Antibody-Drug Conjugates for Cancer Therapy: Chemistry to Clinical Implications



Abstract
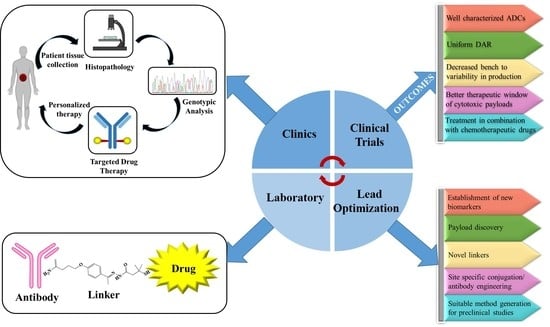
:
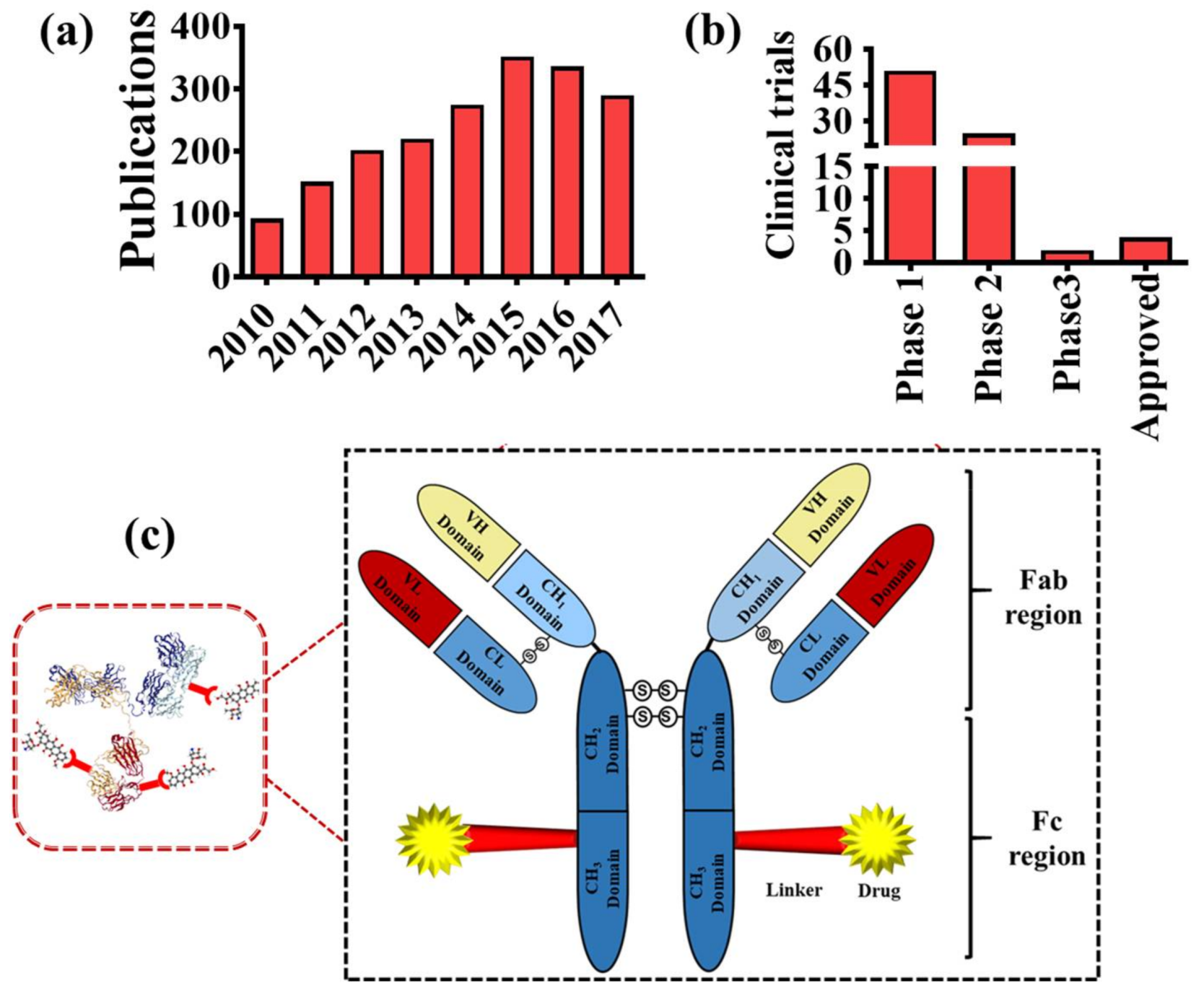
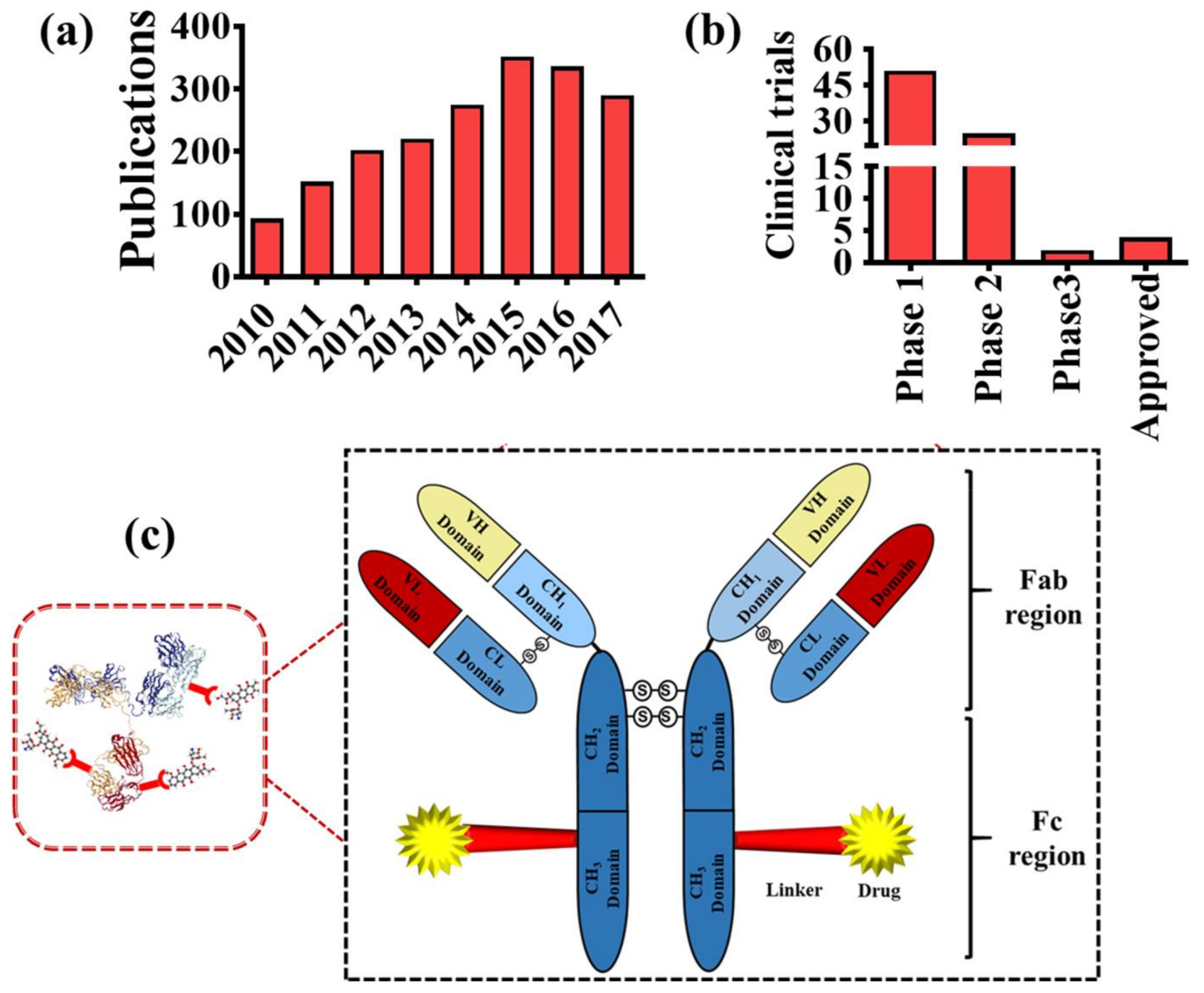
1. Introduction
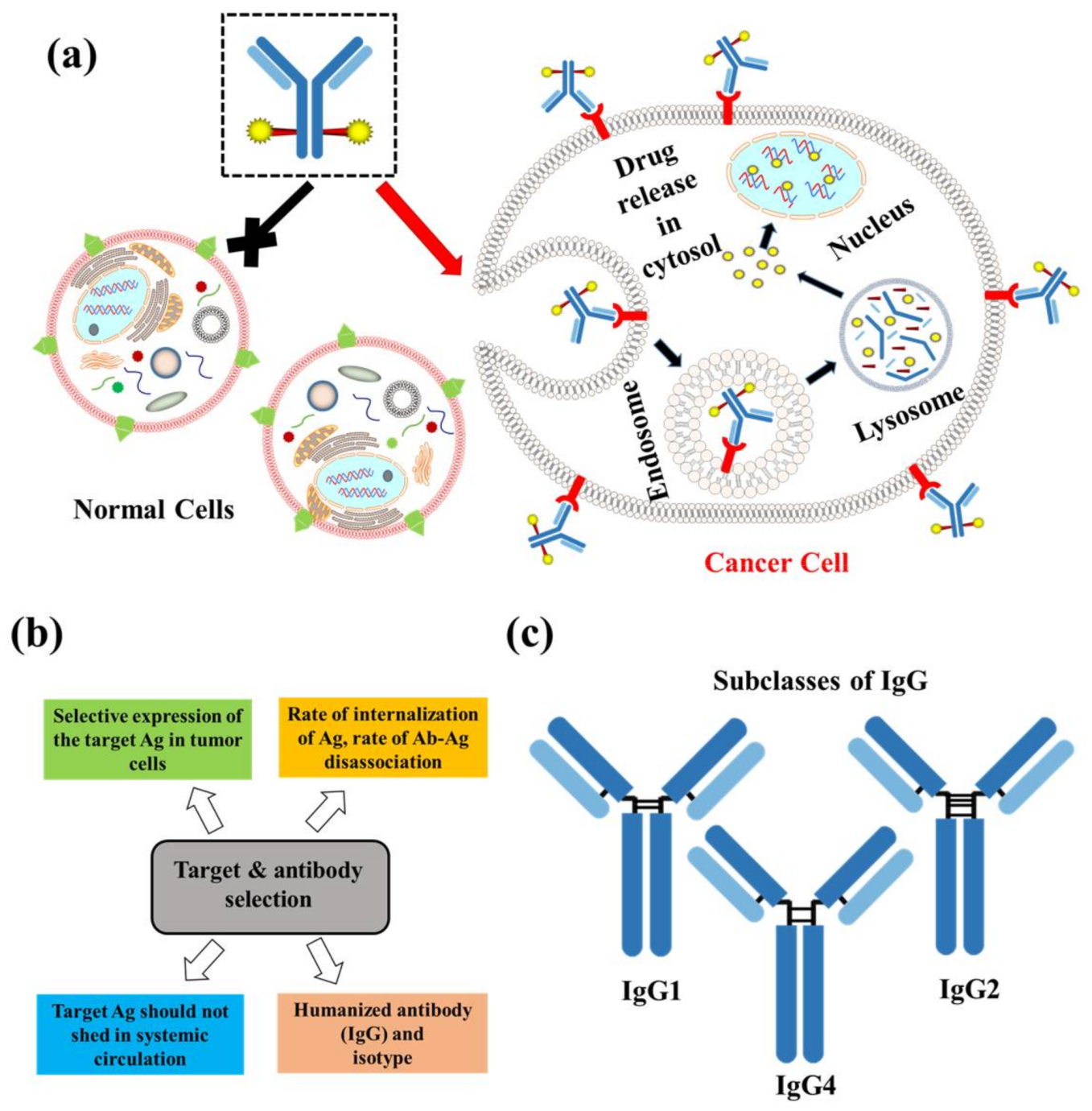
2. Composition of ADCs
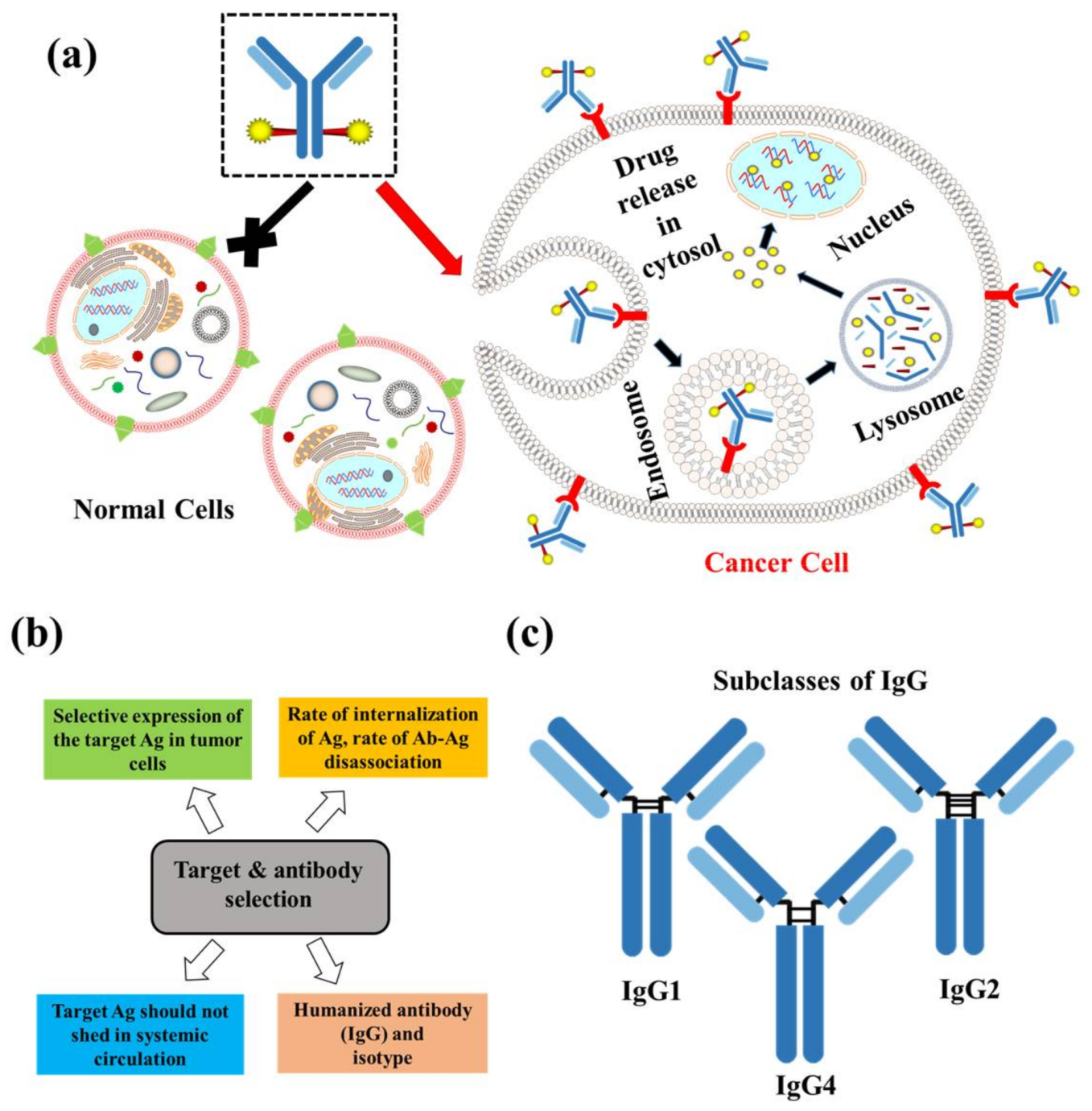
2.1. Target and Antibody
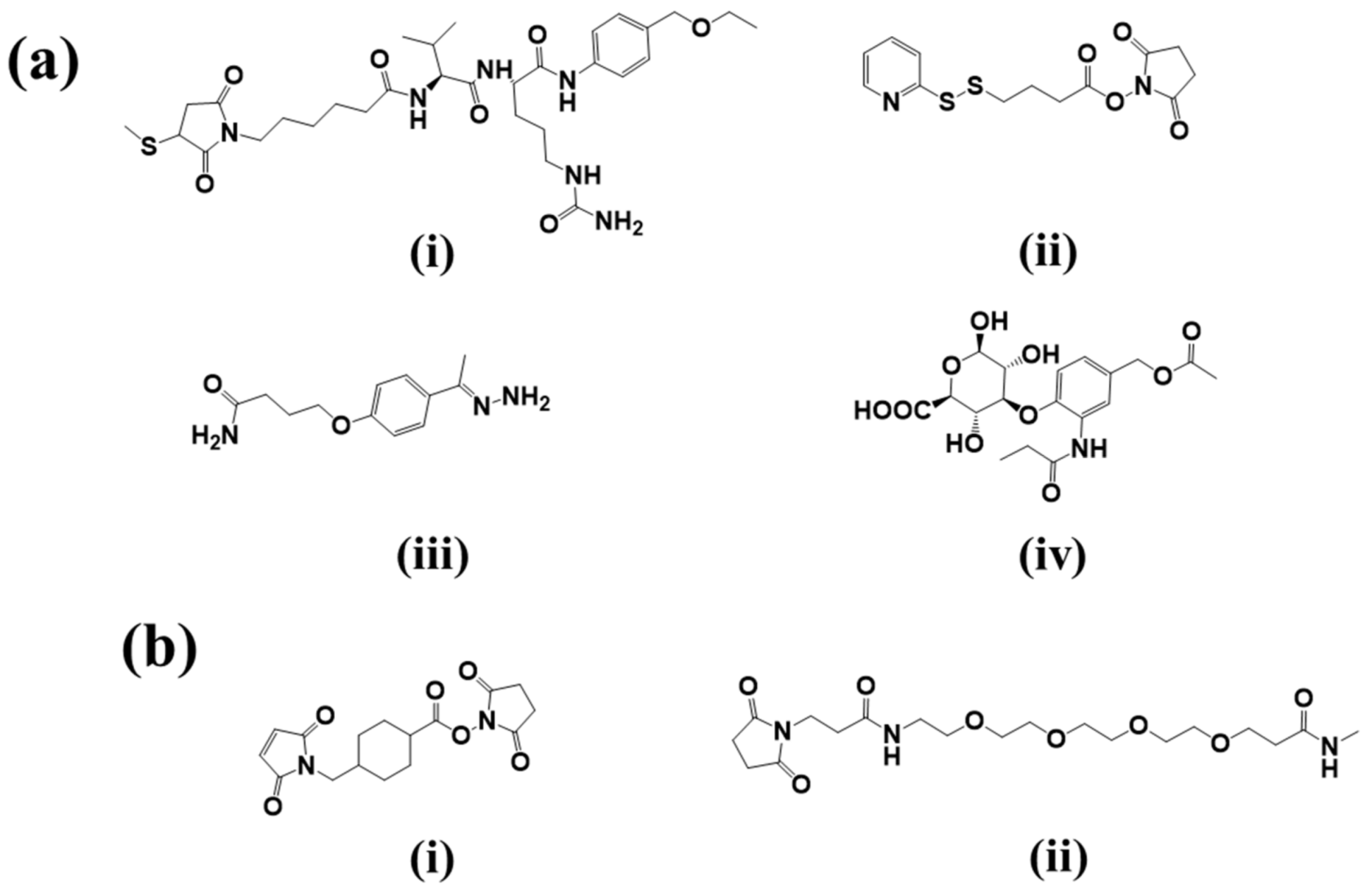
2.2. Linker
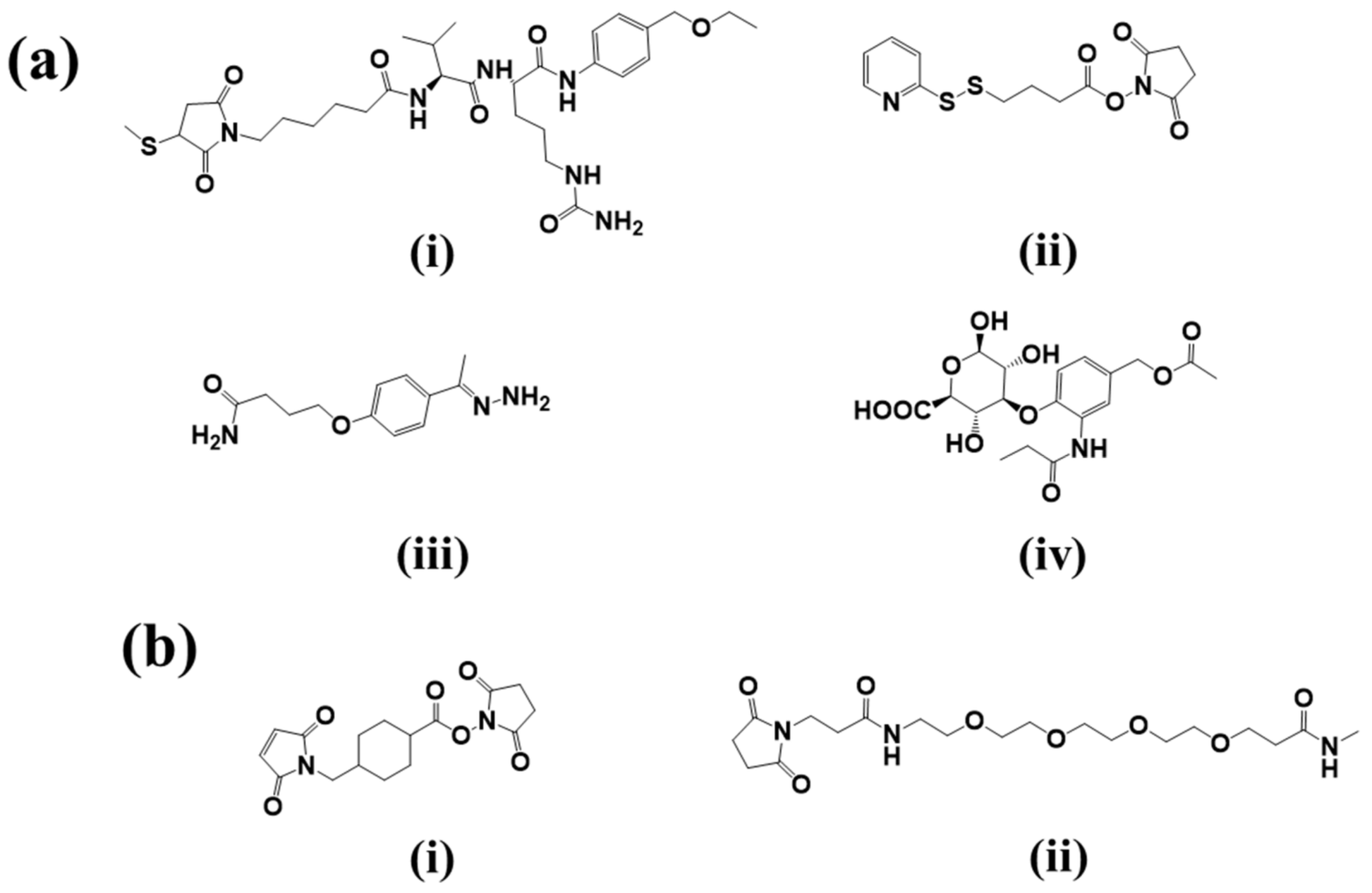
2.2.1. Cleavable Linkers
- Acid-sensitive linkers: Acid-sensitive hydrazone groups in acid-labile linkers remains stable in systemic circulation (pH 7.5) and gets hydrolyzed in lysosomal (pH 4.8) and endosomal (pH 5–6) acidic tumor micro-environment upon internalization in the targeted cells [17]. Withdrawal of gemtuzumab ozogamicin (Mylotarg®) in 2010, an anti-CD33 ADC for treatment of acute myeloid lymphoma, raises concern over the stability of this linker [18]. The heterogeneous nature of the drug conjugate contributed to premature release of payload, which in turn may have contributed to its remarkable toxicity compared to conventional chemotherapy. Currently, inotuzumab ozogamicin and milatuzumab doxorubicin, that are developed with a hydrazone linker.
- Glutathione-sensitive disulfide linkers: Another common example of cleavable linkers is glutathione-sensitive disulfide linkers. Glutathione is a low molecular weight thiol which is present in the cytoplasm (0.5–10 mmol/L) and extracellular environment (2–20 µmol/L in plasma) [19]. In tumor cells elevated levels of thiols are found during stress conditions such as hypoxia [20]. The difference in glutathione concentration in cytoplasm and extracellular environment can be implemented as a selective delivery of the drug payload to target tumor via breakdown of disulfide linkers [21]. Besides glutathione, intercellular protein disulfide isomerase (PDI) is also capable to reduce disulfide bonds. Two cysteine residues in the active site of this enzyme governs the thiol-disulfide exchange reactions with or within substrates [22]. Maytansinoid drug conjugates have been widely employed for disulfide bonds with an average DAR of 3–4 [23].
- Lysosomal protease-sensitive peptide linkers: Tumor cells have higher expression of lysosomal proteases like cathepsin B than normal cells. Cathepsin B-sensitive peptide linker conjugated ADCs selectively binds to and get internalized into tumor cells via receptor mediated endocytosis [24]. Proteases are inactivated in serum in presence of a high pH and different serum protease inhibitors [24]. This makes the peptide linker stable in systemic circulation and only to be cleaved upon internalization in tumors. In case of the FDA approved Adecetris®, cathepsin B- sensitive valine-citruline linker is found to be superior to hydrazone linker. The valine-citruline linker connects the bridge between p-aminobenzylcarbamate-monomethyl auristatin E (MMAE) and anti-CD30-mAb [5].
- β-glucuronide linker: β-Glucuronidase-sensitive linkers have been successfully used in a handful of glucuronide prodrugs [25]. Lysosomes and tumor necrotic areas are rich in β-glucuronidase which is active at lysosomal pH and inactive at physiological pH [26]. This selective site of action allows for a selective release of cytotoxic payloads through cleavage of the glycosidic bond of β-glucuronidase-sensitive β-glucuronide linkers. Further, the hydrophilic nature of this linker provides aqueous solubility for hydrophobic payloads and decreases aggregation of ADCs [27]. A highly hydrophobic CBI payload was conjugated to h1F6 and cAC10 mAbs utilizing β-glucuronide linker with an average DAR ~4–5 [27]. Such ADC compositions were found to be mostly monomeric in nature compared to extremely aggregated PABC-dipeptide based CBI conjugates [27]. Psymberin/irciniastatin A, a phenolic cytotoxic payload-based ADC was developed with N,N′-dimethylethylene diamine self-immolative spacers and a β-glucuronide linker for targeting CD-30-positive and CD-70-positive malignancies [28]. This development led to the possibility of developing phenolic warhead-based ADCs as many anti-cancer drugs have phenol functional groups. Another β-glucuronidase-sensitive linker based ADC has recently been developed utilizing tertiary amine functional group of payloads (tubulysins and auristatin E) as the conjugation site to the linker [29]. Tertiary-ammonium based linkers provide an excellent strategy for conjugating payloads without affecting their activity [29].
2.2.2. Non-Cleavable Linkers
2.2.3. Rational Linker Design to Overcome Resistance
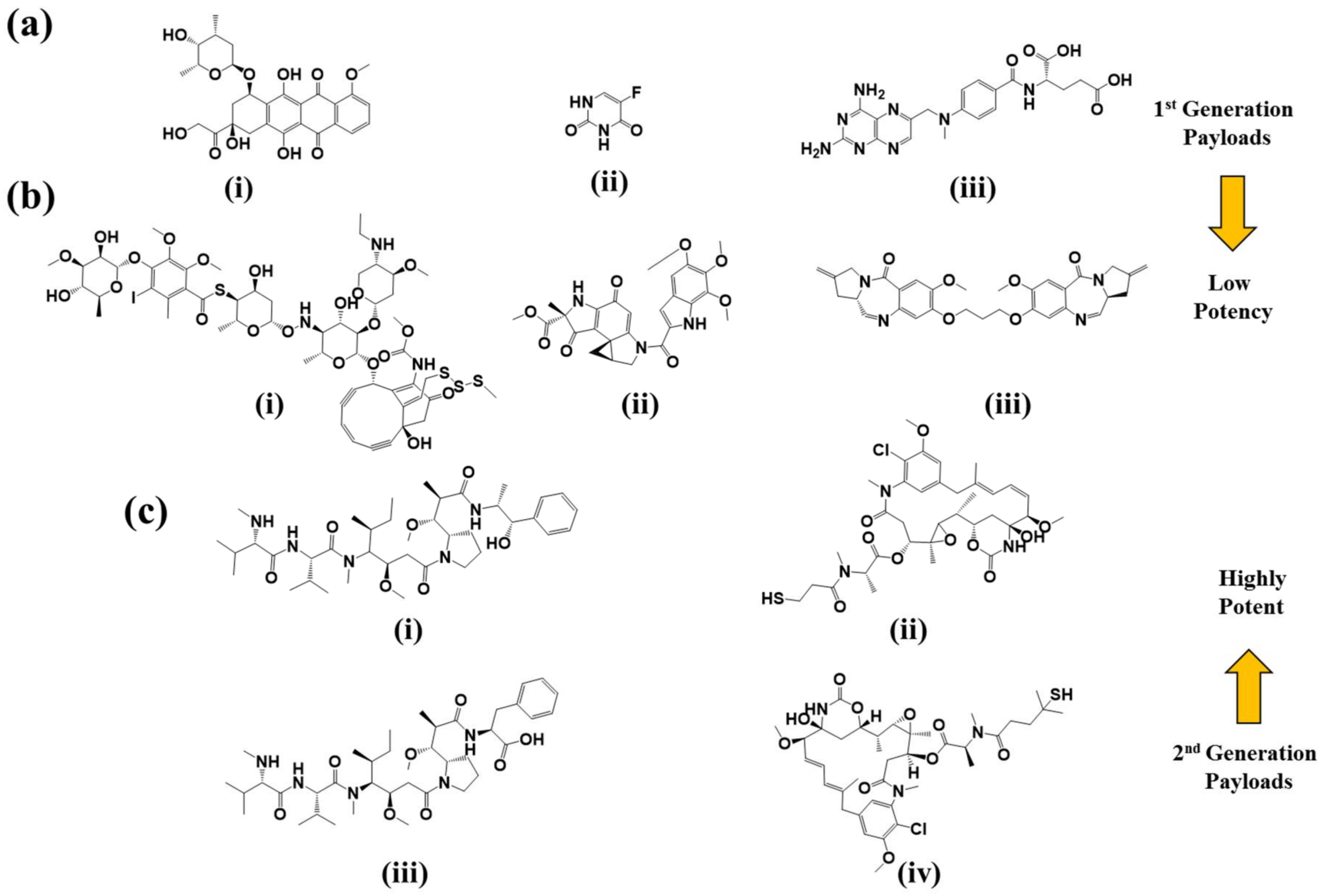
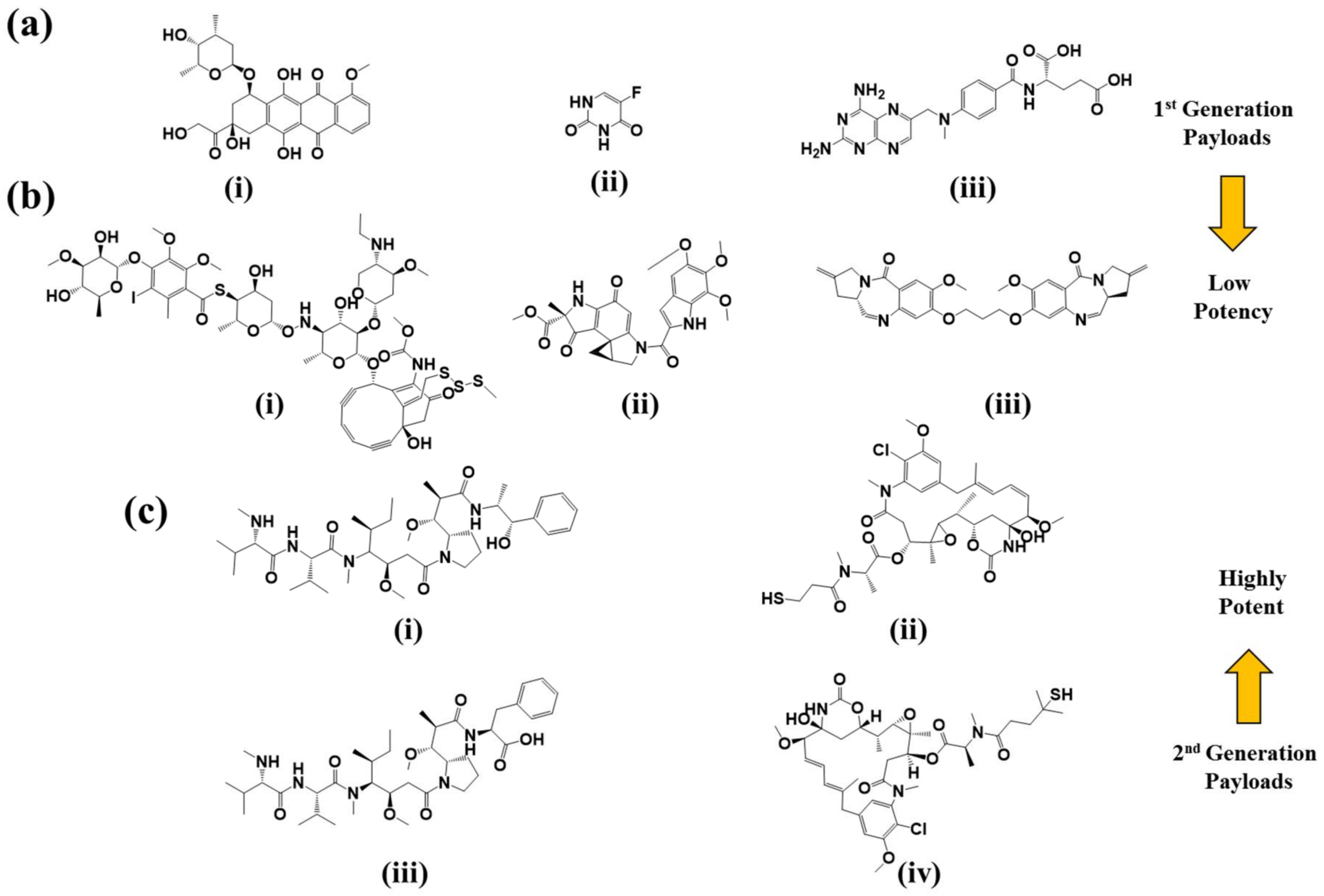
2.3. Payloads
2.3.1. DNA Damaging Agents
2.3.2. Tubulin Polymerization Inhibitors
3. Conjugation
3.1. Via Side Chain Cystine Residues
3.2. Via Side Chain Lysine Residues
3.3. Drug Antibody Ratio (DAR)
3.4. Site Specific Conjugation
3.4.1. Engineering of Side Chain Cysteine Residues
3.4.2. Incorporation of Unnatural Amino Acids (unAA)
3.4.3. Enzymatic Site-Specific Conjugation Processes
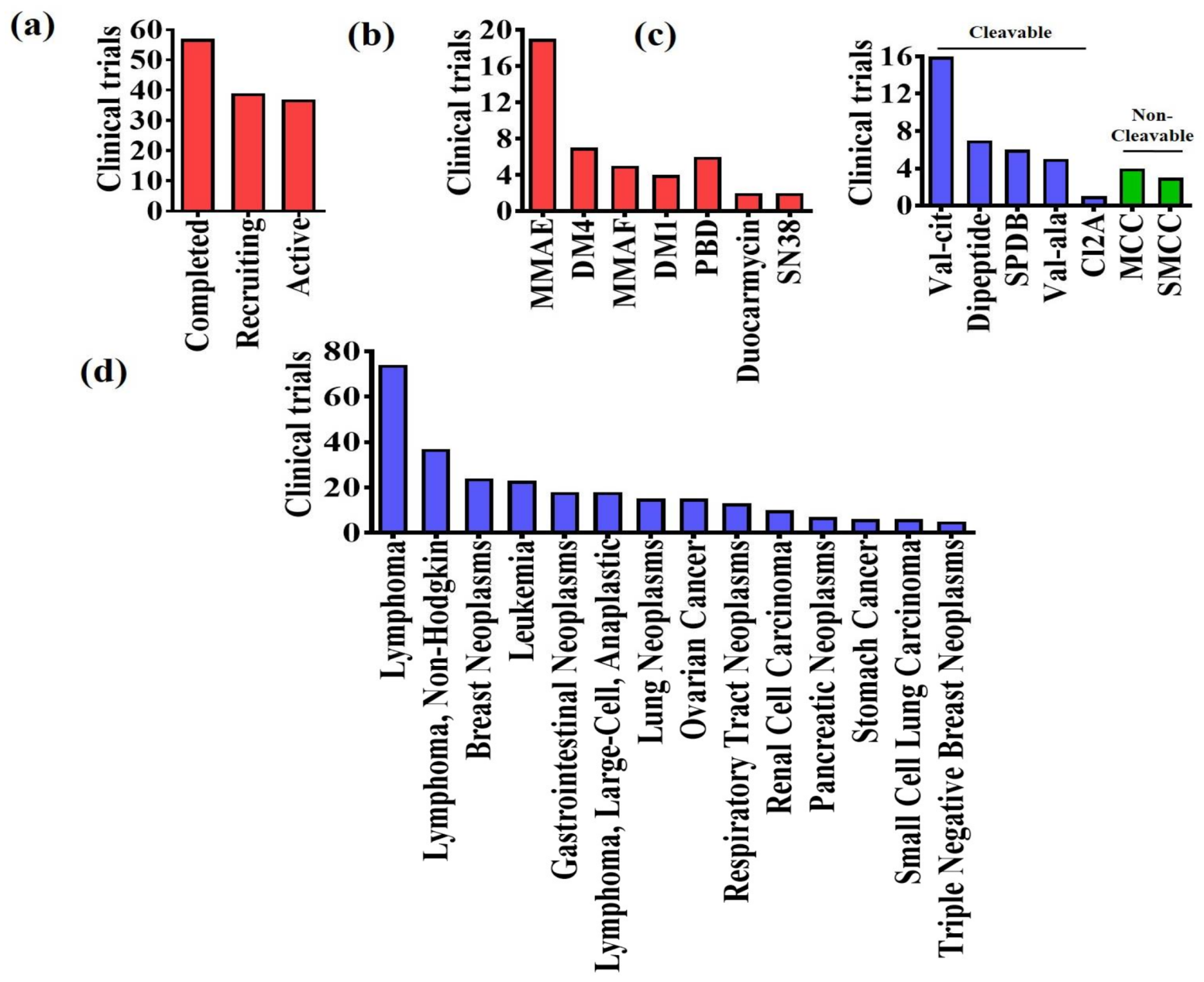
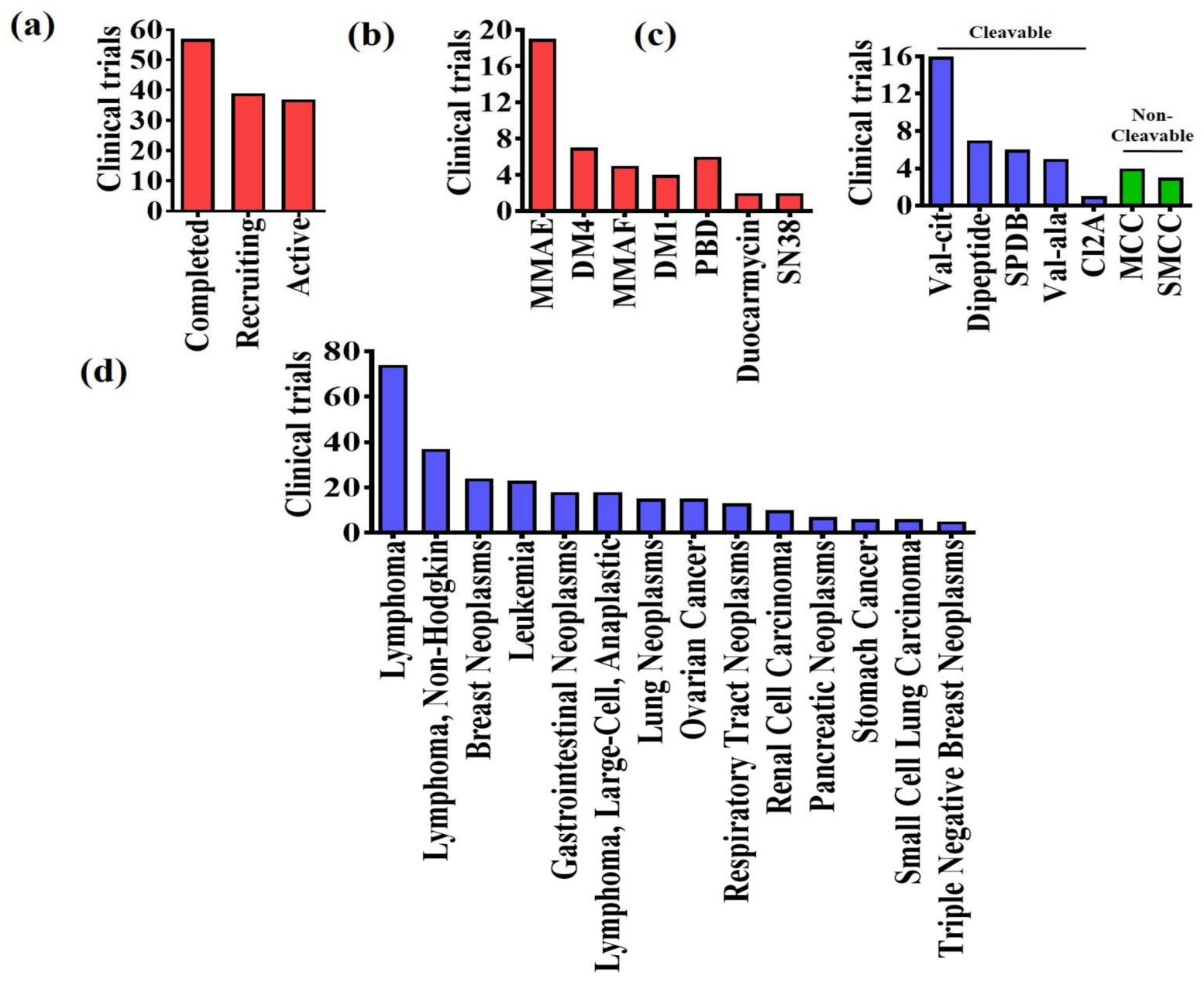
4. Clinical Trials
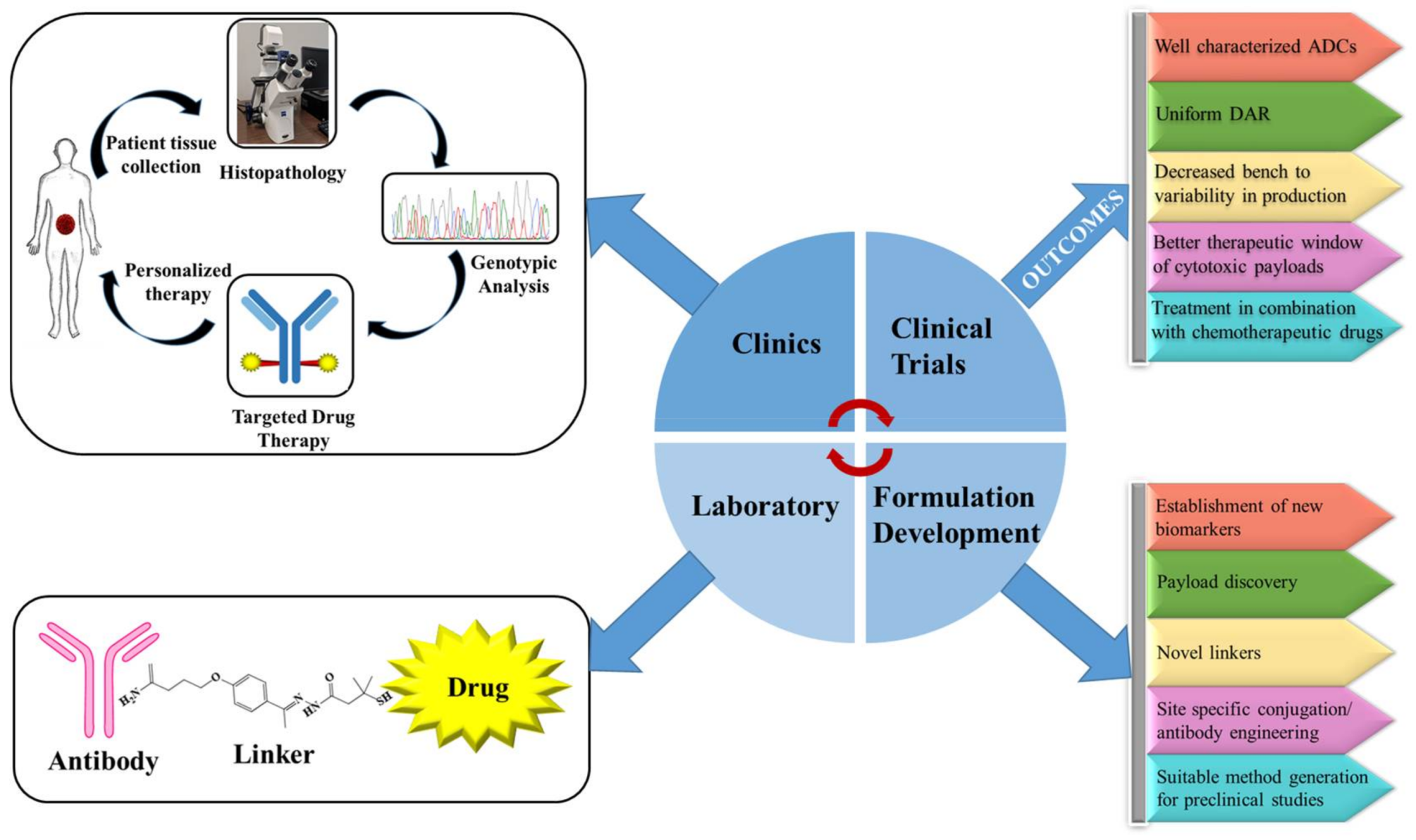
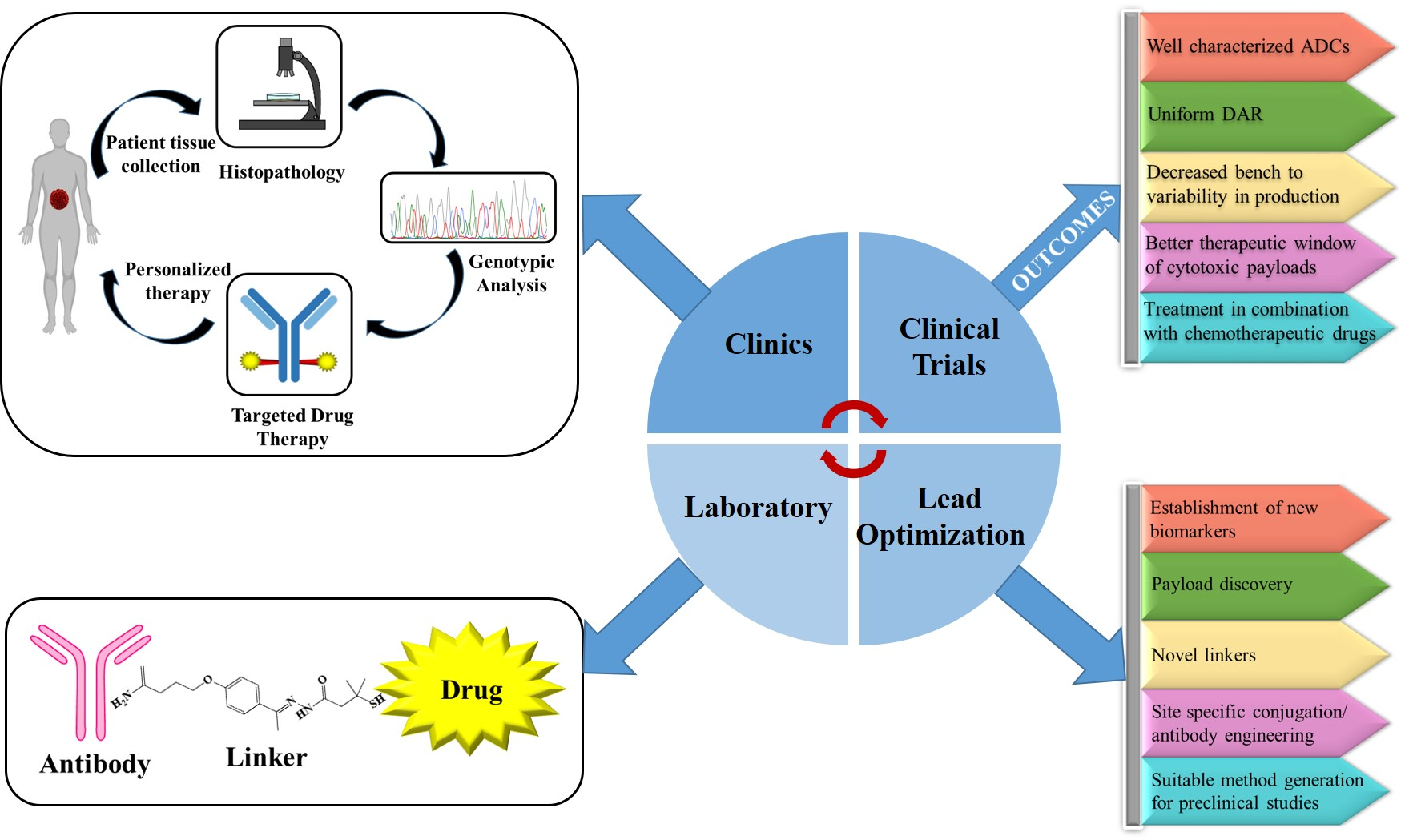
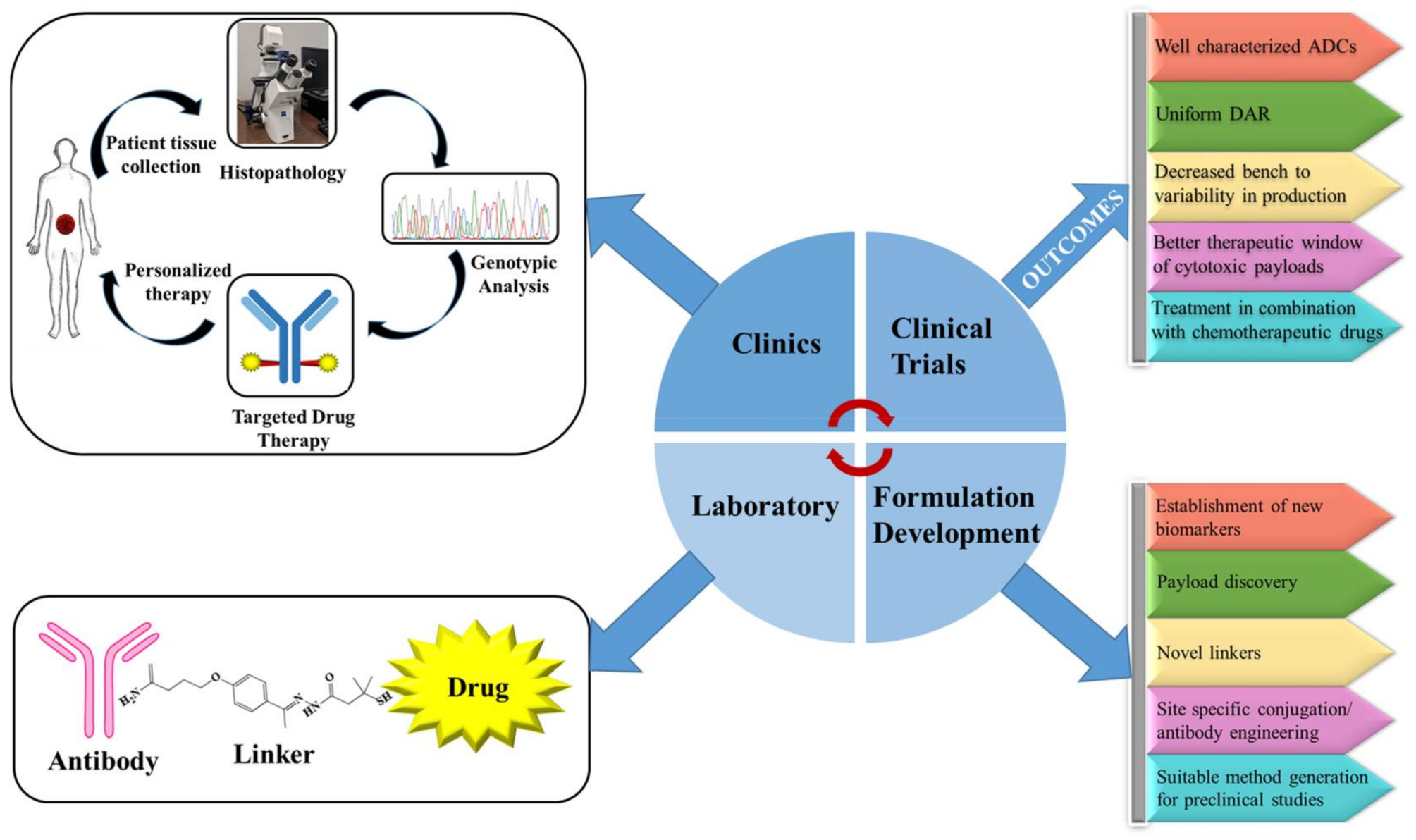
5. Future Directions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- WHO: Cancer World Health Organization. Available online: http://www.who.int/mediacentre/factsheets/fs297/en/ (accessed on 5 April 2018).
- Peer, D.; Karp, J.M.; Hong, S.; Farokhzad, O.C.; Margalit, R.; Langer, R. Nanocarriers as an emerging platform for cancer therapy. Nat. Nanotechnol. 2007, 2, 751–760. [Google Scholar] [CrossRef] [PubMed]
- Gottesman, M.M.; Fojo, T.; Bates, S.E. Multidrug resistance in cancer: Role of atp-dependent transporters. Nat. Rev. Cancer 2002, 2, 48–58. [Google Scholar] [CrossRef] [PubMed]
- Strebhardt, K.; Ullrich, A. Paul ehrlich’s magic bullet concept: 100 years of progress. Nat. Rev. Cancer 2008, 8, 473–480. [Google Scholar] [CrossRef] [PubMed]
- Panowski, S.; Bhakta, S.; Raab, H.; Polakis, P.; Junutula, J.R. Site-specific antibody drug conjugates for cancer therapy. mAbs 2014, 6, 34–45. [Google Scholar] [CrossRef] [PubMed]
- Chari, R.V.J.; Miller, M.L.; Widdison, W.C. Antibody–drug conjugates: An emerging concept in cancer therapy. Angew. Chem. Int. Ed. 2014, 53, 3796–3827. [Google Scholar] [CrossRef] [PubMed]
- Carter, P.J.; Senter, P.D. Antibody-drug conjugates for cancer therapy. Cancer J. 2008, 14, 154–169. [Google Scholar] [CrossRef] [PubMed]
- Milstein, G.K.C. Continuous cultures of fused cells secreting antibody of predefined specificity. Nature 1975, 256, 495–497. [Google Scholar]
- Rudnick, S.I.; Lou, J.; Shaller, C.C.; Tang, Y.; Klein-Szanto, A.J.P.; Weiner, L.M.; Marks, J.D.; Adams, G.P. Influence of affinity and antigen internalization on the uptake and penetration of anti-her2 antibodies in solid tumors. Cancer Res. 2011, 71, 2250–2259. [Google Scholar] [CrossRef] [PubMed]
- Lin, K.; Tibbitts, J. Pharmacokinetic considerations for antibody drug conjugates. Pharm. Res. 2012, 29, 2354–2366. [Google Scholar] [CrossRef] [PubMed]
- Tolcher, A.W.; Ochoa, L.; Hammond, L.A.; Patnaik, A.; Edwards, T.; Takimoto, C.; Smith, L.; de Bono, J.; Schwartz, G.; Mays, T.; et al. Cantuzumab mertansine, a maytansinoid immunoconjugate directed to the canag antigen: A phase I, pharmacokinetic, and biologic correlative study. J. Clin. Oncol. 2003, 21, 211–222. [Google Scholar] [CrossRef] [PubMed]
- Pastuskovas, C.V.; Mallet, W.; Clark, S.; Kenrick, M.; Majidy, M.; Schweiger, M.; Van Hoy, M.; Tsai, S.P.; Bennett, G.; Shen, B.-Q. Effect of immune complex formation on the distribution of a novel antibody to the ovarian tumor antigen CA125. Drug Metab. Dispos. 2010, 38, 2309–2319. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.-R.; Song, A.; Bergelson, S.; Arroll, T.; Parekh, B.; May, K.; Chung, S.; Strouse, R.; Mire-Sluis, A.; Schenerman, M. Advances in the assessment and control of the effector functions of therapeutic antibodies. Nat. Rev. Drug Discov. 2011, 10, 101–111. [Google Scholar] [CrossRef] [PubMed]
- Sapra, P.; Hooper, A.T.; O’Donnell, C.J.; Gerber, H.P. Investigational antibody drug conjugates for solid tumors. Expert Opin. Investig. Drugs 2011, 20, 1131–1149. [Google Scholar] [CrossRef] [PubMed]
- Junttila, T.T.; Li, G.; Parsons, K.; Phillips, G.L.; Sliwkowski, M.X. Trastuzumab-DM1 (T-DM1) retains all the mechanisms of action of trastuzumab and efficiently inhibits growth of lapatinib insensitive breast cancer. Breast Cancer Res. Treat. 2011, 128, 347–356. [Google Scholar] [CrossRef] [PubMed]
- Sharkey, R.M.; Goldenberg, D.M. Targeted therapy of cancer: New prospects for antibodies and immunoconjugates. CA-A Cancer J. Clin. 2006, 56, 226–243. [Google Scholar] [CrossRef]
- Pillay, C.S.; Elliott, E.; Dennison, C. Endolysosomal proteolysis and its regulation. Biochem. J. 2002, 363, 417–429. [Google Scholar] [CrossRef] [PubMed]
- Alley, S.C.; Okeley, N.M.; Senter, P.D. Antibody-drug conjugates: Targeted drug delivery for cancer. Curr. Opin. Chem. Biol. 2010, 14, 529–537. [Google Scholar] [CrossRef] [PubMed]
- Griffith, O.W. Biologic and pharmacologic regulation of mammalian glutathione synthesis. Free Radic. Biol. Med. 1999, 27, 922–935. [Google Scholar] [CrossRef]
- Balendiran, G.K.; Dabur, R.; Fraser, D. The role of glutathione in cancer. Cell Biochem. Funct. 2004, 22, 343–352. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.; Fang, Y.-Z.; Yang, S.; Lupton, J.R.; Turner, N.D. Glutathione metabolism and its implications for health. J. Nutr. 2004, 134, 489–492. [Google Scholar] [CrossRef] [PubMed]
- Appenzeller-Herzog, C.; Ellgaard, L. The human pdi family: Versatility packed into a single fold. BBA-Mol. Cell. Res. 2008, 1783, 535–548. [Google Scholar] [CrossRef] [PubMed]
- Chari, R.V.J. Targeted cancer therapy: Conferring specificity to cytotoxic drugs. Acc. Chem. Res. 2008, 41, 98–107. [Google Scholar] [CrossRef] [PubMed]
- Dubowchik, G.M.; Firestone, R.A. Cathepsin b-sensitive dipeptide prodrugs. 1. A model study of structural requirements for efficient release of doxorubicin. Bioorg. Med. Chem. Lett. 1998, 8, 3341–3346. [Google Scholar] [CrossRef]
- Tranoy-Opalinski, I.; Legigan, T.; Barat, R.; Clarhaut, J.; Thomas, M.; Renoux, B.; Papot, S. Beta-glucuronidase-responsive prodrugs for selective cancer chemotherapy: An update. Eur. J. Med. Chem. 2014, 74, 302–313. [Google Scholar] [CrossRef] [PubMed]
- Michelle de, G.; Epie, B.; Hans, W.S.; Hidde, J.H.; Herbert, M.P. Beta-glucuronidase-mediated drug release. Curr. Pharm. Des. 2002, 8, 1391–1403. [Google Scholar]
- Jeffrey, S.C.; Andreyka, J.B.; Bernhardt, S.X.; Kissler, K.M.; Kline, T.; Lenox, J.S.; Moser, R.F.; Nguyen, M.T.; Okeley, N.M.; Stone, I.J.; et al. Development and properties of β-glucuronide linkers for monoclonal antibody−drug conjugates. Bioconj. Chem. 2006, 17, 831–840. [Google Scholar] [CrossRef] [PubMed]
- Jeffrey, S.C.; De Brabander, J.; Miyamoto, J.; Senter, P.D. Expanded utility of the β-glucuronide linker: ADCs that deliver phenolic cytotoxic agents. ACS Med. Chem. Lett. 2010, 1, 277–280. [Google Scholar] [CrossRef] [PubMed]
- Burke, P.J.; Hamilton, J.Z.; Pires, T.A.; Setter, J.R.; Hunter, J.H.; Cochran, J.H.; Waight, A.B.; Gordon, K.A.; Toki, B.E.; Emmerton, K.K.; et al. Development of novel quaternary ammonium linkers for antibody–drug conjugates. Mol. Cancer Ther. 2016, 15, 938–945. [Google Scholar] [CrossRef] [PubMed]
- Senter, P.D.; Sievers, E.L. The discovery and development of brentuximab vedotin for use in relapsed hodgkin lymphoma and systemic anaplastic large cell lymphoma. Nat. Biotechnol. 2012, 30, 631–637. [Google Scholar] [CrossRef] [PubMed]
- Diamantis, N.; Banerji, U. Antibody-drug conjugates—An emerging class of cancer treatment. Br. J. Cancer 2016, 114, 362–367. [Google Scholar] [CrossRef] [PubMed]
- LoRusso, P.M.; Weiss, D.; Guardino, E.; Girish, S.; Sliwkowski, M.X. Trastuzumab emtansine: A unique antibody-drug conjugate in development for human epidermal growth factor receptor 2–positive cancer. Clin. Cancer Res. 2011, 17, 6437–6447. [Google Scholar] [CrossRef] [PubMed]
- Kovtun, Y.V.; Audette, C.A.; Mayo, M.F.; Jones, G.E.; Doherty, H.; Maloney, E.K.; Erickson, H.K.; Sun, X.; Wilhelm, S.; Ab, O.; et al. Antibody-maytansinoid conjugates designed to bypass multidrug resistance. Cancer Res. 2010, 70, 2528–2537. [Google Scholar] [CrossRef] [PubMed]
- Doronina, S.O.; Mendelsohn, B.A.; Bovee, T.D.; Cerveny, C.G.; Alley, S.C.; Meyer, D.L.; Oflazoglu, E.; Toki, B.E.; Sanderson, R.J.; Zabinski, R.F.; et al. Enhanced activity of monomethylauristatin f through monoclonal antibody delivery: Effects of linker technology on efficacy and toxicity. Bioconj. Chem. 2006, 17, 114–124. [Google Scholar] [CrossRef] [PubMed]
- Loganzo, F.; Sung, M.; Gerber, H.-P. Mechanisms of resistance to antibody–drug conjugates. Mol. Cancer Ther. 2016, 15, 2825–2834. [Google Scholar] [CrossRef] [PubMed]
- Parslow, A.; Parakh, S.; Lee, F.-T.; Gan, H.; Scott, A. Antibody–drug conjugates for cancer therapy. Biomedicines 2016, 4, 14. [Google Scholar] [CrossRef] [PubMed]
- Linenberger, M.L.; Hong, T.; Flowers, D.; Sievers, E.L.; Gooley, T.A.; Bennett, J.M.; Berger, M.S.; Leopold, L.H.; Appelbaum, F.R.; Bernstein, I.D. Multidrug-resistance phenotype and clinical responses to gemtuzumab ozogamicin. Blood 2001, 98, 988–994. [Google Scholar] [CrossRef] [PubMed]
- Shefet-Carasso, L.; Benhar, I. Antibody-targeted drugs and drug resistance--challenges and solutions. Drug Resist. Updates 2015, 18, 36–46. [Google Scholar] [CrossRef] [PubMed]
- Zhao, R.Y.; Wilhelm, S.D.; Audette, C.; Jones, G.; Leece, B.A.; Lazar, A.C.; Goldmacher, V.S.; Singh, R.; Kovtun, Y.; Widdison, W.C.; et al. Synthesis and evaluation of hydrophilic linkers for antibody-maytansinoid conjugates. J. Med. Chem. 2011, 54, 3606–3623. [Google Scholar] [CrossRef] [PubMed]
- Ducry, L.; Stump, B. Antibody−drug conjugates: Linking cytotoxic payloads to monoclonal antibodies. Bioconj. Chem. 2010, 21, 5–13. [Google Scholar] [CrossRef] [PubMed]
- Trail, P.; Willner, D.; Lasch, S.; Henderson, A.; Hofstead, S.; Casazza, A.; Firestone, R.; Hellstrom, I.; Hellstrom, K. Cure of xenografted human carcinomas by Br96-doxorubicin immunoconjugates. Science 1993, 261, 212–215. [Google Scholar] [CrossRef] [PubMed]
- Tolcher, A.W.; Sugarman, S.; Gelmon, K.A.; Cohen, R.; Saleh, M.; Isaacs, C.; Young, L.; Healey, D.; Onetto, N.; Slichenmyer, W. Randomized phase ii study of br96-doxorubicin conjugate in patients with metastatic breast cancer. J. Clin. Oncol. 1999, 17, 478–484. [Google Scholar] [CrossRef] [PubMed]
- Maiese, W.M.; Lechevalier, M.P.; Lechevalier, H.A.; Korshalla, J.; Kuck, N.; Fantini, A.; Wildey, M.J.; Thomas, J.; Greenstein, M. Calicheamicins, a novel family of antitumor antibiotics: Taxonomy, fermentation and biological properties. J. Antibiot. 1989, 42, 558–563. [Google Scholar] [CrossRef] [PubMed]
- Polakis, P. Antibody drug conjugates for cancer therapy. Pharmacol. Rev. 2016, 68, 3–19. [Google Scholar] [CrossRef] [PubMed]
- Jones, R.R.; Bergman, R.G. P-benzyne. Generation as an intermediate in a thermal isomerization reaction and trapping evidence for the 1,4-benzenediyl structure. J. Am. Chem. Soc. 1972, 94, 660–661. [Google Scholar] [CrossRef]
- Watanabe, C.M.; Supekova, L.; Schultz, P.G. Transcriptional effects of the potent enediyne anti-cancer agent calicheamicin gamma(i)(1). Chem. Biol. 2002, 9, 245–251. [Google Scholar] [CrossRef]
- De Vries, J.F.; Zwaan, C.M.; De Bie, M.; Voerman, J.S.; den Boer, M.L.; van Dongen, J.J.; van der Velden, V.H. The novel calicheamicin-conjugated CD22 antibody inotuzumab ozogamicin (CMC-544) effectively kills primary pediatric acute lymphoblastic leukemia cells. Leukemia 2012, 26, 255–264. [Google Scholar] [CrossRef] [PubMed]
- Elgersma, R.C.; Coumans, R.G.; Huijbregts, T.; Menge, W.M.; Joosten, J.A.; Spijker, H.J.; de Groot, F.M.; van der Lee, M.M.; Ubink, R.; van den Dobbelsteen, D.J.; et al. Design, synthesis, and evaluation of linker-duocarmycin payloads: Toward selection of HER2-targeting antibody-drug conjugate SYD985. Mol. Pharm. 2015, 12, 1813–1835. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Khullar, A.; Chou, S.; Sacramo, A.; Gerratana, B. Biosynthesis of sibiromycin, a potent antitumor antibiotic. Appl. Environ. Microbiol. 2009, 75, 2869–2878. [Google Scholar] [CrossRef] [PubMed]
- Bouchard, H.; Viskov, C.; Garcia-Echeverria, C. Antibody-drug conjugates—A new wave of cancer drugs. Bioorg. Med. Chem. Lett. 2014, 24, 5357–5363. [Google Scholar] [CrossRef] [PubMed]
- Mantaj, J.; Jackson, P.J.; Rahman, K.M.; Thurston, D.E. From anthramycin to pyrrolobenzodiazepine (PBD)-containing antibody-drug conjugates (ADCs). Angew. Chem. Int. Ed. Engl. 2017, 56, 462–488. [Google Scholar] [CrossRef] [PubMed]
- Amador, M.L.; Jimeno, J.; Paz-Ares, L.; Cortes-Funes, H.; Hidalgo, M. Progress in the development and acquisition of anticancer agents from marine sources. Ann. Oncol. 2003, 14, 1607–1615. [Google Scholar] [CrossRef] [PubMed]
- Pettit, G.R.; Srirangam, J.K.; Barkoczy, J.; Williams, M.D.; Boyd, M.R.; Hamel, E.; Pettit, R.K.; Hogan, F.; Bai, R.; Chapuis, J.C.; et al. Antineoplastic agents 365. Dolastatin 10 sar probes. Anticancer Drug Des. 1998, 13, 243–277. [Google Scholar] [PubMed]
- Pettit, R.K.; Pettit, G.R.; Hazen, K.C. Specific activities of dolastatin 10 and peptide derivatives against cryptococcus neoformans. Antimicrob. Agents Chemother. 1998, 42, 2961–2965. [Google Scholar] [PubMed]
- Bai, R.L.; Pettit, G.R.; Hamel, E. Binding of dolastatin 10 to tubulin at a distinct site for peptide antimitotic agents near the exchangeable nucleotide and vinca alkaloid sites. J. Biol. Chem. 1990, 265, 17141–17149. [Google Scholar] [PubMed]
- Katz, J.; Janik, J.E.; Younes, A. Brentuximab vedotin (SGN-35). Clin. Cancer Res. 2011, 17, 6428–6436. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.F.; Moore, K.N.; Birrer, M.J.; Berlin, S.; Matulonis, U.A.; Infante, J.R.; Wolpin, B.; Poon, K.A.; Firestein, R.; Xu, J.; et al. Phase I study of safety and pharmacokinetics of the anti-MUC16 antibody-drug conjugate DMUC5754A in patients with platinum-resistant ovarian cancer or unresectable pancreatic cancer. Ann. Oncol. 2016, 27, 2124–2130. [Google Scholar] [CrossRef] [PubMed]
- Tai, Y.T.; Mayes, P.A.; Acharya, C.; Zhong, M.Y.; Cea, M.; Cagnetta, A.; Craigen, J.; Yates, J.; Gliddon, L.; Fieles, W.; et al. Novel anti-b-cell maturation antigen antibody-drug conjugate (GSK2857916) selectively induces killing of multiple myeloma. Blood 2014, 123, 3128–3138. [Google Scholar] [CrossRef] [PubMed]
- Francisco, J.A.; Cerveny, C.G.; Meyer, D.L.; Mixan, B.J.; Klussman, K.; Chace, D.F.; Rejniak, S.X.; Gordon, K.A.; DeBlanc, R.; Toki, B.E.; et al. cAC10-vcMMAE, an anti-CD30-monomethyl auristatin E conjugate with potent and selective antitumor activity. Blood 2003, 102, 1458–1465. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Emmerton, K.K.; Jonas, M.; Zhang, X.; Miyamoto, J.B.; Setter, J.R.; Nicholas, N.D.; Okeley, N.M.; Lyon, R.P.; Benjamin, D.R.; et al. Intracellular released payload influences potency and bystander-killing effects of antibody-drug conjugates in preclinical models. Cancer Res. 2016, 76, 2710–2719. [Google Scholar] [CrossRef] [PubMed]
- Kupchan, S.M.; Komoda, Y.; Court, W.A.; Thomas, G.J.; Smith, R.M.; Karim, A.; Gilmore, C.J.; Haltiwanger, R.C.; Bryan, R.F. Maytansine, a novel antileukemic ansa macrolide from maytenus ovatus. J. Am. Chem. Soc. 1972, 94, 1354–1356. [Google Scholar] [CrossRef] [PubMed]
- Kupchan, S.M.; Komoda, Y.; Branfman, A.R.; Sneden, A.T.; Court, W.A.; Thomas, G.J.; Hintz, H.P.; Smith, R.M.; Karim, A.; Howie, G.A.; et al. The maytansinoids. Isolation, structural elucidation, and chemical interrelation of novel ansa macrolides. J. Org. Chem. 1977, 42, 2349–2357. [Google Scholar] [CrossRef] [PubMed]
- Mandelbaum-Shavit, F.; Wolpert-DeFilippes, M.K.; Johns, D.G. Binding of maytansine to rat brain tubulin. Biochem. Biophys. Res. Commun. 1976, 72, 47–54. [Google Scholar] [CrossRef]
- Widdison, W.C.; Wilhelm, S.D.; Cavanagh, E.E.; Whiteman, K.R.; Leece, B.A.; Kovtun, Y.; Goldmacher, V.S.; Xie, H.; Steeves, R.M.; Lutz, R.J.; et al. Semisynthetic maytansine analogues for the targeted treatment of cancer. J. Med. Chem. 2006, 49, 4392–4408. [Google Scholar] [CrossRef] [PubMed]
- Blanc, V.; Bousseau, A.; Caron, A.; Carrez, C.; Lutz, R.J.; Lambert, J.M. Sar3419: An anti-CD19-maytansinoid immunoconjugate for the treatment of B-cell malignancies. Clin. Cancer Res. 2011, 17, 6448–6458. [Google Scholar] [CrossRef] [PubMed]
- Lindell, T.J.; Weinberg, F.; Morris, P.W.; Roeder, R.G.; Rutter, W.J. Specific inhibition of nuclear RNA polymerase II by alpha-amanitin. Science 1970, 170, 447–449. [Google Scholar] [CrossRef] [PubMed]
- Anderl, J.; Müller, C.; Heckl-Östreicher, B.; Wehr, R. Abstract 3616: Highly potent antibody-amanitin conjugates cause tumor-selective apoptosis. Cancer Res. 2011, 71, 3616. [Google Scholar] [CrossRef]
- Moldenhauer, G.; Salnikov, A.V.; Luttgau, S.; Herr, I.; Anderl, J.; Faulstich, H. Therapeutic potential of amanitin-conjugated anti-epithelial cell adhesion molecule monoclonal antibody against pancreatic carcinoma. J. Natl. Cancer Inst. 2012, 104, 622–634. [Google Scholar] [CrossRef] [PubMed]
- Hechler, T.; Kulke, M.; Mueller, C.; Pahl, A.; Anderl, J. Abstract 664: Amanitin-based antibody-drug conjugates targeting the prostate-specific membrane antigen. Cancer Res. 2014, 74. [Google Scholar] [CrossRef]
- Hamblett, K.J.; Senter, P.D.; Chace, D.F.; Sun, M.M.; Lenox, J.; Cerveny, C.G.; Kissler, K.M.; Bernhardt, S.X.; Kopcha, A.K.; Zabinski, R.F.; et al. Effects of drug loading on the antitumor activity of a monoclonal antibody drug conjugate. Clin. Cancer Res. 2004, 10, 7063–7070. [Google Scholar] [CrossRef] [PubMed]
- Doronina, S.O.; Toki, B.E.; Torgov, M.Y.; Mendelsohn, B.A.; Cerveny, C.G.; Chace, D.F.; DeBlanc, R.L.; Gearing, R.P.; Bovee, T.D.; Siegall, C.B.; et al. Development of potent monoclonal antibody auristatin conjugates for cancer therapy. Nat. Biotechnol. 2003, 21, 778–784. [Google Scholar] [CrossRef] [PubMed]
- McAuley, A.; Jacob, J.; Kolvenbach, C.G.; Westland, K.; Lee, H.J.; Brych, S.R.; Rehder, D.; Kleemann, G.R.; Brems, D.N.; Matsumura, M. Contributions of a disulfide bond to the structure, stability, and dimerization of human igg1 antibody ch3 domain. Protein Sci. 2008, 17, 95–106. [Google Scholar] [CrossRef] [PubMed]
- Sun, M.M.; Beam, K.S.; Cerveny, C.G.; Hamblett, K.J.; Blackmore, R.S.; Torgov, M.Y.; Handley, F.G.; Ihle, N.C.; Senter, P.D.; Alley, S.C. Reduction-alkylation strategies for the modification of specific monoclonal antibody disulfides. Bioconj. Chem. 2005, 16, 1282–1290. [Google Scholar] [CrossRef] [PubMed]
- Schroeder, D.D.; Tankersley, D.L.; Lundblad, J.L. A new preparation of modified immune serum globulin (human) suitable for intravenous administration. I. Standardization of the reduction and alkylation reaction. Vox Sang. 1981, 40, 373–382. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, P.; Bertozzi, C.R. Site-specific antibody–drug conjugates: The nexus of bioorthogonal chemistry, protein engineering, and drug development. Bioconj. Chem. 2015, 26, 176–192. [Google Scholar] [CrossRef] [PubMed]
- Hamann, P.R.; Hinman, L.M.; Hollander, I.; Beyer, C.F.; Lindh, D.; Holcomb, R.; Hallett, W.; Tsou, H.R.; Upeslacis, J.; Shochat, D.; et al. Gemtuzumab ozogamicin, a potent and selective anti-CD33 antibody-calicheamicin conjugate for treatment of acute myeloid leukemia. Bioconjug. Chem. 2002, 13, 47–58. [Google Scholar] [CrossRef] [PubMed]
- Burnett, A.K.; Hills, R.K.; Milligan, D.; Kjeldsen, L.; Kell, J.; Russell, N.H.; Yin, J.A.; Hunter, A.; Goldstone, A.H.; Wheatley, K. Identification of patients with acute myeloblastic leukemia who benefit from the addition of gemtuzumab ozogamicin: Results of the mrc aml15 trial. J. Clin. Oncol. 2011, 29, 369–377. [Google Scholar] [CrossRef] [PubMed]
- Luo, Q.; Chung, H.H.; Borths, C.; Janson, M.; Wen, J.; Joubert, M.K.; Wypych, J. Structural characterization of a monoclonal antibody-maytansinoid immunoconjugate. Anal. Chem. 2016, 88, 695–702. [Google Scholar] [CrossRef] [PubMed]
- Adem, Y.T.; Schwarz, K.A.; Duenas, E.; Patapoff, T.W.; Galush, W.J.; Esue, O. Auristatin antibody drug conjugate physical instability and the role of drug payload. Bioconj. Chem. 2014, 25, 656–664. [Google Scholar] [CrossRef] [PubMed]
- Moussa, E.M.; Panchal, J.P.; Moorthy, B.S.; Blum, J.S.; Joubert, M.K.; Narhi, L.O.; Topp, E.M. Immunogenicity of therapeutic protein aggregates. J. Pharm. Sci. 2016, 105, 417–430. [Google Scholar] [CrossRef] [PubMed]
- McDonagh, C.F.; Turcott, E.; Westendorf, L.; Webster, J.B.; Alley, S.C.; Kim, K.; Andreyka, J.; Stone, I.; Hamblett, K.J.; Francisco, J.A.; et al. Engineered antibody-drug conjugates with defined sites and stoichiometries of drug attachment. Protein Eng. Des. Sel. 2006, 19, 299–307. [Google Scholar] [CrossRef] [PubMed]
- Yurkovetskiy, A.V.; Yin, M.; Bodyak, N.; Stevenson, C.A.; Thomas, J.D.; Hammond, C.E.; Qin, L.; Zhu, B.; Gumerov, D.R.; Ter-Ovanesyan, E.; et al. A polymer-based antibody–vinca drug conjugate platform: Characterization and preclinical efficacy. Cancer Res. 2015, 75, 3365–3372. [Google Scholar] [CrossRef] [PubMed]
- Krop, I.; Winer, E.P. Trastuzumab emtansine: A novel antibody-drug conjugate for HER2-positive breast cancer. Clin. Cancer Res. 2014, 20, 15–20. [Google Scholar] [CrossRef] [PubMed]
- Tsuchikama, K.; An, Z. Antibody-drug conjugates: Recent advances in conjugation and linker chemistries. Protein Cell 2018, 9, 33–46. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y. Drug-to-antibody ratio (DAR) by UV/vis spectroscopy. Methods Mol. Med. 2013, 1045, 267–273. [Google Scholar]
- Wang, L.; Amphlett, G.; Blättler, W.A.; Lambert, J.M.; Zhang, W. Structural characterization of the maytansinoid–monoclonal antibody immunoconjugate, HUN901–DM1, by mass spectrometry. Protein Sci. 2005, 14, 2436–2446. [Google Scholar] [CrossRef] [PubMed]
- Hudecz, F.; Garnett, M.C.; Khan, T.; Baldwin, R.W. The influence of synthetic conditions on the stability of methotrexate-monoclonal antibody conjugates determined by reversed phase high performance liquid chromatography. Biomed. Chromatogr. 1992, 6, 128–132. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, J. Drug-to-antibody ratio (DAR) and drug load distribution by hydrophobic interaction chromatography and reversed phase high-performance liquid chromatography. Methods Mol. Biol. 2013, 1045, 275–283. [Google Scholar] [PubMed]
- Chen, T.; Zhang, K.; Gruenhagen, J.; Medley, C.D.; Hydrophobic interaction chromatography for antibody drug conjugate drug distribution analysis. Am. Pharm. Rev. 2015. Available online: https://www.americanpharmaceuticalreview.com/Featured-Articles/177927-Hydrophobic-Interaction-Chromatography-for-Antibody-Drug-Conjugate-Drug-Distribution-Analysis/ (accessed on 3 April 2018).
- Basa, L. Drug-to-antibody ratio (DAR) and drug load distribution by lc-esi-ms. Methods Mol. Biol. 2013, 1045, 285–293. [Google Scholar] [PubMed]
- Huang, R.Y.C.; Chen, G. Characterization of antibody–drug conjugates by mass spectrometry: Advances and future trends. Drug Discov. Today 2016, 21, 850–855. [Google Scholar] [CrossRef] [PubMed]
- Wagner-Rousset, E.; Janin-Bussat, M.C.; Colas, O.; Excoffier, M.; Ayoub, D.; Haeuw, J.F.; Rilatt, I.; Perez, M.; Corvaia, N.; Beck, A. Antibody-drug conjugate model fast characterization by lc-ms following ides proteolytic digestion. mAbs 2014, 6, 273–285. [Google Scholar] [CrossRef] [PubMed]
- Junutula, J.R.; Raab, H.; Clark, S.; Bhakta, S.; Leipold, D.D.; Weir, S.; Chen, Y.; Simpson, M.; Tsai, S.P.; Dennis, M.S.; et al. Site-specific conjugation of a cytotoxic drug to an antibody improves the therapeutic index. Nat. Biotechnol. 2008, 26, 925–932. [Google Scholar] [CrossRef] [PubMed]
- Axup, J.Y.; Bajjuri, K.M.; Ritland, M.; Hutchins, B.M.; Kim, C.H.; Kazane, S.A.; Halder, R.; Forsyth, J.S.; Santidrian, A.F.; Stafin, K.; et al. Synthesis of site-specific antibody-drug conjugates using unnatural amino acids. Proc. Natl. Acad. Sci. USA 2012, 109, 16101–16106. [Google Scholar] [CrossRef] [PubMed]
- Jeger, S.; Zimmermann, K.; Blanc, A.; Grunberg, J.; Honer, M.; Hunziker, P.; Struthers, H.; Schibli, R. Site-specific and stoichiometric modification of antibodies by bacterial transglutaminase. Angew. Chem. Int. Ed. Engl. 2010, 49, 9995–9997. [Google Scholar] [CrossRef] [PubMed]
- Junutula, J.R.; Bhakta, S.; Raab, H.; Ervin, K.E.; Eigenbrot, C.; Vandlen, R.; Scheller, R.H.; Lowman, H.B. Rapid identification of reactive cysteine residues for site-specific labeling of antibody-fabs. J. Immunol. Methods 2008, 332, 41–52. [Google Scholar] [CrossRef] [PubMed]
- Junutula, J.R.; Flagella, K.M.; Graham, R.A.; Parsons, K.L.; Ha, E.; Raab, H.; Bhakta, S.; Nguyen, T.; Dugger, D.L.; Li, G.; et al. Engineered thio-trastuzumab-dm1 conjugate with an improved therapeutic index to target human epidermal growth factor receptor 2-positive breast cancer. Clin. Cancer Res. 2010, 16, 4769–4778. [Google Scholar] [CrossRef] [PubMed]
- Yao, H.; Jiang, F.; Lu, A.; Zhang, G. Methods to design and synthesize antibody-drug conjugates (adcs). Int. J. Mol. Sci. 2016, 17, 194. [Google Scholar] [CrossRef] [PubMed]
- Hallam, T.J.; Smider, V.V. Unnatural amino acids in novel antibody conjugates. Future Med. Chem. 2014, 6, 1309–1324. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.C.; Schultz, P.G. Adding new chemistries to the genetic code. Annu. Rev. Biochem. 2010, 79, 413–444. [Google Scholar] [CrossRef] [PubMed]
- Tian, F.; Lu, Y.; Manibusan, A.; Sellers, A.; Tran, H.; Sun, Y.; Phuong, T.; Barnett, R.; Hehli, B.; Song, F.; et al. A general approach to site-specific antibody drug conjugates. Proc. Natl. Acad. Sci. USA 2014, 111, 1766–1771. [Google Scholar] [CrossRef] [PubMed]
- Yokoyama, K.; Nio, N.; Kikuchi, Y. Properties and applications of microbial transglutaminase. Appl. Microbiol. Biotechnol. 2004, 64, 447–454. [Google Scholar] [CrossRef] [PubMed]
- Strop, P.; Liu, S.H.; Dorywalska, M.; Delaria, K.; Dushin, R.G.; Tran, T.T.; Ho, W.H.; Farias, S.; Casas, M.G.; Abdiche, Y.; et al. Location matters: Site of conjugation modulates stability and pharmacokinetics of antibody drug conjugates. Chem. Biol. 2013, 20, 161–167. [Google Scholar] [CrossRef] [PubMed]
- Strop, P.; Tran, T.T.; Dorywalska, M.; Delaria, K.; Dushin, R.; Wong, O.K.; Ho, W.H.; Zhou, D.; Wu, A.; Kraynov, E.; et al. RN927C, a site-specific trop-2 antibody-drug conjugate (ADC) with enhanced stability, is highly efficacious in preclinical solid tumor models. Mol. Cancer Ther. 2016, 15, 2698–2708. [Google Scholar] [CrossRef] [PubMed]
- Carrico, I.S.; Carlson, B.L.; Bertozzi, C.R. Introducing genetically encoded aldehydes into proteins. Nat. Chem. Biol. 2007, 3, 321–322. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, P.; van der Weijden, J.; Sletten, E.M.; Rabuka, D.; Bertozzi, C.R. A pictet-spengler ligation for protein chemical modification. Proc. Natl. Acad. Sci. USA 2013, 110, 46–51. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, P.; Kudirka, R.; Albers, A.E.; Barfield, R.M.; de Hart, G.W.; Drake, P.M.; Jones, L.C.; Rabuka, D. Hydrazino-pictet-spengler ligation as a biocompatible method for the generation of stable protein conjugates. Bioconj. Chem. 2013, 24, 846–851. [Google Scholar] [CrossRef] [PubMed]
- Starodub, A.N.; Ocean, A.J.; Shah, M.A.; Vahdat, L.T.; Chuang, E.; Guarino, M.J.; Picozzi, V.J.; Thomas, S.S.; Maliakal, P.P.; Govindan, S.V.; et al. Abstract CT206: SN-38 antibody-drug conjugate (ADC) targeting Trop-2, IMMU-132, as a novel platform for the therapy of diverse metastatic solid cancers: Initial clinical results. Cancer Res. 2014, 74, CT206. [Google Scholar] [CrossRef]
- Goldenberg, D.M.; Cardillo, T.M.; Govindan, S.V.; Rossi, E.A.; Sharkey, R.M. Trop-2 is a novel target for solid cancer therapy with sacituzumab govitecan (immu-132), an antibody-drug conjugate (ADC). Oncotarget 2015, 6, 22496–22512. [Google Scholar] [CrossRef] [PubMed]
- Bardia, A.; Vahdat, L.T.; Diamond, J.R.; Starodub, A.; Moroose, R.L.; Isakoff, S.J.; Ocean, A.J.; Berlin, J.; Messersmith, W.A.; Thomas, S.S.; et al. Therapy of refractory/relapsed metastatic triple-negative breast cancer (TNBC) with an anti-Trop-2-SN-38 antibody-drug conjugate (ADC), sacituzumab govitecan (IMMU-132): Phase I/II clinical experience. J. Clin. Oncol. 2015, 33, 1016. [Google Scholar] [CrossRef]
- Elnakat, H.; Ratnam, M. Distribution, functionality and gene regulation of folate receptor isoforms: Implications in targeted therapy. Adv. Drug Deliv. Rev. 2004, 56, 1067–1084. [Google Scholar] [CrossRef] [PubMed]
- Ab, O.; Whiteman, K.R.; Bartle, L.M.; Sun, X.; Singh, R.; Tavares, D.; LaBelle, A.; Payne, G.; Lutz, R.J.; Pinkas, J.; et al. Imgn853, a folate receptor-α (FRα)-targeting antibody-drug conjugate, exhibits potent targeted antitumor activity against fralpha-expressing tumors. Mol. Cancer Ther. 2015, 14, 1605–1613. [Google Scholar] [CrossRef] [PubMed]
- Erickson, H.K.; Park, P.U.; Widdison, W.C.; Kovtun, Y.V.; Garrett, L.M.; Hoffman, K.; Lutz, R.J.; Goldmacher, V.S.; Blattler, W.A. Antibody-maytansinoid conjugates are activated in targeted cancer cells by lysosomal degradation and linker-dependent intracellular processing. Cancer Res. 2006, 66, 4426–4433. [Google Scholar] [CrossRef] [PubMed]
- Ponte, J.F.; Ab, O.; Lanieri, L.; Lee, J.; Coccia, J.; Bartle, L.M.; Themeles, M.; Zhou, Y.; Pinkas, J.; Ruiz-Soto, R. Mirvetuximab soravtansine (IMGN853), a folate receptor alpha-targeting antibody-drug conjugate, potentiates the activity of standard of care therapeutics in ovarian cancer models. Neoplasia 2016, 18, 775–784. [Google Scholar] [CrossRef] [PubMed]
- Moore, K.N.; Borghaei, H.; O’Malley, D.M.; Jeong, W.; Seward, S.M.; Bauer, T.M.; Perez, R.P.; Matulonis, U.A.; Running, K.L.; Zhang, X.; et al. Phase 1 dose-escalation study of mirvetuximab soravtansine (IMGN853), a folate receptor alpha-targeting antibody-drug conjugate, in patients with solid tumors. Cancer 2017, 123, 3080–3087. [Google Scholar] [CrossRef] [PubMed]
- Moore, K.N.; Martin, L.P.; O’Malley, D.M.; Matulonis, U.A.; Konner, J.A.; Perez, R.P.; Bauer, T.M.; Ruiz-Soto, R.; Birrer, M.J. Safety and activity of mirvetuximab soravtansine (IMGN853), a folate receptor alpha-targeting antibody-drug conjugate, in platinum-resistant ovarian, fallopian tube, or primary peritoneal cancer: A phase i expansion study. J. Clin. Oncol. 2017, 35, 1112–1118. [Google Scholar] [CrossRef] [PubMed]
- Tedder, T.F.; Tuscano, J.; Sato, S.; Kehrl, J.H. Cd22, a B lymphocyte-specific adhesion molecule that regulates antigen receptor signaling. Annu. Rev. Immunol. 1997, 15, 481–504. [Google Scholar] [CrossRef] [PubMed]
- Piccaluga, P.P.; Arpinati, M.; Candoni, A.; Laterza, C.; Paolini, S.; Gazzola, A.; Sabattini, E.; Visani, G.; Pileri, S.A. Surface antigens analysis reveals significant expression of candidate targets for immunotherapy in adult acute lymphoid leukemia. Leuk. lymphoma 2011, 52, 325–327. [Google Scholar] [CrossRef] [PubMed]
- DiJoseph, J.F.; Dougher, M.M.; Evans, D.Y.; Zhou, B.-B.; Damle, N.K. Preclinical anti-tumor activity of antibody-targeted chemotherapy with CMC-544 (inotuzumab ozogamicin), a CD22-specific immunoconjugate of calicheamicin, compared with non-targeted combination chemotherapy with cvp or chop. Cancer Chemother. Pharmacol. 2011, 67, 741–749. [Google Scholar] [CrossRef] [PubMed]
- Advani, A.; Coiffier, B.; Czuczman, M.S.; Dreyling, M.; Foran, J.; Gine, E.; Gisselbrecht, C.; Ketterer, N.; Nasta, S.; Rohatiner, A.; et al. Safety, pharmacokinetics, and preliminary clinical activity of inotuzumab ozogamicin, a novel immunoconjugate for the treatment of B-cell non-hodgkin’s lymphoma: Results of a phase i study. J. Clin. Oncol. 2010, 28, 2085–2093. [Google Scholar] [CrossRef] [PubMed]
- Goy, A.; Forero, A.; Wagner-Johnston, N.; Christopher Ehmann, W.; Tsai, M.; Hatake, K.; Ananthakrishnan, R.; Volkert, A.; Vandendries, E.; Ogura, M. A phase 2 study of inotuzumab ozogamicin in patients with indolent b-cell non-hodgkin lymphoma refractory to rituximab alone, rituximab and chemotherapy, or radioimmunotherapy. Br. J. Haematol. 2016, 174, 571–581. [Google Scholar] [CrossRef] [PubMed]
- Teicher, B.A.; Chari, R.V. Antibody conjugate therapeutics: Challenges and potential. Clin. Cancer Res. 2011, 17, 6389–6397. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.K.; Luisi, D.L.; Pak, R.H. Antibody-drug conjugates: Design, formulation and physicochemical stability. Pharm. Res. 2015, 32, 3541–3571. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, P.J.; Oliveira, C.; Granja, P.L.; Sarmento, B. Antibodies and associates: Partners in targeted drug delivery. Pharmacol. Ther. 2017, 177, 129–145. [Google Scholar] [CrossRef] [PubMed]
- Zolot, R.S.; Basu, S.; Million, R.P. Antibody-drug conjugates. Nat. Rev. Drug Discov. 2013, 12, 259–260. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Conjugation | Reactive Groups | Advantages |
|---|---|---|
| Cysteine Residues | Maleimides, haloacetyls, other Michael acceptors | Simple and reproducible method Used in FDA approved Adcetris, widely employed in pipeline candidates, DAR ~0–8 Comparatively less heterogeneous by products than lysine conjugation Easier to characterize pharmacokinetically |
| Lysine Residues | Activated ester functional groups like N-hydroxysuccinimide esters | Though highly heterogeneous, this method is employed in FDA approved Kadcyla®, Mylotarg™, DAR ~3.5 (Kadcyla®), ~2.5 (Mylotarg™) Mostly used to crosslink via non-reducible linkers. |
| Method of Conjugation | Reactive Groups | Advantages | Developer |
|---|---|---|---|
| Engineered side chain cysteine residues (ThioMAb) [93] | Maleimides | Improved clinical safety, tolerability and therapeutic index over conventional conjugates. Controlled and reproducible DAR 2. Compatible for producing in large scale. | Genentech |
| Incorporation of un-natural amino acids (unAA) [94] | Alkoxy-amine | Highly stable and extended half-life in systemic circulation. Improved pharmacological profile compared to conventional ADCs. Ketone group present in unAA provided conjugation site for different alternative payloads like kinase inhibitors, proteasome inhibitors. | Ambrx |
| Enzymatic Site-Specific Conjugation Process [95] | Amine, Indole | DAR 2-4, More stable conjugates than yielded by ThioMAb and oxime ligation. Controlled conjugation site of the payload on the mAb. Better pharmacokinetic profile over conventional conjugates. | Innate Pharma, Glycos, Pfizer. Inc. |
| Name of the Review Article | Focus of the Review | Year of Publication |
|---|---|---|
| Antibody-Drug Conjugates for Cancer Therapy [7] | This article is focused on different key issues like choosing an appropriate target, expression of the target, selecting right mAb isotype. | 2008 |
| Antibody Conjugate Therapeutics: Challenges and Potential [122] | The key consideration behind choosing an appropriate target for ADC developments. | 2011 |
| Pharmacokinetic Considerations for Antibody Drug Conjugates [10] | Different pharmacokinetic considerations to characterize ADCs as well as PK-PD modellings for development of ADCs | 2012 |
| Site-Specific Antibody−Drug Conjugates: The Nexus of Biorthogonal Chemistry, Protein Engineering, and Drug Development [75] | Focuses on methods to synthesize site-specific homogenous ADCs with details of bio-orthogonal chemistries. | 2014 |
| Antibody-Drug Conjugates: Design, Formulation and Physicochemical Stability [123] | Physiochemical characterization, formulation considerations, and factors involved in process control. | 2015 |
| Methods to Design and Synthesize Antibody-Drug Conjugates (ADCs) [98] | Accounts for different conjugation methods and the chemistry behind in the field of ADCs. | 2016 |
| Mechanisms of Resistance to Antibody–Drug Conjugates [35] | Resistance of various ADCs and possible mechanism. | 2016 |
| Antibodies and associates: Partners in targeted drug delivery [124] | Engineering antibodies and their subsequent use in different targeted drug delivery systems. | 2017 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dan, N.; Setua, S.; Kashyap, V.K.; Khan, S.; Jaggi, M.; Yallapu, M.M.; Chauhan, S.C. Antibody-Drug Conjugates for Cancer Therapy: Chemistry to Clinical Implications. Pharmaceuticals 2018, 11, 32. https://doi.org/10.3390/ph11020032
Dan N, Setua S, Kashyap VK, Khan S, Jaggi M, Yallapu MM, Chauhan SC. Antibody-Drug Conjugates for Cancer Therapy: Chemistry to Clinical Implications. Pharmaceuticals. 2018; 11(2):32. https://doi.org/10.3390/ph11020032
Chicago/Turabian StyleDan, Nirnoy, Saini Setua, Vivek K. Kashyap, Sheema Khan, Meena Jaggi, Murali M. Yallapu, and Subhash C. Chauhan. 2018. "Antibody-Drug Conjugates for Cancer Therapy: Chemistry to Clinical Implications" Pharmaceuticals 11, no. 2: 32. https://doi.org/10.3390/ph11020032
APA StyleDan, N., Setua, S., Kashyap, V. K., Khan, S., Jaggi, M., Yallapu, M. M., & Chauhan, S. C. (2018). Antibody-Drug Conjugates for Cancer Therapy: Chemistry to Clinical Implications. Pharmaceuticals, 11(2), 32. https://doi.org/10.3390/ph11020032