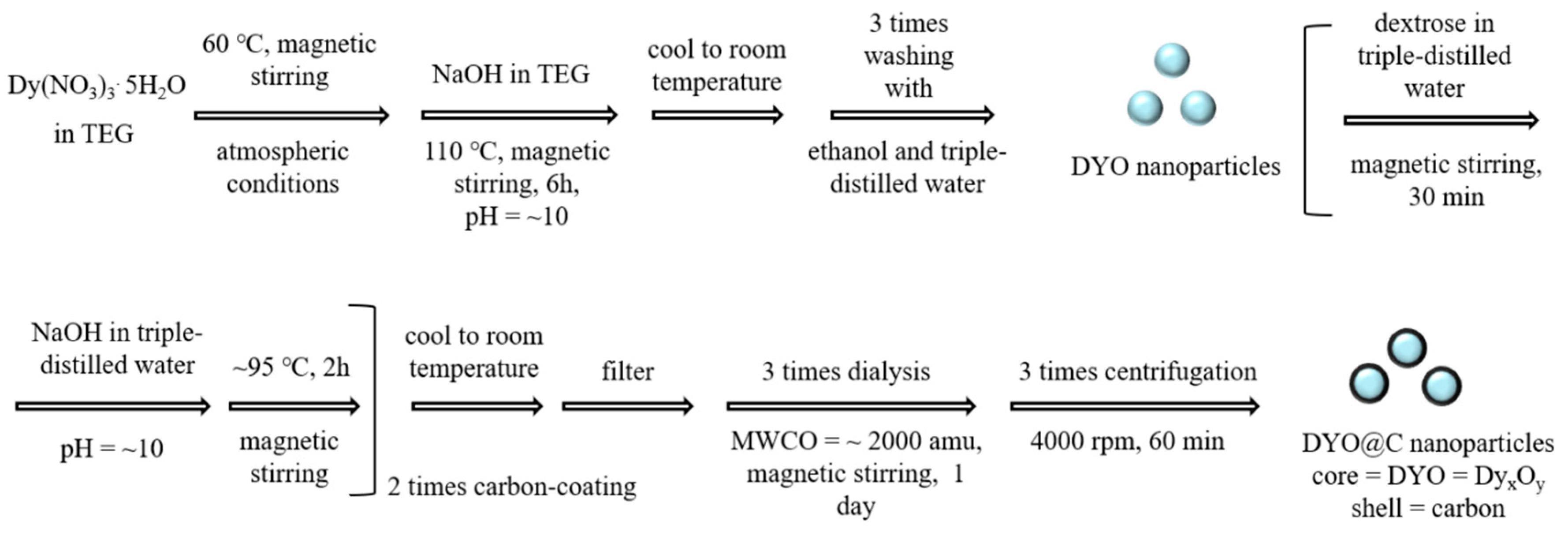
4.2. Synthesis of DYO@C Nanoparticles
4.3. Characterizations
4.4. In Vitro Cell Viability Measurements
4.5. Measurements of Water Proton Spin Relaxation Times and Map Images
4.6. In Vivo T2 MR Image Measurements
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Chang, E.H.; Harford, J.B.; Eaton, M.A.W.; Boisseau, P.M.; Dube, A.; Hayeshi, R.; Swai, H.; Lee, D.S. Nanomedicine: Past, present and future-A global perspective. Biochem. Biophys. Res. Commun. 2015, 468, 511–517. [Google Scholar] [CrossRef]
- Soares, S.; Sousa, J.; Pais, A.; Vitorino, C. Nanomedicine: Principles, properties, and regulatory issues. Front. Chem. 2018, 6, 360. [Google Scholar] [CrossRef] [PubMed]
- Jeevanandam, J.; Barhoum, A.; Chan, Y.S.; Dufresne, A.; Danquah, M.K. Review on nanoparticles and nanostructured materials: History, sources, toxicity and regulations. Beilstein J. Nanotechnol. 2018, 9, 1050–1074. [Google Scholar] [CrossRef] [PubMed]
- Khan, I.; Saeed, K.; Khan, I. Nanoparticles: Properties, applications and toxicities. Arab. J. Chem. 2019, 12, 908–931. [Google Scholar] [CrossRef]
- Nune, S.K.; Gunda, P.; Thallapally, P.K.; Lin, Y.-Y.; Forrest, M.L.; Berkland, C.J. Nanoparticles for biomedical imaging. Expert Opin. Drug Deliv. 2009, 6, 1175–1194. [Google Scholar] [CrossRef]
- Choi, H.S.; Frangioni, J.V. Nanoparticles for biomedical imaging: Fundamentals of clinical translation. Mol. Imaging 2010, 9, 291–310. [Google Scholar] [CrossRef]
- Neuwelt, A.; Sidhu, N.; Hu, C.-A.A.; Mlady, G.; Eberhardt, S.C.; Sillerud, L.O. Iron-based superparamagnetic nanoparticle contrast agents for MRI of infection and inflammation. Am. J. Roentgenol. 2015, 204, W302–W313. [Google Scholar] [CrossRef]
- Stephen, Z.R.; Kievit, F.M.; Zhang, M. Magnetic nanoparticles for medical MR imaging. Mater. Today 2011, 14, 330–338. [Google Scholar] [CrossRef]
- Na, H.B.; Song, I.C.; Hyeon, T. Inorganic nanoparticles for MRI contrast agents. Adv. Mater. 2009, 21, 2133–2148. [Google Scholar] [CrossRef]
- Cao, Y.; Xu, L.; Kuang, Y.; Xiong, D.; Pei, R. Gadolinium-based nanoscale MRI contrast agents for tumor imaging. J. Mater. Chem. B 2017, 5, 3431–3461. [Google Scholar] [CrossRef]
- Zhou, Z.; Lu, Z.-R. Gadolinium-based contrast agents for MR cancer imaging. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2013, 5, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Cheng, W.; Ping, Y.; Zhang, Y.; Chuang, K.-H.; Liu, Y. Magnetic Resonance Imaging (MRI) contrast agents for tumor diagnosis. J. Health Eng. 2013, 4, 23–46. [Google Scholar] [CrossRef] [PubMed]
- Wahsner, J.; Gale, E.M.; Rodríguez-Rodríguez, A.; Caravan, P. Chemistry of MRI contrast agents: Current challenges and new frontiers. Chem. Rev. 2018, 119, 957–1057. [Google Scholar] [CrossRef] [PubMed]
- Caravan, P.; Ellison, J.J.; McMurry, T.J.; Lauffer, R.B. Gadolinium (III) chelates as MRI contrast agents: Structure, dynamics, and applications. Chem. Rev. 1999, 99, 2293–2352. [Google Scholar] [CrossRef]
- Reimer, P.; Balzer, T. Ferucarbotran (Resovist): A new clinically approved RES-specific contrast agent for contrast-enhanced MRI of the liver: Properties, clinical development, and applications. Eur. Radiol. 2003, 13, 1266–1276. [Google Scholar] [CrossRef]
- Bloembergen, N.; Purcell, E.M.; Pound, R.V. Relaxation effects in nuclear magnetic resonance absorption. Phys. Rev. 1948, 73, 679–712. [Google Scholar] [CrossRef]
- Norek, M.; Pereira, G.A.D.L.; Geraldes, C.F.G.C.; Denkova, A.; Zhou, W.; Peters, J.A. NMR Transversal relaxivity of suspensions of lanthanide oxide nanoparticles. J. Phys. Chem. C 2007, 111, 10240–10246. [Google Scholar] [CrossRef]
- Norek, M.; Kampert, E.; Zeitler, U.; Peters, J.A. Tunning of the size of Dy2O3 nanoparticles for optimal performance as an MRI contrast agent. J. Am. Chem. Soc. 2008, 130, 5335–5340. [Google Scholar] [CrossRef]
- Marasini, S.; Yue, H.; Ho, S.L.; Jung, K.-H.; Park, J.A.; Cha, H.; Ghazanfari, A.; Ahmad, M.Y.; Liu, S.; Jang, Y.J.; et al. D-glucuronic acid-coated ultrasmall paramagnetic Ln2O3 (Ln = Tb, Dy, and Ho) nanoparticles: Magnetic properties, water proton relaxivities, and fluorescence properties. Eur. J. Inorg. Chem. 2019, 2019, 3832–3839. [Google Scholar] [CrossRef]
- Das, G.K.; Zhang, Y.; D’Silva, L.; Padmanabhan, P.; Heng, B.C.; Loo, J.S.C.; Selvan, S.T.; Bhakoo, K.K.; Tan, T.T.Y. Single-phase Dy2O3:Tb3+ nanocrystals as dual-modal contrast agent for high field magnetic resonance and optical imaging. Chem. Mater. 2011, 23, 2439–2446. [Google Scholar] [CrossRef]
- Kattel, K.; Park, J.Y.; Xu, W.; Kim, H.G.; Lee, E.J.; Alam Bony, B.; Heo, W.C.; Lee, J.J.; Jin, S.; Baeck, J.S.; et al. A facile synthesis, in vitro and in vivo MR studies of d-glucuronic acid-coated ultrasmall Ln2O3 (Ln = Eu, Gd, Dy, Ho, and Er) nanoparticles as a new potential MRI contrast agent. ACS Appl. Mater. Interfaces 2011, 3, 3325–3334. [Google Scholar] [CrossRef] [PubMed]
- Kattel, K.; Park, J.Y.; Xu, W.; Kim, H.G.; Lee, E.J.; Alam Bony, B.; Heo, W.C.; Jin, S.; Baeck, J.S.; Chang, Y.; et al. Paramagnetic dysprosium oxide nanoparticles and dysprosium hydroxide nanorods as T2 MRI contrast agents. Biomaterials 2012, 33, 3254–3261. [Google Scholar] [CrossRef] [PubMed]
- Fiorito, S.; Serafino, A.; Andreola, F.; Togna, A.; Togna, G. Toxicity and biocompatibility of carbon nanoparticles. J. Nanosci. Nanotechnol. 2006, 6, 591–599. [Google Scholar] [CrossRef] [PubMed]
- Tegafaw, T.; Oh, I.T.; Cha, H.; Yue, H.; Miao, X.; Ho, S.L.; Ahmad, M.Y.; Marasini, S.; Ghazanfari, A.; Kim, H.-K.; et al. Facile synthesis of stable colloidal suspension of amorphous carbon nanoparticles in aqueous medium and their characterization. J. Phys. Chem. Solids 2018, 120, 96–103. [Google Scholar] [CrossRef]
- Lim, S.Y.; Shen, W.; Gao, Z. Carbon quantum dots and their applications. Chem. Soc. Rev. 2015, 44, 362–381. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.M.; Duan, H.; Rhyner, M.N.; Ruan, G.; Nie, S. A systematic examination of surface coatings on the optical and chemical properties of semiconductor quantum dots. Phys. Chem. Chem. Phys. 2006, 8, 3895–3903. [Google Scholar] [CrossRef]
- Pons, T.; Uyeda, H.T.; Medintz, A.I.L.; Mattoussi, H. Hydrodynamic dimensions, electrophoretic mobility, and stability of hydrophilic quantum dots. J. Phys. Chem. B 2006, 110, 20308–20316. [Google Scholar] [CrossRef] [PubMed]
- Pellegrino, T.; Manna, L.; Kudera, S.; Liedl, T.; Koktysh, D.; Rogach, A.L.; Keller, S.; Rädler, J.; Natile, A.G.; Parak, W.J. Hydrophobic nanocrystals coated with an amphiphilic polymer shell: A general route to water soluble nanocrystals. Nano Lett. 2004, 4, 703–707. [Google Scholar] [CrossRef]
- Söderlind, F.; Pedersen, H.; Petoral Jr., R.M.; Käll, P.-O.; Uvdal, K. Synthesis and characterization of Gd2O3 nanocrystals functionalized by organic acids. J. Colloid Interface Sci. 2005, 288, 140–148. [Google Scholar]
- Hara, M.; Yoshida, T.; Takagaki, A.; Takata, T.; Kondo, J.N.; Hayashi, S.; Domen, K. A carbon material as a strong protonic acid. Angew. Chem. Int. Ed. 2004, 43, 2955–2958. [Google Scholar] [CrossRef]
- Tsubouchi, N.; Xu, C.; Ohtsuka, Y. Carbon crystallization during high-temperature pyrolysis of coals and the enhancement by calcium. Energy Fuels 2003, 17, 1119–1125. [Google Scholar] [CrossRef]
- X’pert Highscore, Card No. 01-088-2164, PANalytical Software, Version 2.0.1, 2004. Available online: https://www.malvernpanalytical.com/en/products/category/software/x-ray-diffraction-software/highscore-with-plus-option?campaignid=1330618933&adgroupid=69605355538&creative=338921965823&keyword=x%20pert%20highscore&matchtype=p&network=g&device=c&gclid=EAIaIQobChMI-96BuM217AIVDD5gCh21CAQUEAAYASAAEgLo3vD_BwE (accessed on 15 October 2020).
- Kaufman, J.H.; Metin, S.; Saperstein, D.D. Symmetry breaking in nitrogen-doped amorphous carbon: Infrared observation of the Raman-active G and D bands. Phys. Rev. B 1989, 39, 13053–13060. [Google Scholar] [CrossRef] [PubMed]
- Malard, L.M.; Pimenta, M.A.; Dresselhaus, G.; Dresselhaus, M.S. Raman spectroscopy in graphene. Phys. Rep. 2009, 473, 51–87. [Google Scholar] [CrossRef]
- Ţucureanu, V.; Matei, A.; Avram, A.M. FTIR Spectroscopy for carbon family study. Crit. Rev. Anal. Chem. 2016, 46, 502–520. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, M.; Alaam, M.; El-Haes, H.; Jalbout, A.F.; De Leon, A. Analysis of the structure and vibrational spectra of glucose and fructose. Eclética Química J. 2006, 31, 15–21. [Google Scholar] [CrossRef]
- Wiercigroch, E.; Szafraniec, E.; Czamara, K.; Pacia, M.Z.; Majzner, K.; Kochan, K.; Kaczor, A.; Baranska, M.; Malek, K. Raman and infrared spectroscopy of carbohydrates: A review. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2017, 185, 317–335. [Google Scholar] [CrossRef]
- Sun, X.; Li, Y. Colloidal carbon spheres and their core/shell structures with noble-metal nanoparticles. Angew. Chem. Int. Ed. 2004, 43, 597–601. [Google Scholar] [CrossRef]
- Moulder, J.F.; Stickle, W.F.; Sobol, P.E.; Bomben, K.D. Handbook of X-ray Photoelectron Spectroscopy; Chastain, J., Ed.; Perkin-Elmer Corporation: Eden Prairie, MN, USA, 1992. [Google Scholar]
- Díaz, J.; Paolicelli, G.; Ferrer, S.; Comin, F. Separation of the sp3 and sp2 components in the C1s photoemission spectra of amorphous carbon films. Phys. Rev. B 1996, 54, 8064–8069. [Google Scholar] [CrossRef]
- Barlow, A.; Popescu, S.M.; Artyushkova, K.; Scott, O.; Sano, N.; Hedley, J.; Cumpson, P.J. Chemically specific identification of carbon in XPS imaging using Multivariate Auger Feature Imaging (MAFI). Carbon 2016, 107, 190–197. [Google Scholar] [CrossRef]
- Ray, S.C.; Saha, A.; Jana, N.R.; Sarkar, R. Fluorescent carbon nanoparticles: Synthesis, characterization, and bioimaging application. J. Phys. Chem. C 2009, 113, 18546–18551. [Google Scholar] [CrossRef]
- Arajs, S.; Colvin, R.V. Magnetic susceptibility of gadolinium and dysprosium sesquioxides at elevated temperatures. J. Appl. Phys. 1962, 33, 2517. [Google Scholar] [CrossRef]
- Lal, H.B.; Pratap, V.; Kumar, A. Magnetic susceptibility of heavy rare-earth sesquioxides. Pramana 1978, 10, 409–412. [Google Scholar] [CrossRef]
- Greenwood, N.; Earnshaw, A. Chemistry of the Elements; Butterworth-Heinemann: Oxford, UK, 1977; p. 1243. [Google Scholar]
- Jiles, D. Introduction to Magnetism and Magnetic Materials; Chapman & Hall: London, UK, 1991; p. 185. [Google Scholar]
- Hainfeld, J.F.; Slatkin, D.N.; Focella, T.M.; Smilowitz, H.M. Gold nanoparticles: A new X-ray contrast agent. Br. J. Radiol. 2006, 79, 248–253. [Google Scholar] [CrossRef]
- Choi, H.S.; Liu, W.; Misra, P.; Tanaka, E.; Zimmer, J.P.; Ipe, B.I.; Bawendi, M.G.; Frangioni, J.V. Renal clearance of quantum dots. Nat. Biotechnol. 2007, 25, 1165–1170. [Google Scholar] [CrossRef] [PubMed]
- Longmire, M.; Choyke, P.L.; Kobayashi, H. Clearance properties of nano-sized particles and molecules as imaging agents: Considerations and caveats. Nanomedicine 2008, 3, 703–717. [Google Scholar] [CrossRef] [PubMed]
- Chandrasekhar, M.; Sunitha, D.; Dhananjaya, N.; Nagabhushana, H.; Sharma, S.; Nagabhushana, B.; Shivakumara, C.; Chakradhar, R. Structural and phase dependent thermo and photoluminescent properties of Dy(OH)3 and Dy2O3 nanorods. Mater. Res. Bull. 2012, 47, 2085–2094. [Google Scholar] [CrossRef]
- Kofod, N.; Arppe, R.; Sørensen, T.J. Electronic energy levels of dysprosium (III) ions in solution. assigning the emitting state and the intraconfigurational 4f-4f transitions in the Vis–NIR region and photophysical characterization of Dy (III) in water, methanol, and dimethyl sulfoxide. J. Phys. Chem. A 2019, 123, 2734–2744. [Google Scholar] [CrossRef]
- Jung, C.W.; Jacobs, P. Physical and chemical properties of superparamagnetic iron oxide MR contrast agents: Ferumoxides, ferumoxtran, ferumoxsil. Magn. Reson. Imaging 1995, 13, 661–674. [Google Scholar] [CrossRef]
- Saini, S.; Edelman, R.R.; Sharma, P.; Li, W.; Mayo-Smith, W.; Slater, G.J.; Eisenberg, P.J.; Hahn, P.F. Blood-pool MR contrast material for detection and characterization of focal hepatic lessions: Initial clinical experience with ultrasmall superparamagnetic iron oxide (AMI-227). AJR 1995, 164, 1147–1152. [Google Scholar] [CrossRef][Green Version]
- Yue, H.; Marasini, S.; Ahmad, M.Y.; Ho, S.L.; Cha, H.; Liu, S.; Jang, Y.J.; Tegafaw, T.; Ghazanfari, A.; Miao, X.; et al. Carbon-coated ultrasmall gadolinium oxide (Gd2O3@C) nanoparticles: Application to magnetic resonance imaging and fluorescence properties. Colloids Surf. A Physicochem. Eng. Asp. 2020, 586, 124261. [Google Scholar] [CrossRef]
- Weinmann, H.J.; Brasch, R.C.; Press, W.R.; Wesbey, G.E. Characteristics of gadolinium-DTPA complex: A potential NMR contrast agent. Am. J. Roentgenol. 1984, 142, 619–624. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| davg 1 (nm) | aav 2 (nm) | ξavg 3 (mV) | Surface Coating Amount (wt%) | M 4 at 2.0 T (emu/g) | r1 5 (s−1mM−1) | r2 6 (s−1mM−1) | r2/r1 | λabs-max 7 (nm) | λem-max 8 (nm) | η 9 (%) | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| TGA | EA | ||||||||||
| 3.0 ± 0.1 | 22.4 ± 0.1 | −40.0 ± 0.2 | 59.3 | 63.32 | 4.08 | 0.1 | 5.7 | 57 | 260 | 460 | 6.5 |
| Element | Orbital | Observed EBE (eV) | Literature | Ref. |
|---|---|---|---|---|
| C | 1s, C=O | 287.9 | 288.3 | [42] |
| 1s, C–O | 285.8 | 285.6 | [42] | |
| 1s, C=C | 284.5 | 284.3, 284.5, 284.4 | [40,41,42] | |
| 1s, C–H | 283.7 | 281.8 | [42] | |
| O | 1s | 531.6 | 531 | [39] |
| 2s | 25.0 | 23 | [39] | |
| Na | 1s | 1072.0 | 1072.1 | [39] |
| Dy | 3d3/2 | 1334.5 | 1333 | [39] |
| 3d5/2 | 1296.4 | 1296 | [39] | |
| 4d | 155.6 | 152 | [39] | |
| 5p | 25.0 | 23 | [39] | |
| 4f | 10.0 | 8 | [39] |
| Nanoparticle | davg (nm) | aavg (nm) | Coating Material | r1 (s−1mM−1) | r2 (s−1mM−1) | r2/r1 | H (tesla) | T (℃) | Medium | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|
| DYO | 3.0 | 22.4 | Carbon | 0.1 | 5.7 | 57 | 3.0 | 22 | Water | This work |
| Fe3O4/γ-Fe2O3 1 | 4.2 | 60 | Dextran 40000 | 19.4 | 185.8 | 9.6 | 0.47 | 40 | Plasma | [15] |
| Fe3O4 2 | 4.9 | 21 | Dextran T-10 | 22.7 | 53.1 | 2.3 | 0.47 | 39 | Water | [52,53] |
| Gd2O3 | 3.1 | 18.9 | Carbon | 16.3 | 24.1 | 1.5 | 3.0 | 22 | Water | [54] |
| Gd(III)-DTPA 3 | - | - | - | 4.5 | 5.7 | 1.3 | 0.47 | 39 | Water | [55] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yue, H.; Park, J.A.; Ho, S.L.; Ahmad, M.Y.; Cha, H.; Liu, S.; Tegafaw, T.; Marasini, S.; Ghazanfari, A.; Kim, S.; et al. New Class of Efficient T2 Magnetic Resonance Imaging Contrast Agent: Carbon-Coated Paramagnetic Dysprosium Oxide Nanoparticles. Pharmaceuticals 2020, 13, 312. https://doi.org/10.3390/ph13100312
Yue H, Park JA, Ho SL, Ahmad MY, Cha H, Liu S, Tegafaw T, Marasini S, Ghazanfari A, Kim S, et al. New Class of Efficient T2 Magnetic Resonance Imaging Contrast Agent: Carbon-Coated Paramagnetic Dysprosium Oxide Nanoparticles. Pharmaceuticals. 2020; 13(10):312. https://doi.org/10.3390/ph13100312
Chicago/Turabian StyleYue, Huan, Ji Ae Park, Son Long Ho, Mohammad Yaseen Ahmad, Hyunsil Cha, Shuwen Liu, Tirusew Tegafaw, Shanti Marasini, Adibehalsadat Ghazanfari, Soyeon Kim, and et al. 2020. "New Class of Efficient T2 Magnetic Resonance Imaging Contrast Agent: Carbon-Coated Paramagnetic Dysprosium Oxide Nanoparticles" Pharmaceuticals 13, no. 10: 312. https://doi.org/10.3390/ph13100312
APA StyleYue, H., Park, J. A., Ho, S. L., Ahmad, M. Y., Cha, H., Liu, S., Tegafaw, T., Marasini, S., Ghazanfari, A., Kim, S., Chae, K. S., Chang, Y., & Lee, G. H. (2020). New Class of Efficient T2 Magnetic Resonance Imaging Contrast Agent: Carbon-Coated Paramagnetic Dysprosium Oxide Nanoparticles. Pharmaceuticals, 13(10), 312. https://doi.org/10.3390/ph13100312