The Effects of Dabigatran and Rivaroxaban on Markers of Polymorphonuclear Leukocyte Activation


Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals and Reagents
2.2. Preparation of PMNL
2.3. Measurement of Reactive Oxygen Species (ROS)
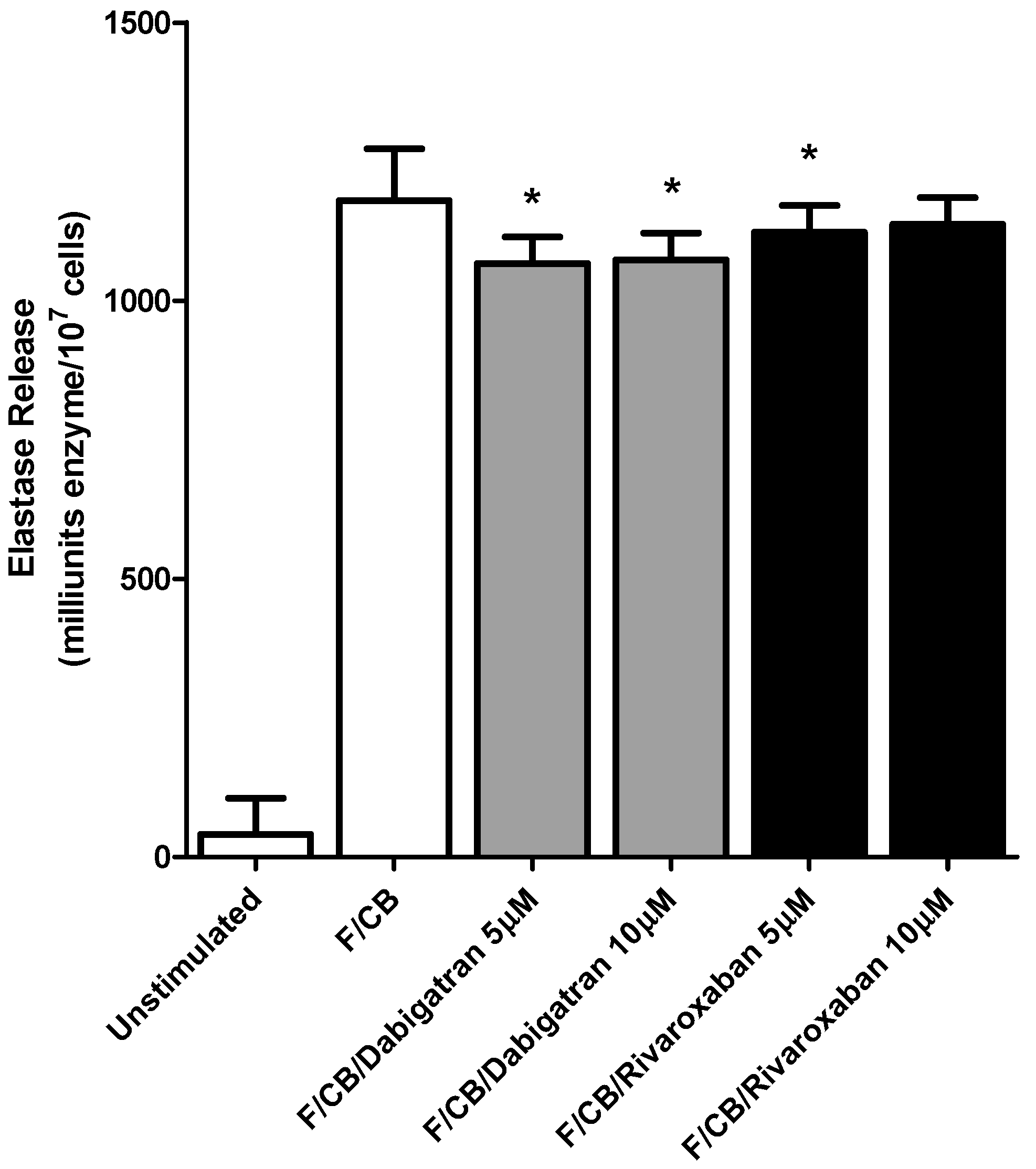
2.4. Elastase Release
2.5. Spectrofluorimetric Measurement of Cytosolic Ca2+
2.6. Spectrofluorimetric Detection of Neutrophil Extracellular Traps (NETs)
2.7. Cell Viability
2.8. Expression and Statistical Analysis of Results
3. Results
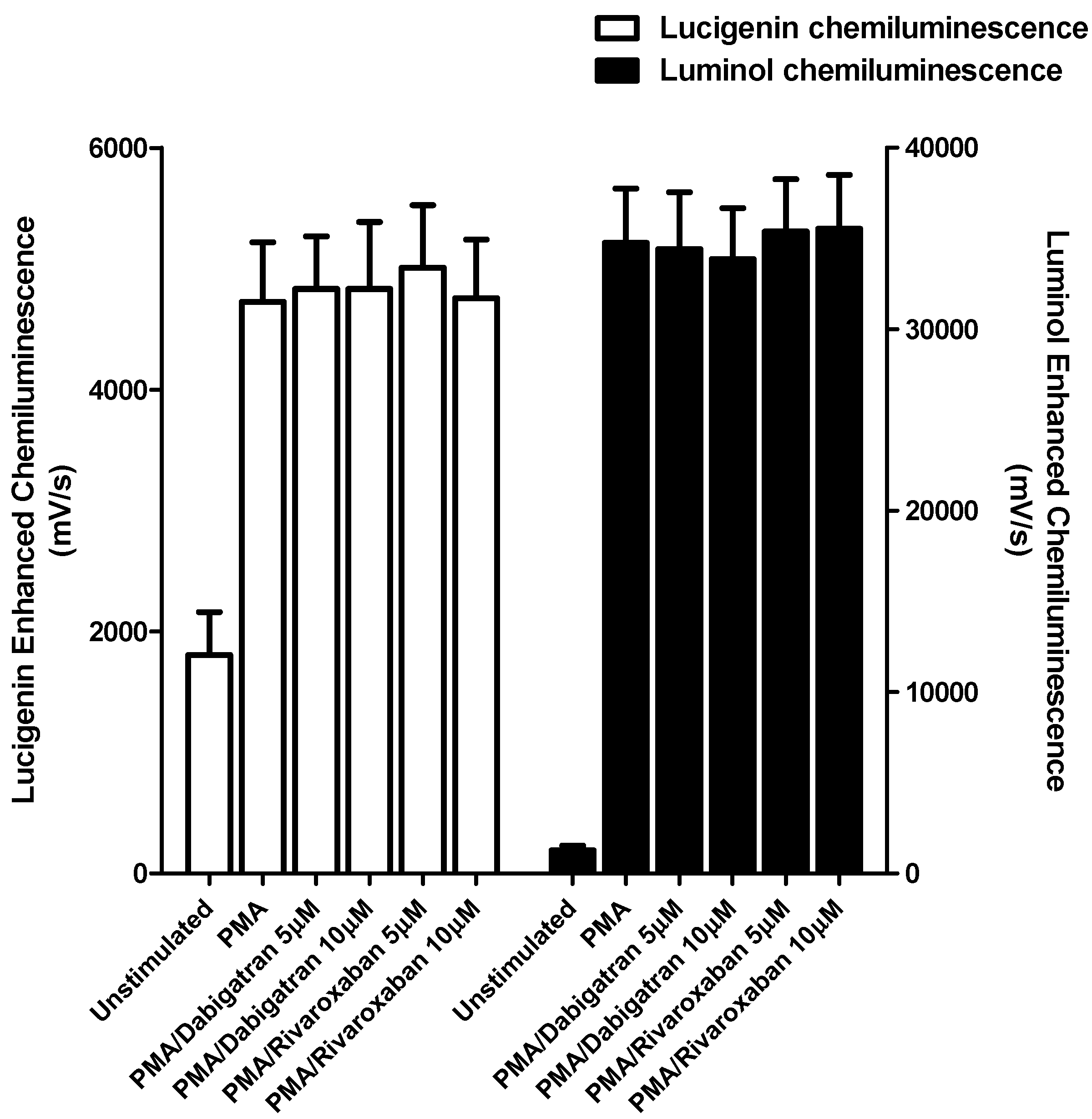
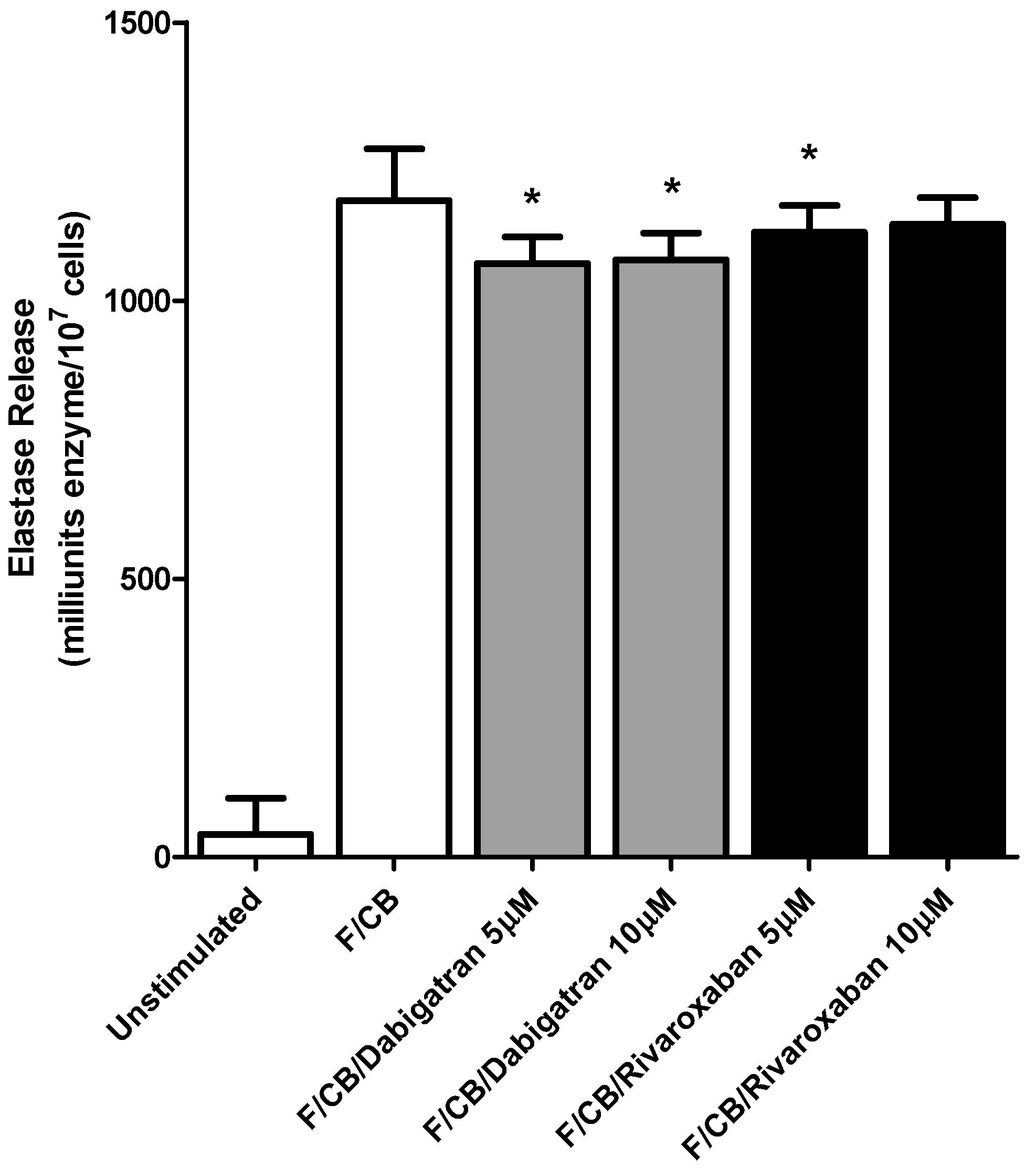
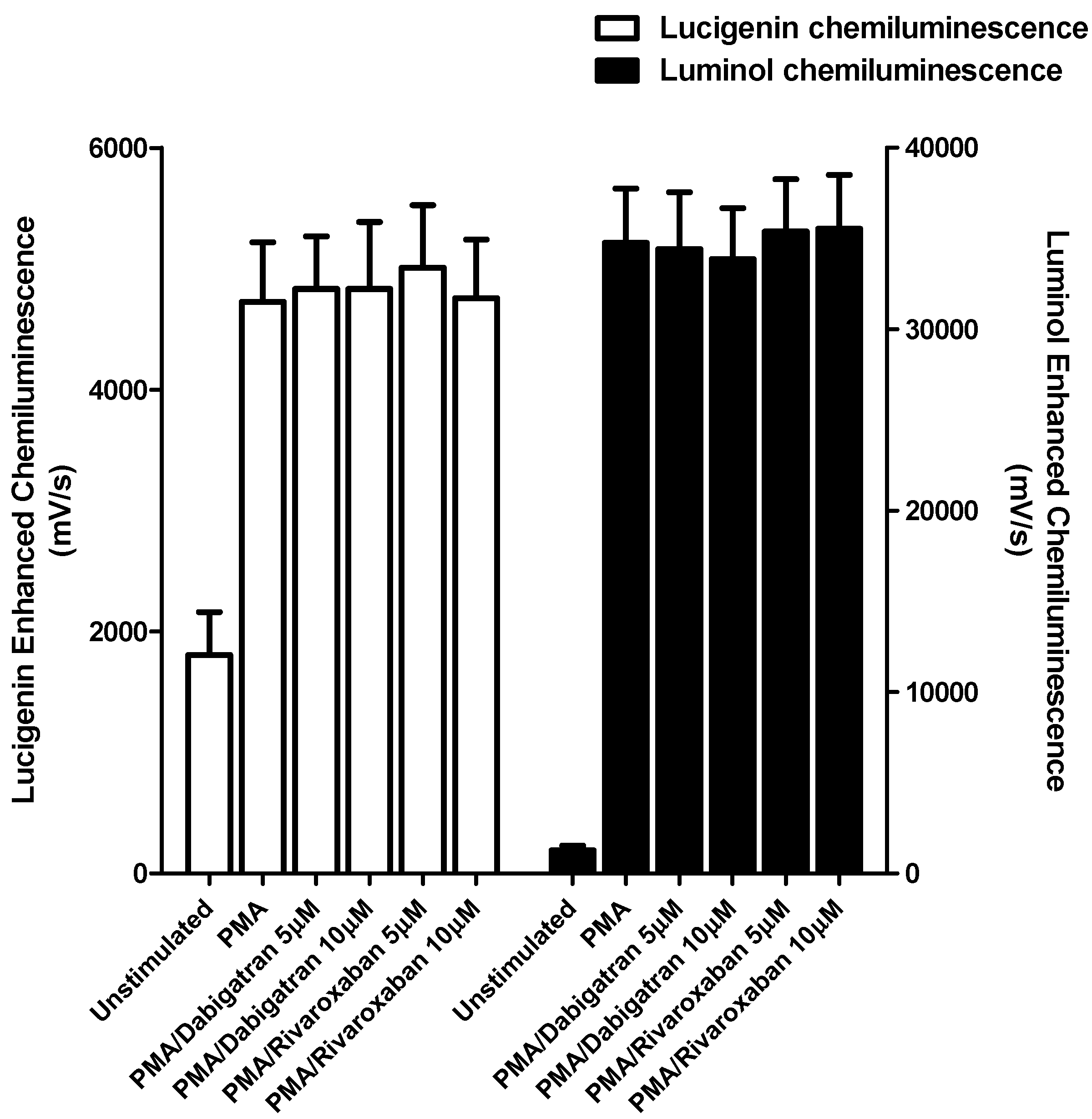
3.1. Generation of Reactive Oxygen Species (ROS) and Release of Elastase
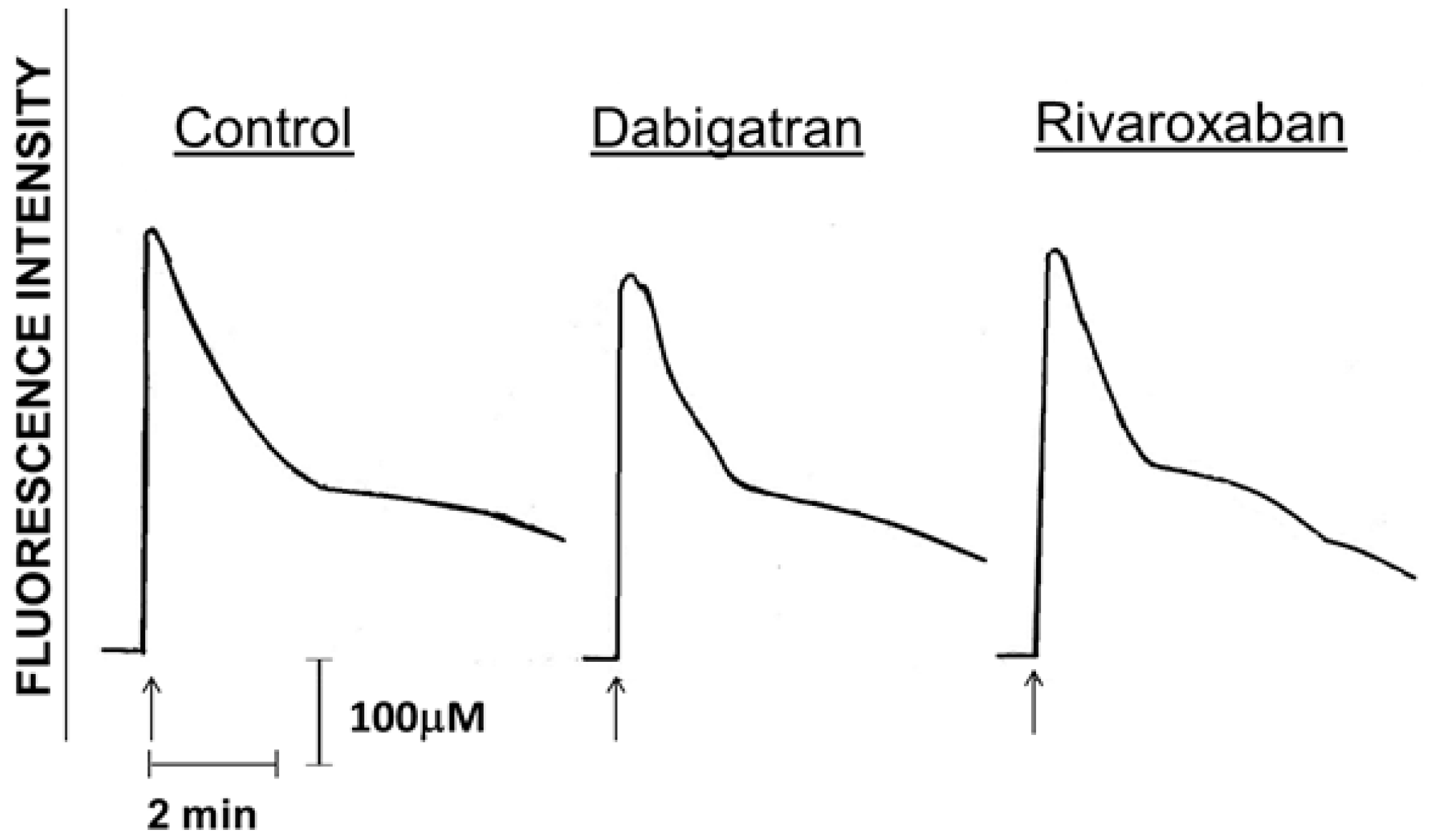
3.2. Cytosolic Ca2+ Fluxes
3.3. NET Formation
3.4. Neutrophil Viability
4. Discussion
Author Contributions
Conflicts of Interest
References
- Ross, R. Atherosclerosis—An inflammatory disease. N. Engl. J. Med. 1999, 340, 115–126. [Google Scholar] [CrossRef] [PubMed]
- Madjid, M.; Willerson, J.T. Inflammatory markers in coronary heart disease. Br. Med. Bull. 2011, 100, 23–38. [Google Scholar] [CrossRef] [PubMed]
- Gould, T.J.; Vu, T.T.; Swystun, L.L.; Dwivedi, D.J.; Mai, S.H.; Weitz, J.I.; Liaw, P.C. Neutrophil extracellular traps promote thrombin generation through platelet-dependent and platelet-independent mechanisms. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 1977–1984. [Google Scholar] [CrossRef] [PubMed]
- Stakos, D.A.; Kambas, K.; Konstantinidis, T.; Mitroulis, I.; Apostolidou, E.; Arelaki, S.; Tsironidou, V.; Giatromanolaki, A.; Skendros, P.; Konstantinides, S.; et al. Expression of functional tissue factor by neutrophil extracellular traps in culprit artery of acute myocardial infarction. Eur. Heart J. 2015, 36, 1405–1414. [Google Scholar] [CrossRef] [PubMed]
- Pfeiler, S.; Stark, K.; Massberg, S.; Engelmann, B. Propagation of thrombosis by neutrophils and extracellular nucleosome networks. Haematologica 2017, 102, 206–213. [Google Scholar] [CrossRef] [PubMed]
- Koupenova, M.; Clancy, L.; Corkrey, H.A.; Freedman, J.E. Circulating platelets as mediators of immunity, inflammation, and thrombosis. Circ. Res. 2018, 122, 337–351. [Google Scholar] [CrossRef] [PubMed]
- Weitz, J.I.; Bates, S.M. New anticoagulants. J. Thromb. Haemost. 2005, 3, 1843–1853. [Google Scholar] [CrossRef] [PubMed]
- Davidson, B.L. The association of direct thrombin inhibitor anticoagulants with cardiac thromboses. Chest 2015, 147, 21–24. [Google Scholar] [CrossRef] [PubMed]
- Connolly, S.J.; Ezekowitz, M.D.; Yusuf, S.; Eikelboom, J.; Oldgren, J.; Parekh, A.; Pogue, J.; Reilly, P.A.; Themeles, E.; Varrone, J.; et al. Dabigatran versus warfarin in patients with atrial fibrillation. N. Engl. J. Med. 2009, 361, 1139–1151. [Google Scholar] [CrossRef] [PubMed]
- Connolly, S.J.; Ezekowitz, M.D.; Yusuf, S.; Reilly, P.A.; Wallentin, L. Randomized Evaluation of Long-Term Anticoagulation Therapy Investigators. Newly identified events in the RE-LY trial. N. Engl. J. Med. 2010, 363, 1875–1876. [Google Scholar] [CrossRef] [PubMed]
- Schulman, S.; Kearon, C.; Kakkar, A.K.; Mismetti, P.; Schellong, S.; Eriksson, H.; Baanstra, D.; Schnee, J.; Goldhaber, S.Z.; RE-COVER Study Group. Dabigatran versus warfarin in the treatment of acute venous thromboembolism. N. Engl. J. Med. 2009, 361, 2342–2352. [Google Scholar] [CrossRef] [PubMed]
- Schulman, S.; Kakkar, A.K.; Goldhaber, S.Z.; Schellong, S.; Eriksson, H.; Mismetti, P.; Christiansen, A.V.; Friedman, J.; Le Maulf, F.; Peter, N.; et al. Treatment of acute venous thromboembolism with dabigatran or warfarin and pooled analysis. Circulation 2014, 129, 764–772. [Google Scholar] [CrossRef] [PubMed]
- Uchino, K.; Hernandez, A.V. Dabigatran association with higher risk of acute coronary events: Meta-analysis of noninferiority randomized controlled trials. Arch. Intern. Med. 2012, 172, 397–402. [Google Scholar] [CrossRef] [PubMed]
- Furugohri, T.; Sugiyama, N.; Morishima, Y.; Shibano, T. Antithrombin-independent thrombin inhibitors, but not direct factor Xa inhibitors, enhance thrombin generation in plasma through inhibition of thrombin-thrombomodulin-protein C system. Thromb. Haemost. 2011, 106, 1076–1083. [Google Scholar] [CrossRef] [PubMed]
- Perzborn, E.; Heitmeier, S.; Buetehorn, U.; Laux, V. Direct thrombin inhibitors, but not the direct factor Xa inhibitor rivaroxaban, increase tissue factor-induced hypercoagulability in vitro and in vivo. J. Thromb. Haemost. 2014, 12, 1054–1065. [Google Scholar] [CrossRef] [PubMed]
- Minkenberg, I.; Ferber, E. Lucigenin-dependent chemiluminescence as a new assay for NADPH-oxidase activity in particulate fractions of human polymorphonuclear leukocytes. J. Immunol. Methods 1984, 71, 61–67. [Google Scholar] [CrossRef]
- Beatty, K.; Robertie, P.; Senior, R.M.; Travis, J. Determination of oxidized alpha-1-proteinase inhibitor in serum. J. Lab. Clin. Med. 1982, 100, 186–192. [Google Scholar] [PubMed]
- Grynkiewicz, G.; Poenie, M.; Tsien, R.Y. A new generation of Ca2+ indicators with greatly improved fluorescence properties. J. Biol. Chem. 1985, 260, 3440–3450. [Google Scholar] [PubMed]
- Berkes, E.; Oehmke, F.; Tinneberg, H.R.; Preissner, K.T.; Saffarzadeh, M. Association of neutrophil extracellular traps with endometriosis-related chronic inflammation. Eur. J. Obstet. Gynecol. Reprod. Biol. 2014, 183, 193–200. [Google Scholar] [CrossRef] [PubMed]
- Anderson, R.; Goolam Mahomed, A. Calcium efflux and influx in f-met-leu-phe (fMLP)-activated human neutrophils are chronologically distinct events. Clin. Exp. Immunol. 1997, 110, 132–138. [Google Scholar] [CrossRef] [PubMed]
- Ten Cate, H. Dabigatran and apolipoprotein B. Heart 2016, 102, 5–6. [Google Scholar] [CrossRef] [PubMed]
- Spronk, H.M.H.; de Jong, A.M.; Crijns, H.J.; Schotten, U.; Van Gelder, I.C.; ten Cate, H. Pleiotropic effects of factor Xa and thrombin: What to expect from novel anticoagulants. Cardiovasc. Res. 2014, 101, 344–351. [Google Scholar] [CrossRef] [PubMed]
- Incampo, F.; Carrieri, C.; Semeraro, N.; Colucci, M. The paradoxical antifibrinolytic effect of dabigatran and argatroban in the presence of soluble thrombomodulin is unrelated to protein C-dependent increase of thrombin generation. Thromb. Res. 2014, 134, 1110–1116. [Google Scholar] [CrossRef] [PubMed]
- Ellinghaus, P.; Perzborn, E.; Hauenschild, P.; Gerdes, C.; Heitmeier, S.; Visser, M.; Summer, H.; Laux, V. Expression of pro-inflammatory genes in human endothelial cells: Comparison of rivaroxaban and dabigatran. Thromb. Res. 2016, 142, 44–51. [Google Scholar] [CrossRef] [PubMed]
- Page, C.; Pitchford, S. Neutrophil and platelet complexes and their relevance to neutrophil recruitment and activation. Int. Immunopharmacol. 2013, 17, 1176–1184. [Google Scholar] [CrossRef] [PubMed]
- Gerdes, N.; Seijkens, T.; Lievens, D.; Kuijpers, M.J.; Winkels, H.; Projahn, D.; Hartwig, H.; Beckers, L.; Megens, R.T.; Boon, L.; et al. Platelet CD40 exacerbates atherosclerosis by transcellular activation of endothelial cells and leukocytes. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 482–490. [Google Scholar] [CrossRef] [PubMed]
- Achilles, A.; Mohring, A.; Zeus, T.; Kelm, M.; Polzin, A. Dabigatran enhances platelet reactivity and platelet thrombin receptor expression in patients with atrial fibrillation: Reply. J. Thromb. Haemost. 2017, 15, 1524–1525. [Google Scholar] [CrossRef] [PubMed]
- Joyce, D.E.; Grinnell, B.W. Recombinant human activated protein C attenuates the inflammatory response in endothelium and monocytes by modulating nuclear factor-kappaB. Crit. Care Med. 2002, 30, S288–S293. [Google Scholar] [CrossRef] [PubMed]
- Nakase, T.; Moroi, J.; Ishikawa, T. Anti-inflammatory and antiplatelet effects of non-vitamin K antagonist oral anticoagulants in acute phase of ischemic stroke patients. Clin. Transl. Med. 2018, 7, 2. [Google Scholar] [CrossRef] [PubMed]
- Joseph, P.; Pare, G.; Wallentin, L.; Connolly, S.; Yusuf, S.; Wang, J.; Ezekowitz, M.; Eikelboom, J.; Siegbahn, A.; Reilly, P.; et al. Dabigatran etexilate and reduction in serum apolipoprotein B. Heart 2016, 102, 57–62. [Google Scholar] [CrossRef] [PubMed]
- Caldeira, D.; Ferreira, J.J.; Pinto, F.J.; Costa, J. Safety of non-vitamin K antagonist oral anticoagulants—Coronary risks. Expert Opin. Drug Saf. 2016, 15, 731–740. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
| System | Intracellular Ca2+ Concentrations (µM) | |
|---|---|---|
| Basal | Peak | |
| Control neutrophils | 70.3 ± 4 | 397.0 ± 36 * |
| Neutrophils + 1 µM dabigatran | 68.3 ± 12 | 403.9 ± 36 |
| Neutrophils + 5 µM dabigatran | 70.3 ± 3 | 402.6 ± 36 |
| Neutrophils +10 µM dabigatran | 64.3 ± 12 | 392.8 ± 59 |
| Neutrophils + 1 µM rivaroxaban | 74.8 ± 13 | 421.0 ± 34 |
| Neutrophils + 5 µM rivaroxaban | 82.0 ± 10 | 393.4 ± 32 |
| Neutrophils + 10 µM rivaroxaban | 68.8 ± 7 | 415.0 ± 32 |
| System | NETosis (Metered Fluorescence Units) | |
|---|---|---|
| Spontaneous | PMA-Activated | |
| Control neutrophils | 29.5 ± 4.1 * | 231.8 ± 13.6 |
| Neutrophils + 5 μM dabigatran | 19.5 ± 2.5 | 238.3 ± 15.6 |
| Neutrophils + 10 μM dabigatran | 26.8 ± 5.2 | 254.0 ± 28.5 |
| Neutrophils + 5 μM rivaroxaban | 21.0 ± 5.0 | 250.3 ± 39.7 |
| Neutrophils + 10 μM rivaroxaban | 34.5 ± 7.6 | 260.0 ± 15.7 |
© 2018 by the authors. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Richards, G.A.; Theron, A.; Tintinger, G.; Anderson, R. The Effects of Dabigatran and Rivaroxaban on Markers of Polymorphonuclear Leukocyte Activation. Pharmaceuticals 2018, 11, 46. https://doi.org/10.3390/ph11020046
Richards GA, Theron A, Tintinger G, Anderson R. The Effects of Dabigatran and Rivaroxaban on Markers of Polymorphonuclear Leukocyte Activation. Pharmaceuticals. 2018; 11(2):46. https://doi.org/10.3390/ph11020046
Chicago/Turabian StyleRichards, Guy A., Annette Theron, Gregory Tintinger, and Ronald Anderson. 2018. "The Effects of Dabigatran and Rivaroxaban on Markers of Polymorphonuclear Leukocyte Activation" Pharmaceuticals 11, no. 2: 46. https://doi.org/10.3390/ph11020046
APA StyleRichards, G. A., Theron, A., Tintinger, G., & Anderson, R. (2018). The Effects of Dabigatran and Rivaroxaban on Markers of Polymorphonuclear Leukocyte Activation. Pharmaceuticals, 11(2), 46. https://doi.org/10.3390/ph11020046