Development and Validation of a Rapid High-Performance Liquid Chromatography–Tandem Mass Spectrometric Method for Determination of Folic Acid in Human Plasma


Abstract
:
1. Introduction
2. Materials and Methods
2.1. Materials and Reagents
2.2. Liquid Chromatography Tandem Mass Spectrometry
2.3. Preparation of Standard Stock, Calibration Standards, and Quality Control Samples
2.4. Sample Preparation
2.5. Method Validation
3. Results
3.1. Method Development
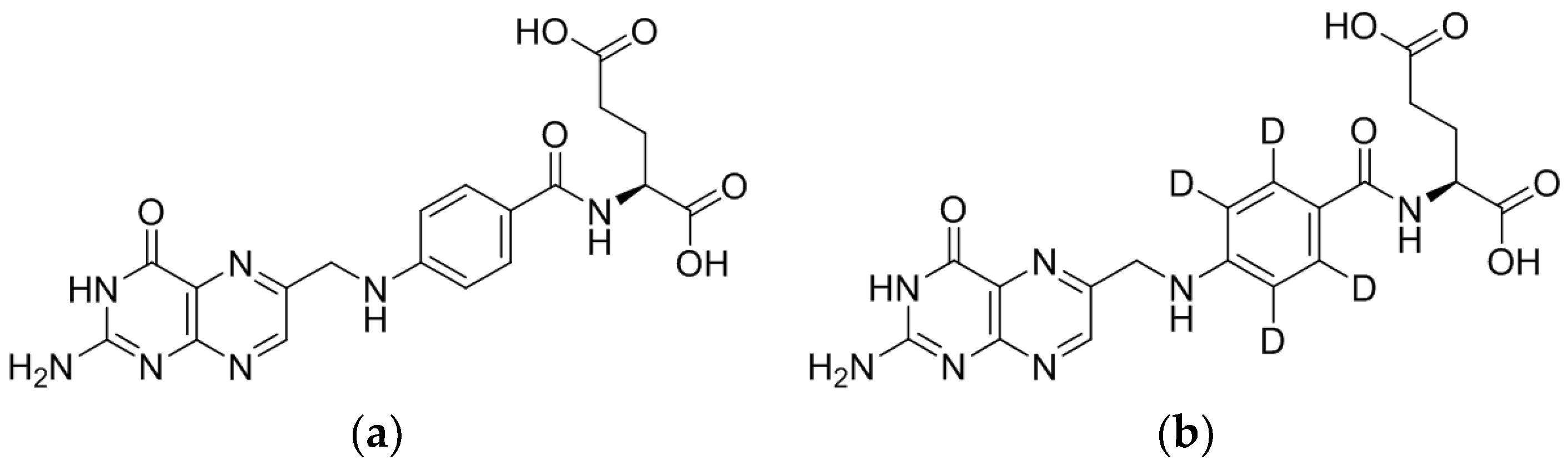
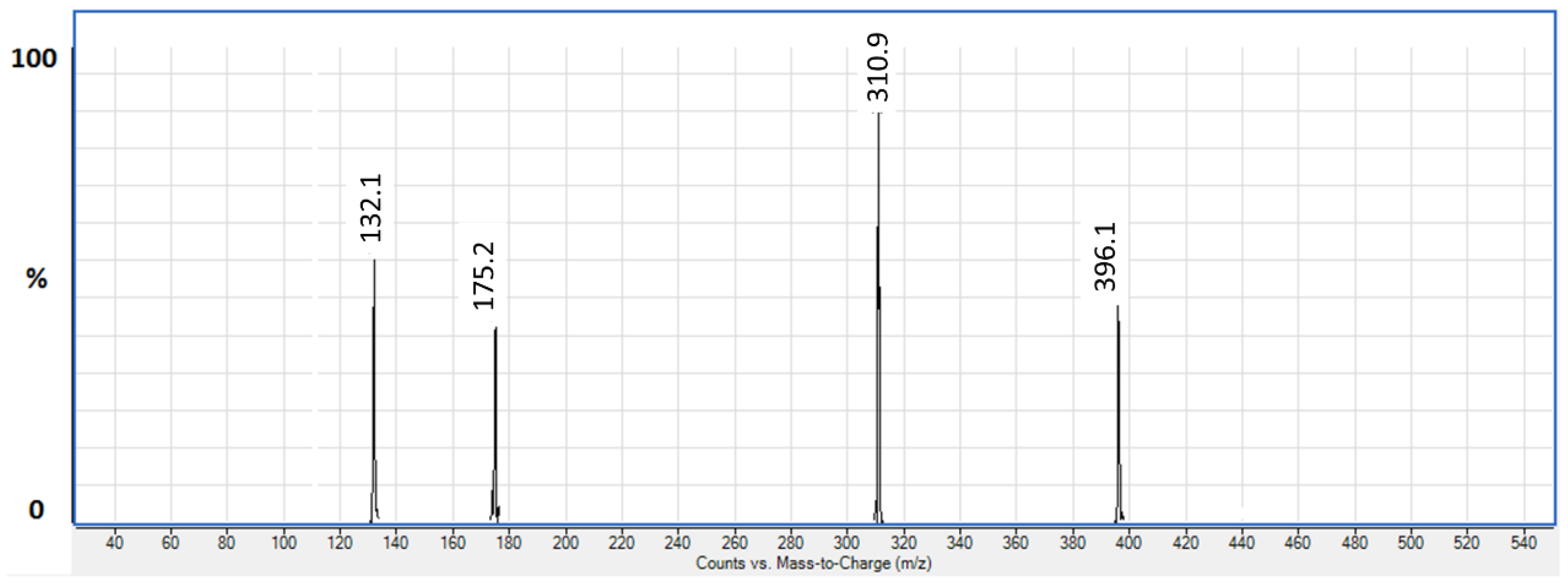
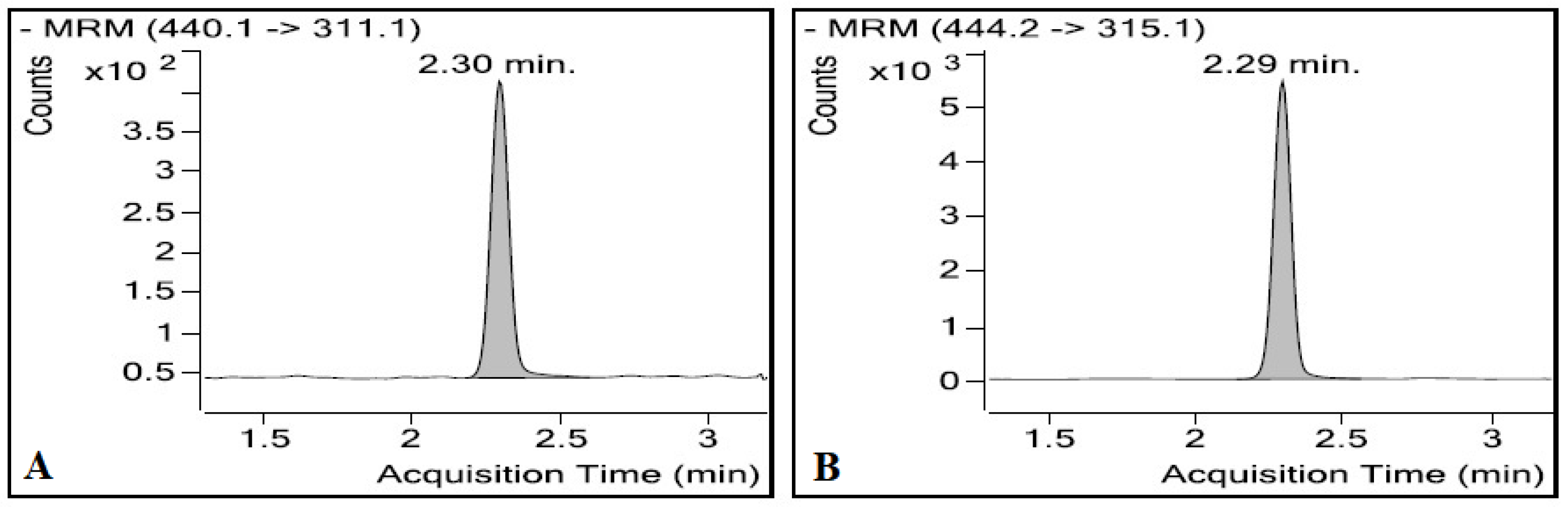
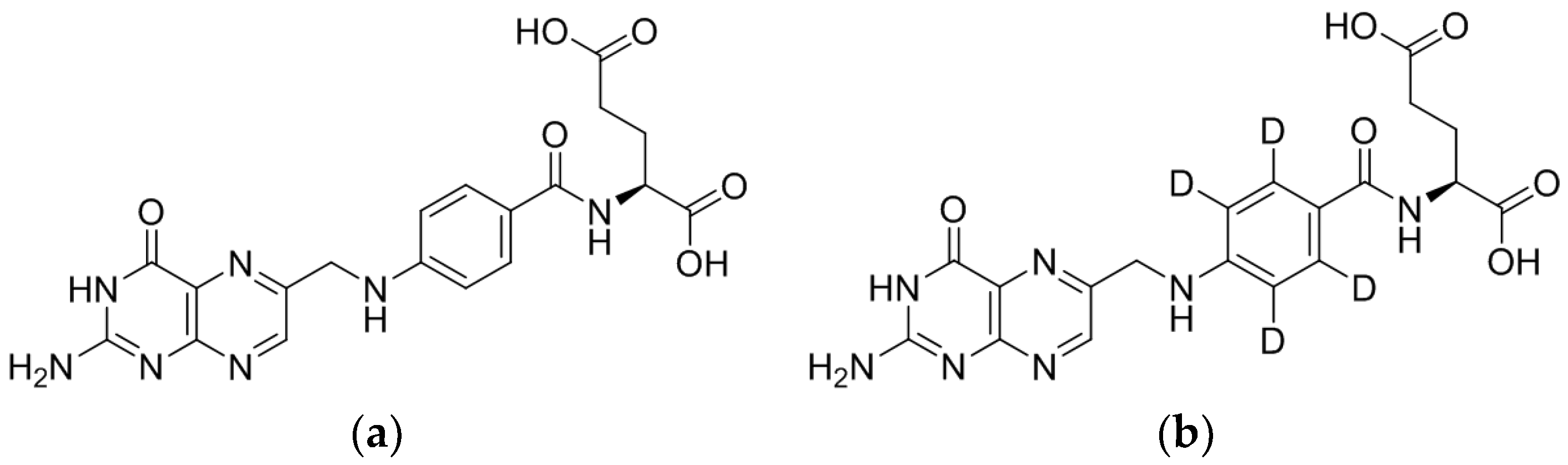
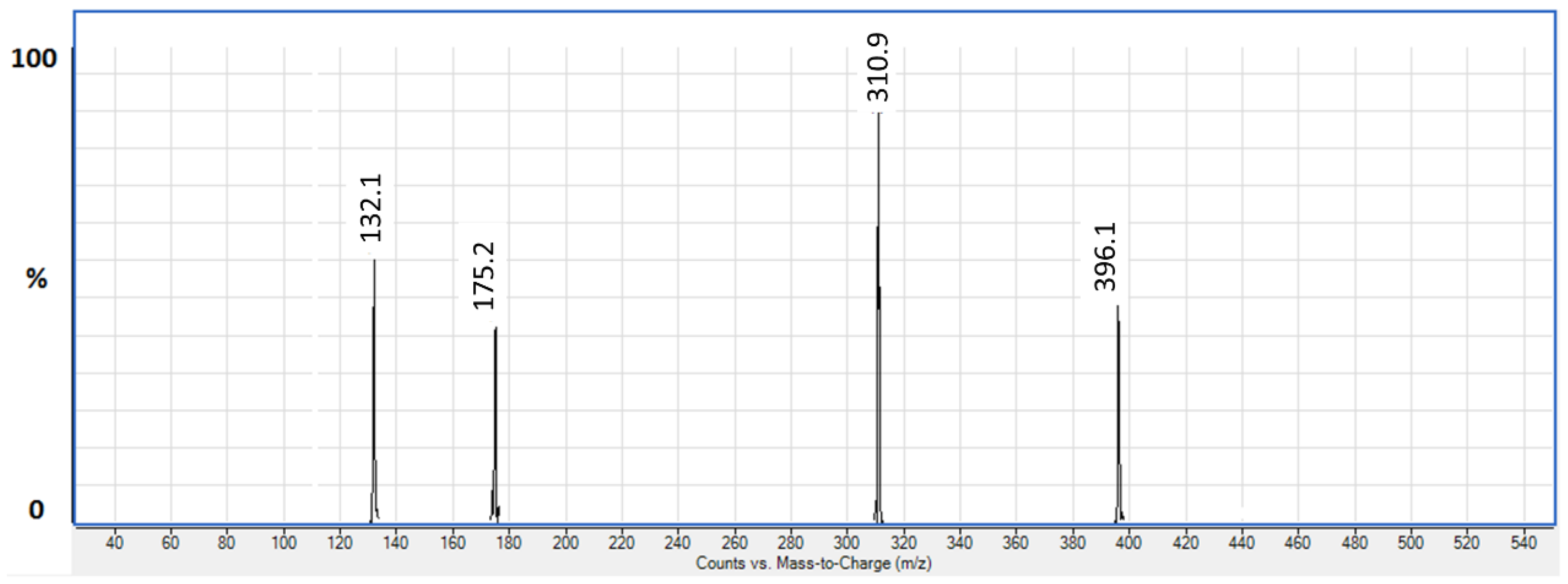
3.1.1. LC/ESI-MS/MS
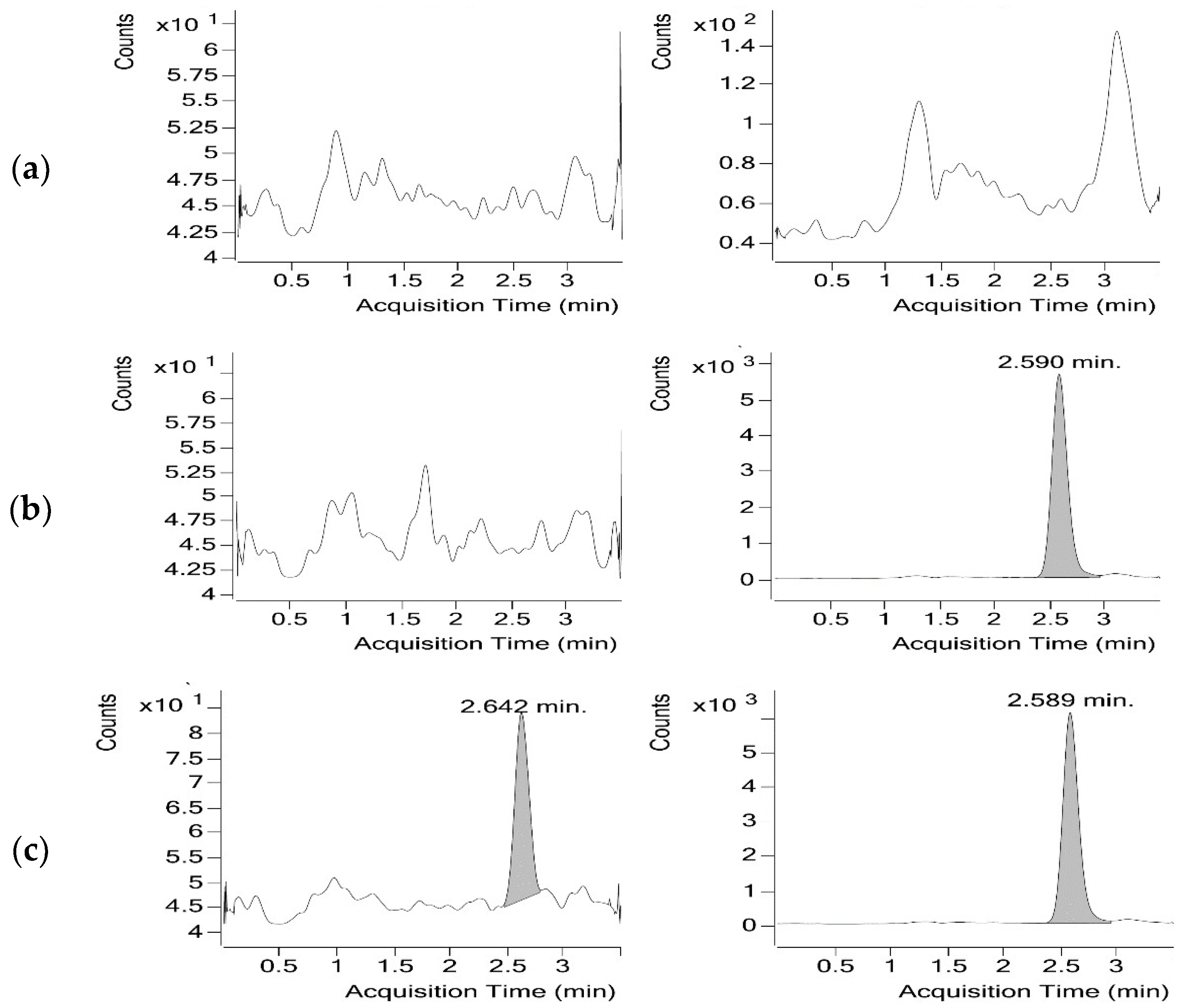
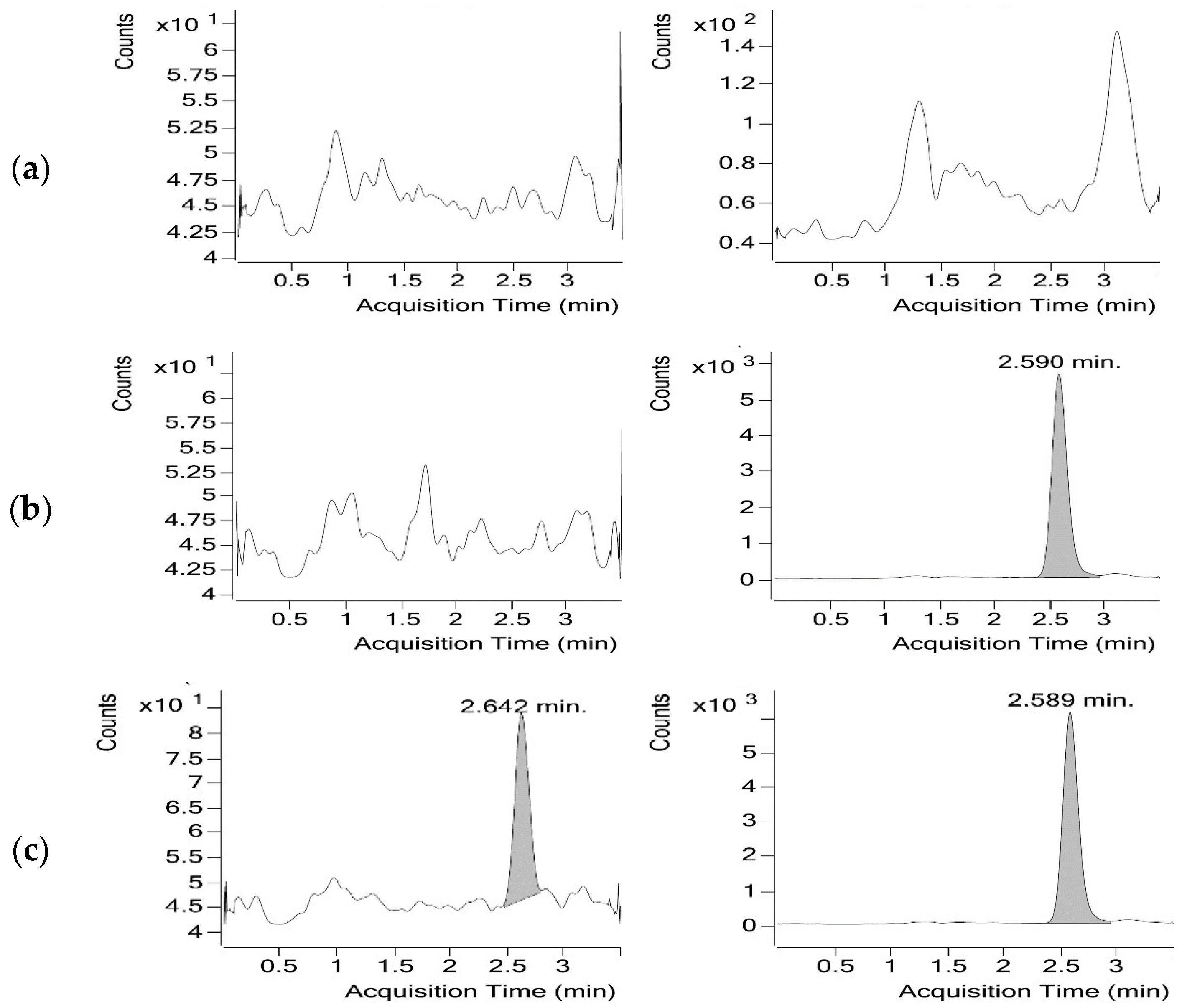
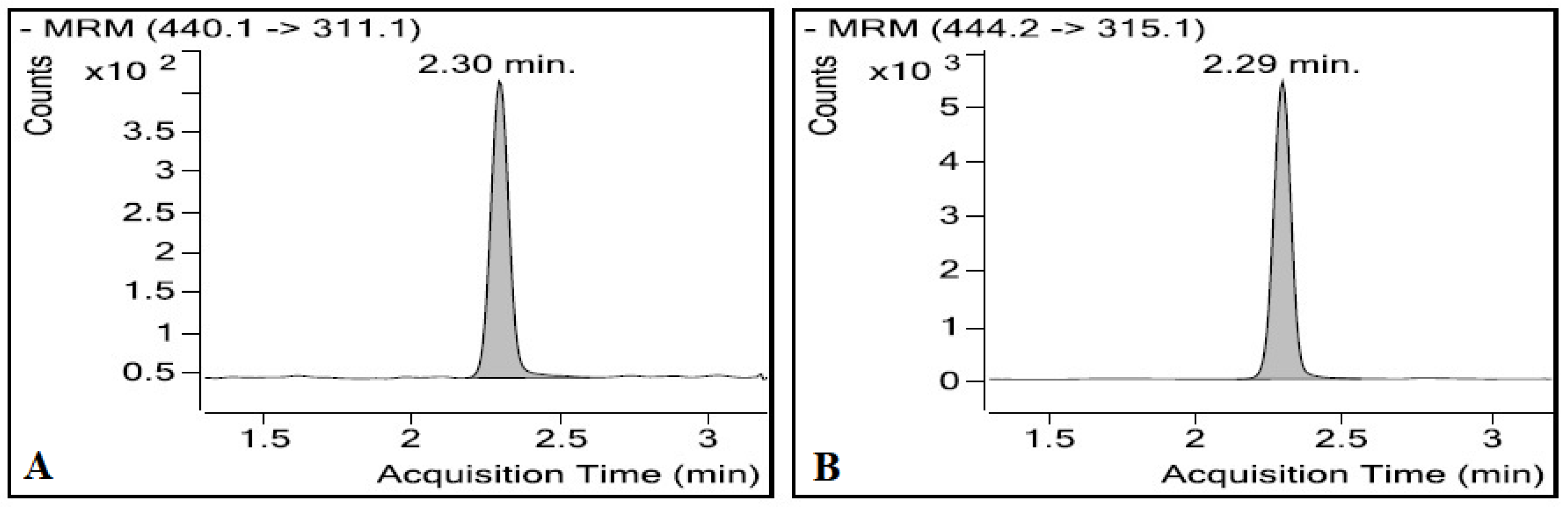
3.1.2. Optimisation of the Chromatographic Conditions
3.1.3. Sample Preparation
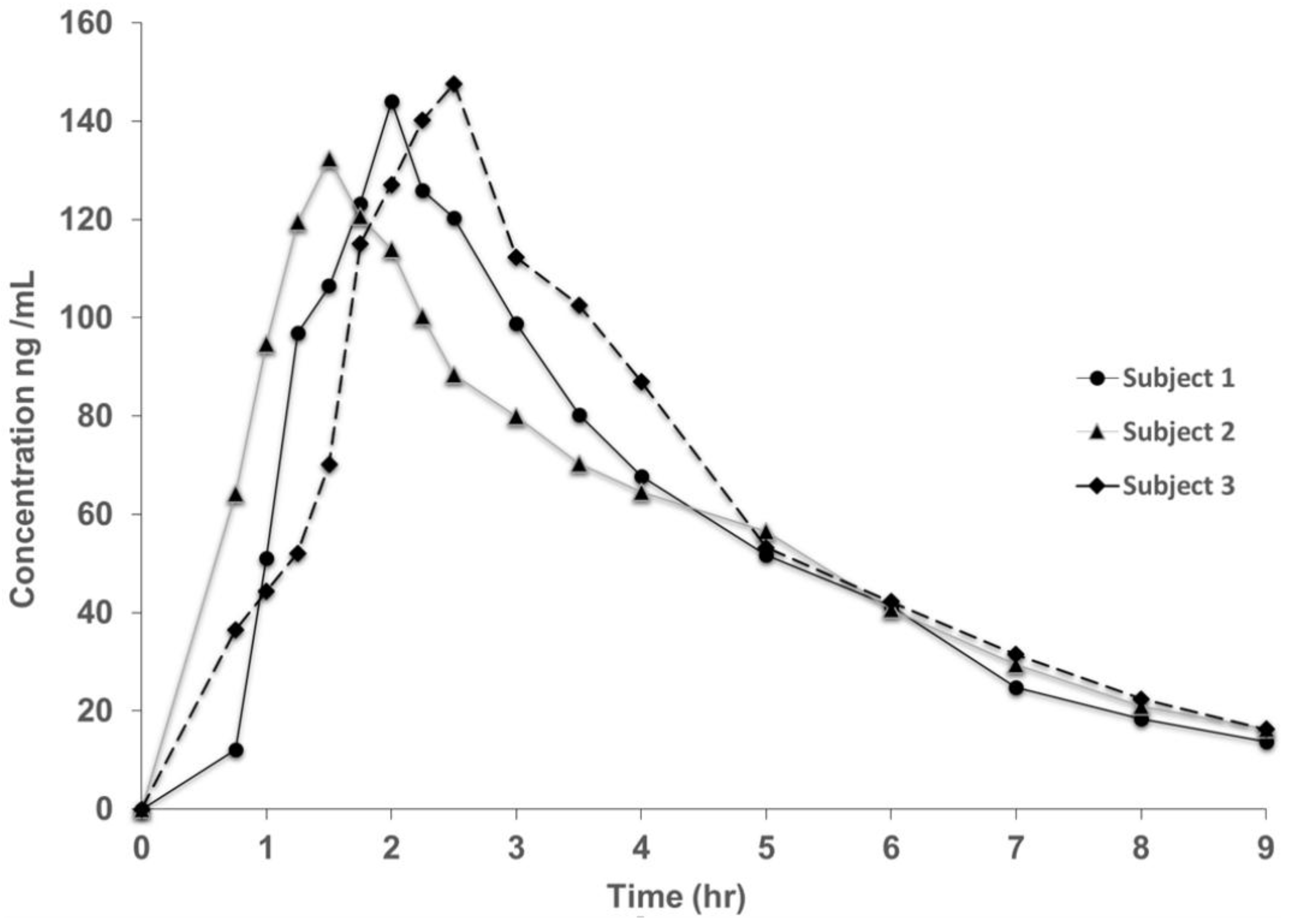
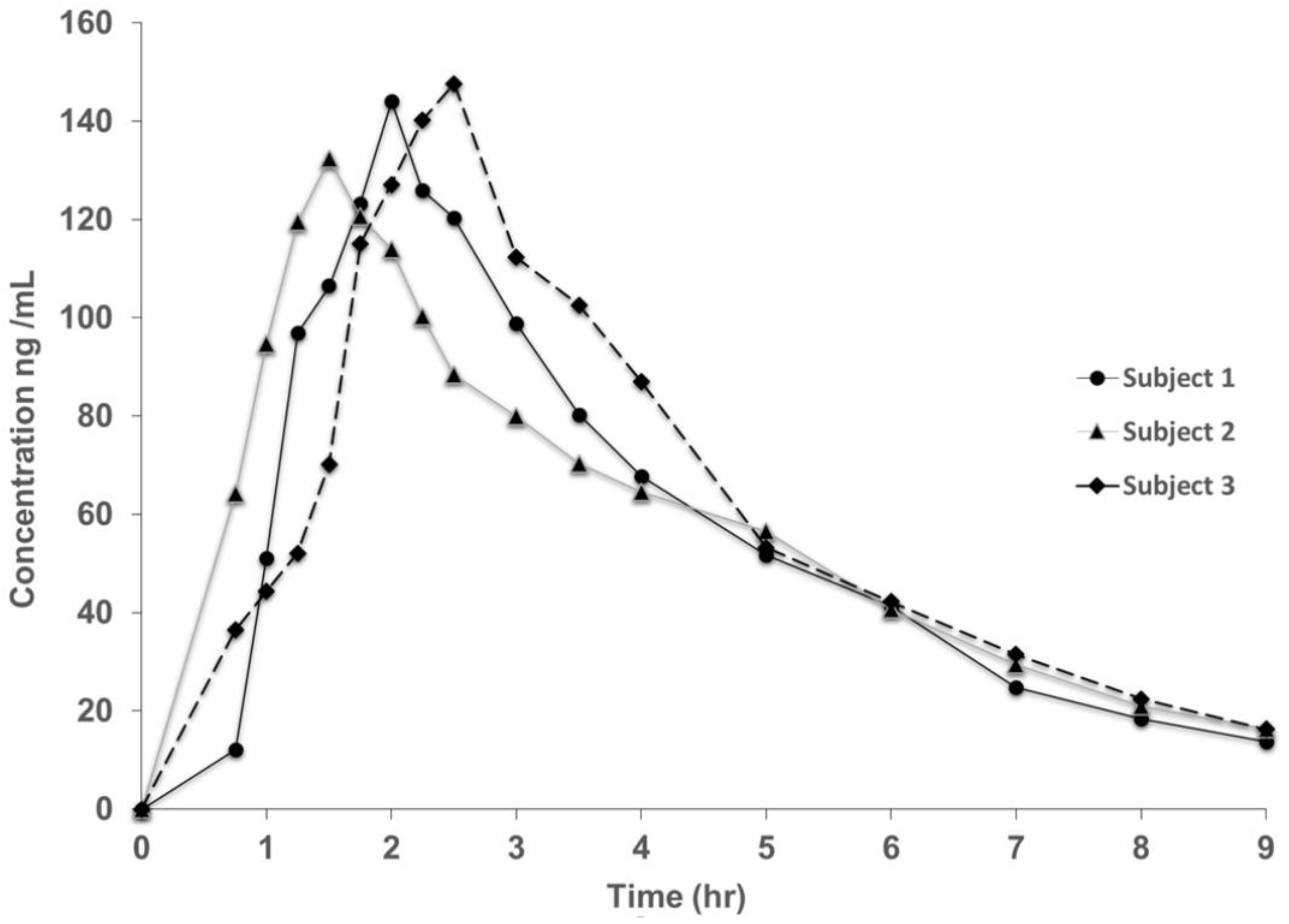
3.2. Method Validation
4. Conclusions
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Cahill, L.E.; El-Sohemy, A. Genetic variation and nutrient metabolism: Folate. In Present Knowledge in Nutrition, 10th ed.; Erdman, J.W., MacDonald, I.A., Zeisel, S.H., Eds.; Wiley-Blackwell: Hoboken, NJ, USA, 2012; pp. 27–37. ISBN 9781119946045. [Google Scholar]
- Nollet, L.M.L.; Toldra, F. Food Analysis by HPLC, 3rd ed.; CRC Press Taylor & Francis: Boca Raton, FL, USA, 2012; p. 1078. ISBN 9781439830840. [Google Scholar]
- Bailey, L.B.; da Silva, V.; West, A.A.; Caudill, M.A. Folate. In Handbook of Vitamins, 5th ed.; Zempleni, J., Suttie, J.W., Gregory, J.F., III, Stover, P.J., Eds.; CRC Press, Tayler & Francis Group: Boca Raton, FL, USA, 2013; pp. 421–446. ISBN 978-1-4665-1556-7. [Google Scholar]
- Institute of Medicine. Dietary Reference Intakes for Thiamin, Riboflavin, Niacin, Vitamin B6, Folate, Vitamin B12, Pantothenic Acid, Biotin, and Choline; The National Academies Press: Washington, DC, USA, 1998; p. 592. ISBN 978-0-309-06554-2. [Google Scholar]
- Pietrzik, K.; Bailey, L.; Shane, B. Folic acid and L-5-methyltetrahydrofolate. Clin. Pharmacokinet. 2010, 49, 535–548. [Google Scholar] [CrossRef] [PubMed]
- Rima, O.; Wolfgang, H. The Emerging Role of Unmetabolized Folic Acid in Human Diseases: Myth or Reality? Curr. Drug Metab. 2012, 13, 1184–1195. [Google Scholar]
- Stabler, S.P. Clinical folate deficiency. In Folate in Health and Disease, 2nd ed.; Bailey, L.B., Ed.; CRC Press: Boca Raton, FL, USA, 2009; pp. 409–428. ISBN 9781420071252. [Google Scholar]
- Hesdorffer, C.S.; Longo, D.L. Drug-induced megaloblastic anemia. N. Engl. J. Med. 2015, 373, 1649–1658. [Google Scholar] [CrossRef] [PubMed]
- Kalin, S.R.; Rimm, E.B. Folate and vascular disease: Epidemiological perspective. In Folate in Health and Disease, 2nd ed.; Bailey, L.B., Ed.; CRC Press: Boca Raton, FL, USA, 2009; pp. 263–290. ISBN 9781420071252. [Google Scholar]
- Zeng, R.; Xu, C.-H.; Xu, Y.-N.; Wang, Y.-L.; Wang, M. The effect of folate fortification on folic acid-based homocysteine-lowering intervention and stroke risk: A meta-analysis. Public Health Nutr. 2015, 18, 1514–1521. [Google Scholar] [CrossRef] [PubMed]
- Keum, N.; Giovannucci, E.L. Folic acid fortification and colorectal cancer risk. Am. J. Prev. Med. 2014, 46, S65–S72. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Xu, X.; Liu, A.; Ulrich, C.M. Folate and cancer: Epidemiological perspective. In Folate in Health and Disease, 2nd ed.; Bailey, L.B., Ed.; CRC Press: Boca Raton, FL, USA, 2009; pp. 206–234. ISBN 9781420071252. [Google Scholar]
- Araújo, J.R.; Martel, F.; Borges, N.; Araújo, J.M.; Keating, E. Folates and aging: Role in mild cognitive impairment, dementia and depression. Ageing Res. Rev. 2015, 22, 9–19. [Google Scholar] [CrossRef] [PubMed]
- Morris, M.S.; Jacques, P.F. Folate and neurological function: Epidemiological perspective. In Folate in Health and Disease, 2nd ed.; Bailey, L.B., Ed.; CRC Press: Boca Raton, FL, USA, 2009; pp. 325–354. ISBN 9781420071252. [Google Scholar]
- Tamura, T.; Picciano, M.F.; McGuire, M.K. Folate in pregnancy and lactation. In Folate in Health and Disease, 2nd ed.; Bailey, L.B., Ed.; CRC Press: Boca Raton, FL, USA, 2009; pp. 112–132. ISBN 9781420071252. [Google Scholar]
- Scholl, T.O.; Johnson, W.G. Folic acid: Influence on the outcome of pregnancy. Am. J. Clin. Nutr. 2000, 71, 1295S–1303S. [Google Scholar] [CrossRef] [PubMed]
- Huhta, J.C.; Linask, K. When should we prescribe high-dose folic acid to prevent congenital heart defects? Curr. Opin. Cardiol. 2015, 30, 125–131. [Google Scholar] [CrossRef] [PubMed]
- Greene, N.D.E.; Copp, A.J. Neural tube defects. Annu. Rev. Neurosci. 2014, 37, 221–242. [Google Scholar] [CrossRef] [PubMed]
- Hobbs, C.A.; Shaw, G.M.; Werler, M.M.; Mosley, B. Folate status and birth defect risk: Epidemiological perspective. In Folate in Health and Disease, 2nd ed.; Bailey, L.B., Ed.; CRC Press: Boca Raton, FL, USA, 2009; pp. 112–132. ISBN 9781420071252. [Google Scholar]
- De-Regil, L.M.; Peña-Rosas, J.P.; Fernández-Gaxiola, A.C.; Rayco-Solon, P. Effects and safety of periconceptional oral folate supplementation for preventing birth defects. Cochrane Database Syst. Rev. 2015. [Google Scholar] [CrossRef] [PubMed]
- Castillo-Lancellotti, C.; Tur, J.A.; Uauy, R. Impact of folic acid fortification of flour on neural tube defects: A systematic review. Public Health Nutr. 2013, 16, 901–911. [Google Scholar] [CrossRef] [PubMed]
- Bailey, S.W.; Ayling, J.E. The extremely slow and variable activity of dihydrofolate reductase in human liver and its implications for high folic acid intake. Proc. Natl. Acad. Sci. USA 2009, 106, 15424–15429. [Google Scholar] [CrossRef] [PubMed]
- Wright, A.J.A.; Dainty, J.R.; Finglas, P.M. Folic acid metabolism in human subjects revisited: Potential implications for proposed mandatory folic acid fortification in the UK. Br. J. Nutr. 2007, 98, 667–675. [Google Scholar] [CrossRef] [PubMed]
- Mason, J.B.; Dickstein, A.; Jacques, P.F.; Haggarty, P.; Selhub, J.; Dallal, G.; Rosenberg, I.H. A temporal association between folic acid fortification and an increase in colorectal cancer rates may be illuminating important biological principles: A hypothesis. Cancer Epidemiol. Biomarkers Prev. 2007, 16, 1325–1329. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.-I. Folate and colorectal cancer: An evidence-based critical review. Mol. Nutr. Food Res. 2007, 51, 267–292. [Google Scholar] [CrossRef] [PubMed]
- Williams, E.A. Folate, colorectal cancer and the involvement of DNA methylation. Proc. Nutr. Soc. 2012, 71, 592–597. [Google Scholar] [CrossRef] [PubMed]
- Mudryj, A.N.; de Groh, M.; Aukema, H.M.; Yu, N. Folate intakes from diet and supplements may place certain Canadians at risk for folic acid toxicity. Br. J. Nutr. 2016, 116, 1236–1245. [Google Scholar] [CrossRef] [PubMed]
- Cuskelly, G.J.; Mooney, K.M.; Young, I.S. Folate and vitamin B12: Friendly or enemy nutrients for the elderly: Symposium on ‘Micronutrients through the life cycle’. Proc. Nutr. Soc. 2007, 66, 548–558. [Google Scholar] [CrossRef] [PubMed]
- Selhub, J.; Rosenberg, I.H. Excessive folic acid intake and relation to adverse health outcome. Biochimie 2016, 126, 71–78. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.I. Folate: A magic bullet or a double edged sword for colorectal cancer prevention? Gut 2006, 55, 1387–1389. [Google Scholar] [CrossRef] [PubMed]
- Mason, J.B. Unraveling the complex relationship between folate and cancer risk. BioFactors 2011, 37, 253–260. [Google Scholar] [CrossRef] [PubMed]
- Figueiredo, J.C.; Grau, M.V.; Haile, R.W.; Sandler, R.S.; Summers, R.W.; Bresalier, R.S.; Burke, C.A.; McKeown-Eyssen, G.E.; Baron, J.A. Folic acid and risk of prostate cancer: Results from a randomized clinical trial. JNCI, J. Natl. Cancer Inst. 2009, 101, 432–435. [Google Scholar] [CrossRef] [PubMed]
- O'Broin, S.; Kelleher, B. Microbiological assay on microtitre plates of folate in serum and red cells. J. Clin. Pathol. 1992, 45, 344–347. [Google Scholar] [CrossRef] [PubMed]
- Iyer, R.; Tomar, S.K. Determination of folate/folic acid level in milk by microbiological assay, immuno assay and high performance liquid chromatography. J. Dairy Res. 2013, 80, 233–239. [Google Scholar] [CrossRef] [PubMed]
- Pfeiffer, C.M.; Fazili, Z.; Zhang, M. Folate analytical methodology. In Folate in Health and Disease, 2nd ed.; Bailey, L.B., Ed.; CRC Press: Boca Raton, FL, USA, 2009; pp. 517–574. ISBN 9781420071252. [Google Scholar]
- Osseyi, E.S.; Wehling, R.L.; Albrecht, J.A. Liquid chromatographic method for determining added folic acid in fortified cereal products1. J. Chromatogr. A 1998, 826, 235–240. [Google Scholar] [CrossRef]
- Ichinose, N.; Tsuneyoshi, T.; Kato, M.; Suzuki, T.; Ikeda, S. Fluorescent high-performance liquid chromatography of folic acid and its derivatives using permanganate as a fluorogenic reagent. Fresenius J. Anal. Chem. 1993, 346, 841–846. [Google Scholar] [CrossRef]
- Kalmbach, R.; Paul, L.; Selhub, J. Determination of unmetabolized folic acid in human plasma using affinity HPLC. Am. J. Clin. Nutr. 2011, 94, 343S–347S. [Google Scholar] [CrossRef] [PubMed]
- Pawlosky, R.J.; Flanagan, V.P. A quantitative stable-isotope lC−MS method for the determination of folic acid in fortified foods. J. Agric. Food Chem. 2001, 49, 1282–1286. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.-H.; Jiang, L.-Y.; Zhao, L.-T.; Zhang, Q.-Y.; Ding, L. Simultaneous quantitation of folic acid and 5-methyltetrahydrofolic acid in human plasma by HPLC–MS/MS and its application to a pharmacokinetic study. J. Pharm. Anal. 2015, 5, 269–275. [Google Scholar] [CrossRef] [PubMed]
- Kirsch, S.H.; Knapp, J.-P.; Herrmann, W.; Obeid, R. Quantification of key folate forms in serum using stable-isotope dilution ultra performance liquid chromatography–tandem mass spectrometry. J. Chromatogr. B 2010, 878, 68–75. [Google Scholar] [CrossRef] [PubMed]
- Fazili, Z.; Whitehead, R.D.; Paladugula, N.; Pfeiffer, C.M. A high-throughput LC-MS/MS method suitable for population biomonitoring measures five serum folate vitamers and one oxidation product. Anal. Bioanal. Chem. 2013, 405, 4549–4560. [Google Scholar] [CrossRef] [PubMed]
- Garbis, S.D.; Melse-Boonstra, A.; West, C.E.; van Breemen, R.B. Determination of folates in human plasma using hydrophilic interaction chromatography−tandem mass spectrometry. Anal. Chem. 2001, 73, 5358–5364. [Google Scholar] [CrossRef] [PubMed]
- FDA Guidance. Guidance for Industry. Bioanalytical Method Validation. 2013. Available online: https://www.fda.gov/downloads/Drugs/guidancecomplianceregulatoryinformation/guidances/ucm368107.pdf (accessed on 30 September 2017).
- Álvarez-Sánchez, B.; Priego-Capote, F.; Mata-Granados, J.M.; Luque de Castro, M.D. Automated determination of folate catabolites in human biofluids (urine, breast milk and serum) by on-line SPE–HILIC–MS/MS. J. Chromatogr. A 2010, 1217, 4688–4695. [Google Scholar] [CrossRef] [PubMed]
- Rychlik, M.; Netzel, M.; Pfannebecker, I.; Frank, T.; Bitsch, I. Application of stable isotope dilution assays based on liquid chromatography–tandem mass spectrometry for the assessment of folate bioavailability. J. Chromatogr. B 2003, 792, 167–176. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameters | Linear Range (ng/mL) | Slope Mean ± SD | Intercept Mean ± SD | Correlation Coefficient Mean ± SD |
|---|---|---|---|---|
| Plasma sample | 13.17–3657 | 0.00044 ± 0.00001 | −0.0009 ± 0.0006 | 0.995 ± 0.004 |
| QC Level | Nominal Conc. (ng/mL) | Intrabatch (n = 6; Single Batch) | Interbatch (n = 30; 6 From Each Batch) | ||||
|---|---|---|---|---|---|---|---|
| Measured Mean Conc. (ng/mL) | CV% | Accuracy% | Measured Mean Conc. (ng/mL) | CV% | Accuracy% | ||
| LLOQ QC | 13.17 | 12.49 | 19.79 | 94.88 | 12.88 | 13.15 | 97.84 |
| QCL | 39.50 | 39.83 | 13.78 | 100.83 | 38.30 | 11.00 | 96.97 |
| QCM-1 | 731.5 | 665.2 | 2.58 | 90.94 | 660.3 | 3.56 | 90.26 |
| QCM-2 | 1829 | 1800 | 3.96 | 98.41 | 1813. | 3.89 | 99.14 |
| QCH | 2743 | 2863 | 2.20 | 104.36 | 2871 | 3.14 | 104.67 |
| QC Level | Nominal Conc. (ng/mL) | Measured Mean Conc. (ng/mL) (n = 6) | CV% | Accuracy% |
|---|---|---|---|---|
| LLOQ QC | 13.17 | 12.62 | 9.92 | 95.82 |
| QCL | 39.51 | 35.72 | 7.05 | 90.43 |
| QCM-1 | 731.5 | 655.9 | 2.45 | 89.67 |
| QCM-2 | 1829 | 1838 | 4.15 | 100.50 |
| QCH | 2743 | 2950 | 2.10 | 107.56 |
| QC Level | Area Response (Replicate, n = 6) | Mean Recovery % | |
|---|---|---|---|
| Extracted Sample Analyte Response Ratio | Untreated Standard Analyte Response Ratio | ||
| QCL | 0.0145 | 0.0156 | 92.95 |
| QCM-1 | 0.2775 | 0.3408 | 81.43 |
| QCM-2 | 0.7504 | 0.9479 | 79.16 |
| QCH | 1.2797 | 1.4823 | 86.33 |
| I.S. | 0.7818 | 0.6748 | 115.86 |
| Stability Test | Nominal Conc. (ng/mL) ± SD | Mean Stability Sample Conc. (ng/mL) ± SD | Change % |
|---|---|---|---|
| Stability in blood at room temperature for 1 h | 51.8 ± 0.8 | 49.4 ± 3.5 | −4.69 |
| 3934 ± 78 | 3815 ± 30 | −3.01 | |
| Long term stability at −20 °C for 9 day | 35.6 ± 3.4 | 36.3 ± 1.6 | 1.94 |
| 2757 ± 57 | 2778.864 ± 38 | 0.79 | |
| Long term stability at −70 °C for 9 day | 35.6 ± 3.4 | 37.8 ± 2.1 | 5.99 |
| 2757 ± 57 | 2761 ± 80 | 0.14 | |
| Freeze-thaw stability at −20 °C | 33.8 ± 2.5 | 36.7 ± 2.3 | 8.72 |
| 2866 ± 81 | 2830 ± 45 | −1.26 | |
| Freeze-thaw stability at −70 °C | 33.8 ± 2.5 | 37.9 ± 2.4 | 12.19 |
| 2866 ± 81 | 2872 ± 91 | 0.20 | |
| Autosampler stability at room temperature for 123 h | 39.8 ± 3.7 | 40.9 ± 5.5 | 2.90 |
| 2862 ± 73 | 2864 ± 109 | 0.09 | |
| Autosampler stability at 4 °C for 124 h | 39.7 ± 3.6 | 35.9 ± 3.0 | −9.6 |
| 2862 ± 73 | 2890 ± 93 | 0.97 | |
| Dry extract stability at room temperature for 125 h | 39.7 ± 3.6 | 37.9 ± 3.6 | −4.71 |
| 2862 ± 73 | 2711 ± 69 | −5.27 | |
| Dry extract stability at −20 °C for 120 h | 39.7 ± 3.6 | 38.4 ± 4.0 | −3.47 |
| 2862 ± 73 | 2787 ± 49 | −2.63 | |
| Bench-top stability at room temperature for 24 h | 33.8 ± 2.5 | 34.1 ± 4.0 | 1.01 |
| 2866 ± 81 | 2905 ± 64 | 1.35 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zayed, A.; Bustami, R.; Alabsi, W.; El-Elimat, T. Development and Validation of a Rapid High-Performance Liquid Chromatography–Tandem Mass Spectrometric Method for Determination of Folic Acid in Human Plasma. Pharmaceuticals 2018, 11, 52. https://doi.org/10.3390/ph11020052
Zayed A, Bustami R, Alabsi W, El-Elimat T. Development and Validation of a Rapid High-Performance Liquid Chromatography–Tandem Mass Spectrometric Method for Determination of Folic Acid in Human Plasma. Pharmaceuticals. 2018; 11(2):52. https://doi.org/10.3390/ph11020052
Chicago/Turabian StyleZayed, Aref, Rana Bustami, Wafaa Alabsi, and Tamam El-Elimat. 2018. "Development and Validation of a Rapid High-Performance Liquid Chromatography–Tandem Mass Spectrometric Method for Determination of Folic Acid in Human Plasma" Pharmaceuticals 11, no. 2: 52. https://doi.org/10.3390/ph11020052
APA StyleZayed, A., Bustami, R., Alabsi, W., & El-Elimat, T. (2018). Development and Validation of a Rapid High-Performance Liquid Chromatography–Tandem Mass Spectrometric Method for Determination of Folic Acid in Human Plasma. Pharmaceuticals, 11(2), 52. https://doi.org/10.3390/ph11020052