Roles of Heat Shock Proteins in Apoptosis, Oxidative Stress, Human Inflammatory Diseases, and Cancer

 and
and
Abstract
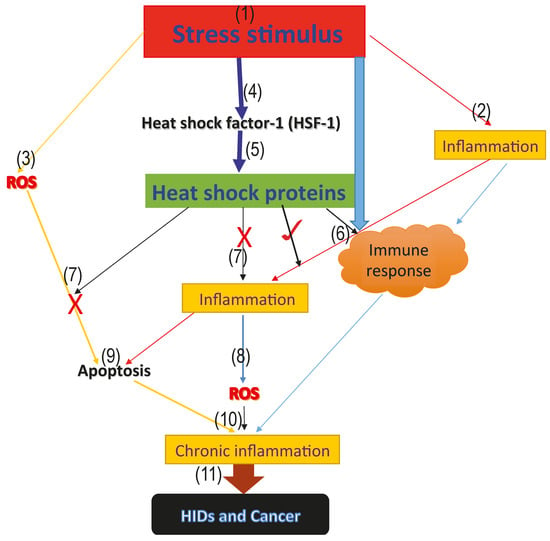
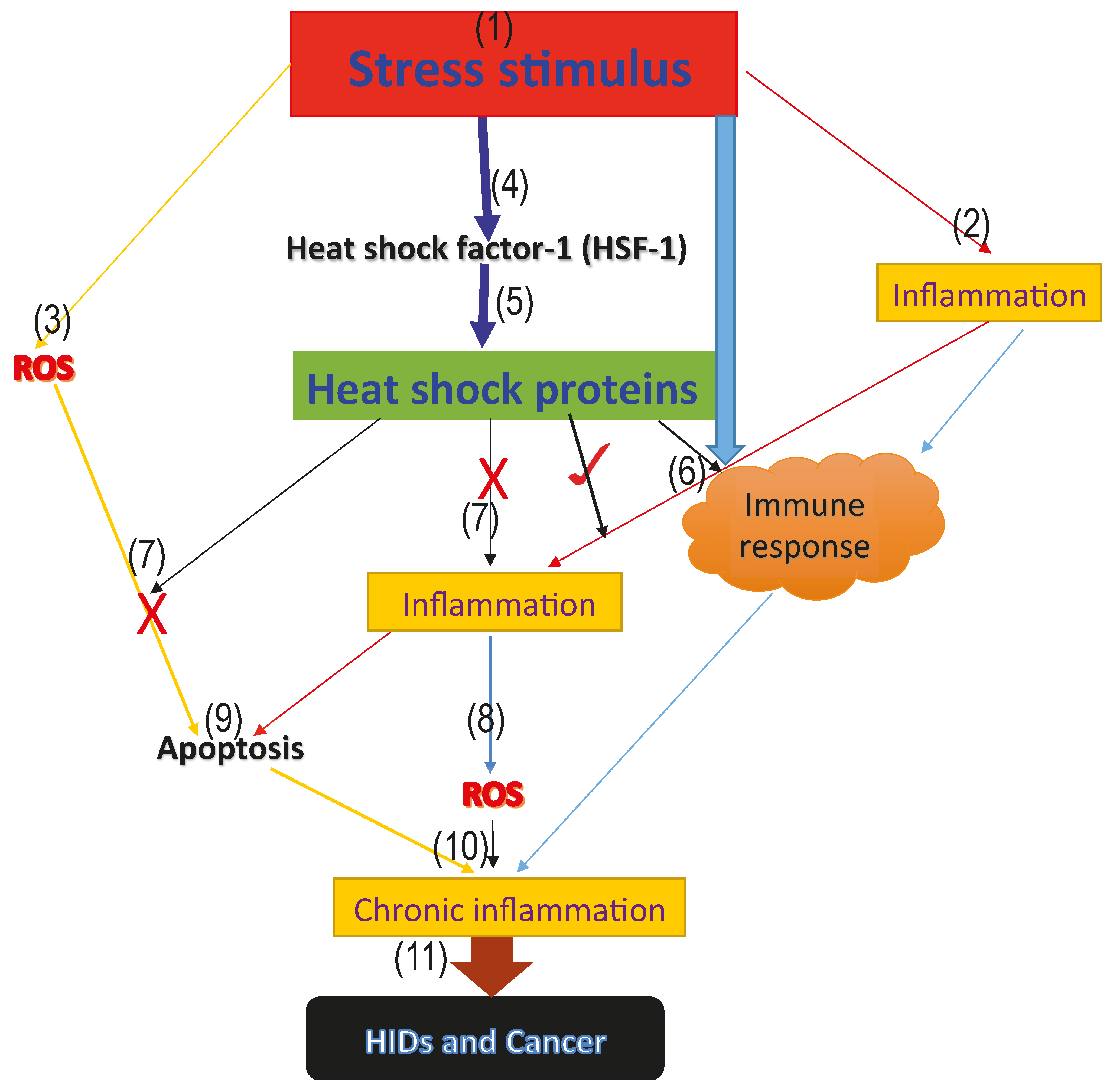
1. Introduction
2. Role of HSPs in Apoptosis
3. HSPs and Oxidative Stress
4. HSPs in Human Inflammatory Diseases (HIDs) and Cancer
4.1. Acute Respiratory Distress Syndrome
4.2. Rheumatoid Arthritis
4.3. Asthma
4.4. Cancer
5. Association between Heat Shock Proteins, Oxidative Stress, Apoptosis, Human Inflammatory Diseases (HIDs), and Cancer
6. Future Direction in the Use of HSPs as Therapeutic Candidates
7. Conclusions
Conflicts of Interest
References
- Srivastava, P. Roles of heat-shock proteins in innate and adaptive immunity. Nat. Rev. Immunol. 2002, 2, 185. [Google Scholar] [CrossRef] [PubMed]
- Asea, A. Chaperokine-induced signal transduction pathways. Exerc. Immunol. Rev. 2003, 9, 25–33. [Google Scholar] [PubMed]
- Searle, S.; McCrossan, M.V.; Smith, D.F. Expression of a mitochondrial stress protein in the protozoan parasite Leishmania major. J. Cell Sci. 1993, 104, 1091–1100. [Google Scholar] [PubMed]
- Zügel, U.; Kaufmann, E. Role of Heat Shock Proteins in Protection from and Pathogenesis of Infectious Diseases. Clin. Microbiol. Rev. 1999, 12, 19–39. [Google Scholar] [PubMed]
- Jee, H. Size dependent classification of heat shock proteins: A mini-review. J. Exerc. Rehabil. 2016, 12, 255–259. [Google Scholar] [CrossRef] [PubMed]
- Sharma, G.; Nath, A.; Prasad, S.; Singh, N.; Dubey, P.; Saikumar, G. Expression and Characterization of Constitutive Heat Shock Proteins 70.1 (HSPA-1A) Gene in In Vitro produced and In Vivo-Derived Buffalo (Bubalis) Embryos. Reprod. Domest. Anim. 2012, 47, 975–983. [Google Scholar] [CrossRef] [PubMed]
- De Maio, A. Heat shock proteins: Facts, thoughts, and dreams. Shock 1999, 11, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Shiber, A.; Ravid, T. Chaperoning proteins for destruction: Diverse roles of HSP70 chaperones and their co-chaperones in targeting misfolded proteins to the proteasome. Biomolecules 2014, 4, 704–724. [Google Scholar] [CrossRef] [PubMed]
- Daugaard, M.; Kirkegaard-Sørensen, T.; Ostenfeld, M.S.; Aaboe, M.; Høyer-Hansen, M.; Ørntoft, T.F.; Rohde, M.; Jäättelä, M. Lens epithelium-derived growth factor is an HSP70-2 regulated guardian of lysosomal stability in human cancer. Cancer Res. 2007, 67, 2559–2567. [Google Scholar] [CrossRef] [PubMed]
- Singh, O.V.; Pollard, H.B.; Zeitlin, P.L. Chemical rescue of ΔF508-CFTR mimics genetic repair in cystic fibrosis bronchial epithelial cells. Mol. Cell. Proteom. 2008, 7, 1099–1110. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Liu, R.; Huang, W.A. 14-mer peptide from HSP70 protein is the critical epitope which enhances NK activity against tumor cells in vivo. Immunol. Investig. 2007, 36, 233–246. [Google Scholar] [CrossRef] [PubMed]
- De Maio, A. Extracellular heat shock proteins, cellular export vesicles, and the Stress Observation System: A form of communication during injury, infection, and cell damage. Cell Stress Chaperones 2011, 16, 235–249. [Google Scholar] [CrossRef] [PubMed]
- De Maio, A.; Santoro, M.G.; Tanguay, R.M.; Hightower, L.E. Ferruccio Ritossa’s scientific legacy 50 years after his discovery of the heat shock response: A new view of biology, a new society, and a new journal. Cell Stress Chaperones 2012, 17, 139–143. [Google Scholar] [CrossRef] [PubMed]
- Garbuz, D.G.; Astakhova, L.N.; Zatsepina, O.G.; Arkhipova, I.R.; Nudler, E.; Evgen’ev, M.B. Functional organization of hsp70 cluster in camel (Camelus dromedarius) and other mammals. PLoS ONE 2011, 6, 27205. [Google Scholar] [CrossRef] [PubMed]
- Lanneau, D.; Wettstein, G.; Bonniaud, P.; Garrido, C. Heat Shock Proteins: Cell Protection through Protein Triage. Sci. World J. 2010, 10, 1543–1552. [Google Scholar] [CrossRef] [PubMed]
- Sung, Y.Y.; MacRae, T.H. Heat shock proteins and disease control in aquatic organisms. J. Aquac. Res. Dev. S 2011, 2. [Google Scholar] [CrossRef]
- Wan, T.; Zhou, X.; Chen, G.; An, H.; Chen, T.; Zhang, W.; Liu, S.; Jiang, Y.; Yang, F.; Wu, Y.; et al. Novel heat shock protein HSP70L1 activates dendritic cells and acts as a Th1 polarizing adjuvant. Blood 2004, 103, 1747–1754. [Google Scholar] [CrossRef] [PubMed]
- Juo, L.Y.; Liao, W.C.; Shih, Y.L.; Yang, B.Y.; Liu, A.B.; Yan, Y.T. HSPB7 interacts with dimerized FLNC and its absence results in progressive myopathy in skeletal muscles. J. Cell Sci. 2016, 129, 1661–1670. [Google Scholar] [CrossRef] [PubMed]
- Dubé, V.; Grigull, J.; DeSouza, L.V.; Ghanny, S.; Colgan, T.J.; Romaschin, A.D.; Siu, K.M. Verification of endometrial tissue biomarkers previously discovered using mass spectrometry-based proteomics by means of immunohistochemistry in a tissue microarray format. J. Proteom. Res. 2007, 6, 2648–2655. [Google Scholar] [CrossRef] [PubMed]
- Meyer, A.S.; Gillespie, J.R.; Walther, D.; Millet, I.S.; Doniach, S.; Frydman, J. Closing the folding chamber of the eukaryotic chaperonin requires the transition state of ATP hydrolysis. Cell 2003, 113, 369–381. [Google Scholar] [CrossRef]
- Vidyasagar, A.; Wilson, N.A.; Djamali, A. Heat shock protein 27 (HSP27): Biomarker of disease and therapeutic target. Fibrogenesis Tissue Repair 2012, 5, 7. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Qian, X.; Sha, B. Heat shock protein 40: Structural studies and their functional implications. Protein Pept. Lett. 2009, 16, 606–612. [Google Scholar] [CrossRef] [PubMed]
- Belles, C.; Kuhl, A.; Nosheny, R.; Carding, S.R. Plasma membrane expression of heat shock protein 60 in vivo in response to infection. Infect. Immun. 1999, 67, 4191–4200. [Google Scholar] [PubMed]
- Tuttle, J.A.; Castle, P.C.; Metcalfe, A.J.; Midgley, A.W.; Taylor, L.; Lewis, M.P. Downhill running and exercise in hot environments increase leukocyte Hsp72 (HSPA1A) and Hsp90α (HSPC1) gene transcripts. J. Appl. Physiol. 2015, 118, 996–1005. [Google Scholar] [CrossRef] [PubMed]
- Krobitsch, S.; Brandau, S.; Hoyer, C.; Schmetz, C.; Hübel, A.; Clos, J. Leishmania donovani heat shock protein 100 characterization and function in amastigote stage differentiation. J. Biol. Chem. 1998, 273, 6488–6494. [Google Scholar] [CrossRef] [PubMed]
- Zuo, D.; Subjeck, J.; Wang, X.Y. Unfolding the role of large heat shock proteins: New insights and therapeutic implications. Front. Immunol. 2016, 7, 75. [Google Scholar] [CrossRef] [PubMed]
- Van Eden, W.; Van der Zee, R.; Prakken, B. Heat-shock proteins induce T-cell regulation of chronic inflammation. Nat. Rev. Immunol. 2005, 5, 318. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Wan, T.; Zhou, X.; Wang, B.; Yang, F.; Li, N.; Chen, G.; Dai, S.; Liu, S.; Zhang, M.; et al. Hsp70-like Protein 1 fusion protein enhances induction of carcinoembryonic antigen–specific CD8+ CTL response by dendritic cell vaccine. Cancer Res. 2005, 65, 4947–4954. [Google Scholar] [CrossRef] [PubMed]
- Colaco, C.A.; Bailey, C.R.; Walker, K.B.; Keeble, J. Heat shock proteins: Stimulators of innate and acquired immunity. BioMed. Res. Int. 2013, 2013. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, R.; Rasheed, Z.; Ahsan, H. Biochemical and cellular toxicology of peroxynitrite: Implications in cell death and autoimmune phenomenon. Immunopharmacol. Immunotoxicol. 2009, 31, 388–396. [Google Scholar] [CrossRef] [PubMed]
- Wong, R.S. Apoptosis in cancer: From pathogenesis to treatment. J. Exp. Clin. Cancer Res. 2011, 30, 87. [Google Scholar] [CrossRef] [PubMed]
- Elmore, S. Apoptosis: A review of programmed cell death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef] [PubMed]
- Lowe, S.W.; Lin, A.W. Apoptosis in cancer. Carcinogenesis 2000, 21, 485–495. [Google Scholar] [CrossRef] [PubMed]
- Beere, H.M. The stress of dying’: The role of heat shock proteins in the regulation of apoptosis. J. Cell Sci. 2004, 117, 2641–2651. [Google Scholar] [CrossRef] [PubMed]
- Wolf, B.B.; Green, D.R. Suicidal tendencies: Apoptotic cell death by caspase family proteinases. J. Biol. Chem. 1999, 274, 20049–20052. [Google Scholar] [CrossRef] [PubMed]
- McIlwain, D.R.; Berger, T.; Mak, T.W. Caspase functions in cell death and disease. Cold Spring Harb. Perspect. Biol. 2013, 5, a008656. [Google Scholar] [CrossRef] [PubMed]
- Novara, G.; Galfano, A.; Berto, R.B.; Ficarra, V.; Navarrete, R.V.; Artibani, W. Inflammation, apoptosis, and BPH: What is the evidence? Eur. Urol. Suppl. 2006, 5, 401–409. [Google Scholar] [CrossRef]
- Takayama, S.; Reed, J.C.; Homma, S. Heat-shock proteins as regulators of apoptosis. Oncogene 2003, 22, 9041–9047. [Google Scholar] [CrossRef] [PubMed]
- Jolly, C.; Morimoto, R.I. Role of the heat shock response and molecular chaperones in oncogenesis and cell death. J. Natl. Cancer Inst. 2000, 92, 1564–1572. [Google Scholar] [CrossRef] [PubMed]
- Leung, A.M.; Redlak, M.J.; Miller, T.A. Role of heat shock proteins in oxygen radical–induced gastric apoptosis. J. Surg. Res. 2015, 193, 135–144. [Google Scholar] [CrossRef] [PubMed]
- Bukau, B.; Weissman, J.; Horwich, A. Molecular chaperones and protein quality control. Cell 2006, 125, 443–451. [Google Scholar] [CrossRef] [PubMed]
- Garrido, C.; Bruey, J.M.; Fromentin, A.; Hammann, A.; Arrigo, A.P.; Solary, E. HSP27 inhibits cytochrome c-dependent activation of procaspase-9. FASEB J. 1999, 13, 2061–2070. [Google Scholar] [PubMed]
- Rane, M.J.; Pan, Y.; Singh, S.; Powell, D.W.; Wu, R.; Cummins, T.; Chen, Q.; McLeish, K.R.; Klein, J.B. Heat shock protein 27 controls apoptosis by regulating Akt activation. J. Biol. Chem. 2003, 278, 27828–27835. [Google Scholar] [CrossRef] [PubMed]
- Ravagnan, L.; Gurbuxani, S.; Susin, S.A.; Maisse, C.; Daugas, E.; Zamzami, N.; Mak, T.; Jäättelä, M.; Penninger, J.M.; Garrido, C.; et al. Heat-shock protein 70 antagonizes apoptosis-inducing factor. Nat. Cell Biol. 2001, 3, 839–843. [Google Scholar] [CrossRef] [PubMed]
- Goto, H.; Yano, S.; Matsumori, Y.; Ogawa, H.; Blakey, D.C.; Sone, S. Sensitization of tumor-associated endothelial cell apoptosis by the novel vascular-targeting agent ZD6126 in combination with cisplatin. Clin. Cancer Res. 2004, 10, 7671–7676. [Google Scholar] [CrossRef] [PubMed]
- Samali, A.; Cai, J.; Zhivotovsky, B.; Jones, D.P.; Orrenius, S. Presence of a pre-apoptotic complex of pro-caspase-3, HSP60 and Hsp10 in the mitochondrial fraction of Jurkat cells. EMBO J. 1999, 18, 2040–2048. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Knowlton, A.A. Cytosolic heat shock protein 60, hypoxia, and apoptosis. Circulation 2002, 106, 2727–2733. [Google Scholar] [CrossRef] [PubMed]
- Lobo, V.; Patil, A.; Phatak, A.; Chandra, N. Free radicals, antioxidants and functional foods: Impact on human health. Pharmacogn. Rev. 2010, 4, 118. [Google Scholar] [CrossRef] [PubMed]
- Ji, L. Oxidative stress during exercise: Implication of antioxidant nutrients. Free Radic. Biol. Med. 1995, 18, 1079–1086. [Google Scholar] [CrossRef]
- Fubini, B.; Hubbard, A. Reactive oxygen species (ROS) and reactive nitrogen species (RNS) generation by silica in inflammation and fibrosis. Free Radic. Biol. Med. 2003, 34, 1507–1516. [Google Scholar] [CrossRef]
- Uttara, B.; Singh, A.V.; Zamboni, P.; Mahajan, R.T. Oxidative stress and neurodegenerative diseases: A review of upstream and downstream antioxidant therapeutic options. Curr. Neuropharmacol. 2009, 7, 65–74. [Google Scholar] [CrossRef] [PubMed]
- Alfadda, A.A.; Sallam, R.M. Reactive oxygen species in health and disease. BioMed. Res. Int. 2012. [Google Scholar] [CrossRef] [PubMed]
- Oyinloye, B.E.; Adenowo, A.F.; Kappo, A.P. Reactive oxygen species, apoptosis, antimicrobial peptides and human inflammatory diseases. Pharmaceuticals 2015, 8, 151–175. [Google Scholar] [CrossRef] [PubMed]
- Lushchak, V.I. Free radicals, reactive oxygen species, oxidative stress and its classification. Chem.-Biol. Int. 2014, 224, 164–175. [Google Scholar] [CrossRef] [PubMed]
- Finkel, T. Signal transduction by reactive oxygen species. J. Cell. Biol. 2011, 194, 7–15. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, C.; Sanz-Alfayate, G.; Agapito, M.T.; Gomez-Nino, A.; Rocher, A.; Obeso, A. Significance of ROS in oxygen sensing in cell systems with sensitivity to physiological hypoxia. Respir. Physiol. Neurobiol. 2002, 132, 17–41. [Google Scholar] [CrossRef]
- Scandalios, J.G. Oxidative stress responses-what have genome-scale studies taught us? Genome Biol. 2002, 3, 1019–1021. [Google Scholar] [CrossRef]
- Mittler, R.; Vanderauwera, S.; Suzuki, N.; Miller, G.; Tognetti, V.B.; Vandepoele, K.; Gollery, M.; Shulaev, V.; Van Breusegem, F. ROS signaling: The new wave? Trends Plant Sci. 2011, 16, 300–309. [Google Scholar] [CrossRef] [PubMed]
- Coyle, J.T.; Puttfarcken, P. Oxidative stress, glutamate, and neurodegenerative disorders. Science 1993, 262, 689–695. [Google Scholar] [CrossRef] [PubMed]
- Schneeberger, K.; Czirják, G.Á.; Voigt, C.C. Inflammatory challenge increases measures of oxidative stress in a free-ranging, long-lived mammal. J. Exp. Biol. 2013, 216, 4514–4519. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Koo, N.; Min, D.B. Reactive oxygen species, aging, and antioxidative nutraceuticals. Compr. Rev. Food Sci. Food Saf. 2004, 3, 21–33. [Google Scholar] [CrossRef]
- Hsieh, H.L.; Yang, C.M. Role of redox signaling in neuroinflammation and neurodegenerative diseases. BioMed. Res. Int. 2013, 2013. [Google Scholar] [CrossRef] [PubMed]
- Mayer, M.P.; Bukau, B. Hsp70 chaperones: Cellular functions and molecular mechanism. Cell. Mol. Life Sci. 2005, 62, 670. [Google Scholar] [CrossRef] [PubMed]
- Jacquier-Sarlin, M.R.; Fuller, K.; Dinh-Xuan, A.T.; Richard, M.J.; Polla, B.S. Protective effects of HSP70 in inflammation. Cell. Mol. Life Sci. 1994, 50, 1031–1038. [Google Scholar] [CrossRef]
- Trott, A.; West, J.D.; Klaić, L.; Westerheide, S.D.; Silverman, R.B.; Morimoto, R.I.; Morano, K.A. Activation of heat shock and antioxidant responses by the natural product celastrol: Transcriptional signatures of a thiol-targeted molecule. Mol. Biol. Cell. 2008, 19, 1104–1112. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.-W.; Biggar, K.K.; Zhang, J.; Tessier, S.N.; Pifferi, F.; Perret, M.; Storey, K.B. Induction of antioxidant and heat shock protein responses during torpor in the gray mouse lemur, Microcebus murinus. Genom. Proteom. Bioinform. 2015, 13, 119–126. [Google Scholar] [CrossRef] [PubMed]
- Ashley, N.T.; Weil, Z.M.; Nelson, R.J. Inflammation: Mechanisms, costs, and natural variation. Ann. Rev. Ecol. Evol. Syst. 2012, 43, 385–406. [Google Scholar] [CrossRef]
- Ferrero-Miliani, L.; Nielsen, O.H.; Andersen, P.S.; Girardin, S.E. Chronic inflammation: Importance of NOD2 and NALP3 in interleukin-1β generation. Clin. Exp. Immunol. 2007, 147, 227–235. [Google Scholar] [CrossRef] [PubMed]
- Villar, J.; Blanco, J.; Añón, J.M.; Santos-Bouza, A.; Blanch, L.; Ambrós, A.; Gandía, F.; Carriedo, D.; Mosteiro, F.; Basaldúa, S.; et al. The ALIEN study: Incidence and outcome of acute respiratory distress syndrome in the era of lung protective ventilation. Intensive Care Med. 2011, 37, 1932–1941. [Google Scholar] [CrossRef] [PubMed]
- Ware, L.B.; Matthay, M.A. The acute respiratory distress syndrome. N. Engl. J. Med. 2000, 342, 1334–1349. [Google Scholar] [CrossRef] [PubMed]
- Rubenfeld, G.D.; Caldwell, E.; Peabody, E.; Weaver, J.; Martin, D.P.; Neff, M.; Stern, E.J.; Hudson, L.D. Incidence and outcomes of acute lung injury. N. Engl. J. Med. 2005, 353, 1685–1693. [Google Scholar] [CrossRef] [PubMed]
- Slutsky, A.S. Hot new therapy for sepsis and the acute respiratory distress syndrome. J. Clin. Investig. 2002, 110, 737. [Google Scholar] [CrossRef] [PubMed]
- González-Reimers, E.; Santolaria-Fernández, F.; Martín-González, M.C.; Fernández-Rodríguez, C.M.; Quintero-Platt, G. Alcoholism: A systemic proinflammatory condition. World J. Gastroenterol. 2014, 20, 14660. [Google Scholar] [CrossRef] [PubMed]
- Hirsh, M.I.; Junger, W.G. Roles of heat shock proteins and γδT cells in inflammation. Am. J. Respir. Cell Mol. Biol. 2008, 39, 509–513. [Google Scholar] [CrossRef] [PubMed]
- Weiss, Y.G.; Bromberg, Z.; Raj, N.; Raphael, J.; Goloubinoff, P.; Ben-Neriah, Y.; Deutschman, C.S. Enhanced heat shock protein 70 expression alters proteasomal degradation of IκB kinase in experimental acute respiratory distress syndrome. Crit. Care Med. 2007, 35, 2128–2138. [Google Scholar] [CrossRef] [PubMed]
- Chu, E.K.; Ribeiro, S.P.; Slutsky, A.S. Heat stress increases survival rates in lipopolysaccharide-stimulated rats. Crit. Care Med. 1997, 25, 1727–1732. [Google Scholar] [CrossRef] [PubMed]
- Bromberg, Z.; Raj, N.; Goloubinoff, P.; Deutschman, C.S.; Weiss, Y.G. Enhanced expression of 70-kilodalton heat shock protein limits cell division in a sepsis-induced model of acute respiratory distress syndrome. Crit. Care Med. 2008, 36, 246. [Google Scholar] [CrossRef] [PubMed]
- Smolen, J.S.; Breedveld, F.C.; Burmester, G.R.; Bykerk, V.; Dougados, M.; Emery, P.; Kvien, T.K.; Navarro-Compán, M.V.; Oliver, S.; Schoels, M.; et al. Treating rheumatoid arthritis to target: 2014 update of the recommendations of an international task force. Ann. Rheum. Dis. 2016, 75, 3–15. [Google Scholar] [CrossRef] [PubMed]
- Aletaha, D.; Neogi, T.; Silman, A.J.; Funovits, J.; Felson, D.T.; Bingham, C.O.; Birnbaum, N.S.; Burmester, G.R.; Bykerk, V.P.; Cohen, M.D.; et al. 2010 rheumatoid arthritis classification criteria: An American College of Rheumatology/European League Against Rheumatism collaborative initiative. Arthritis Rheum. 2010, 62, 2569–2581. [Google Scholar] [CrossRef] [PubMed]
- Klareskog, L.; Malmström, V.; Lundberg, K.; Padyukov, L.; Alfredsson, L. Smoking, citrullination and genetic variability in the immunopathogenesis of rheumatoid arthritis. Semin. Immunol. 2011, 23, 92–98. [Google Scholar] [CrossRef] [PubMed]
- Millar, K.; Lloyd, S.M.; McLean, J.S.; Batty, G.D.; Burns, H.; Cavanagh, J.; Deans, K.A.; Ford, I.; McConnachie, A.; McGinty, A.; et al. Personality, socio-economic status and inflammation: Cross-sectional, population-based study. PLoS ONE 2013, 8, 58256. [Google Scholar] [CrossRef] [PubMed]
- Callahan, L.F.; Pincus, T. Education, self-care, and outcomes of rheumatic diseases: Further challenges to the “biomedical model” paradigm. Arthritis Rheum. 1997, 10, 283–288. [Google Scholar] [CrossRef]
- Schett, G.; Redlich, K.; Xu, Q.; Bizan, P.; Gröger, M.; Tohidast-Akrad, M.; Kiener, H.; Smolen, J.; Steiner, G. Enhanced expression of heat shock protein 70 (HSP70) and heat shock factor 1 (HSF1) activation in rheumatoid arthritis synovial tissue. Differential regulation of HSP70 expression and hsf1 activation in synovial fibroblasts by proinflammatory cytokines, shear stress, and antiinflammatory drugs. J. Clin. Investig. 1998, 102, 302–311. [Google Scholar] [PubMed]
- Kang, E.H.; Kim, D.J.; Lee, E.Y.; Lee, Y.J.; Lee, E.B.; Song, Y.W. Downregulation of heat shock protein 70 protects rheumatoid arthritis fibroblast-like synoviocytes from nitric oxide-induced apoptosis. Arthritis Res. Ther. 2009, 11, 130. [Google Scholar] [CrossRef] [PubMed]
- Van Roon, J.A.; van Eden, W.; van Roy, J.L.; Lafeber, F.J.; Bijlsma, J.W. Stimulation of suppressive T cell responses by human but not bacterial 60-kD heat-shock protein in synovial fluid of patients with rheumatoid arthritis. J. Clin. Investig. 1997, 100, 459–463. [Google Scholar] [CrossRef] [PubMed]
- Pockley, A.G. Heat shock proteins as regulators of the immune response. Lancet 2003, 362, 469–476. [Google Scholar] [CrossRef]
- Anderton, S.M.; Van Der Zee, R.; Prakken, B.; Noordzij, A.; Van Eden, W. Activation of T cells recognizing self 60-kD heat shock protein can protect against experimental arthritis. J. Exp. Med. 1995, 181, 943–952. [Google Scholar] [CrossRef] [PubMed]
- Kaul, G.; Thippeswamy, H. Role of heat shock proteins in diseases and their therapeutic potential. Indian J. Microbiol. 2011, 51, 124–131. [Google Scholar] [CrossRef] [PubMed]
- Salinthone, S.; Ba, M.; Hanson, L.; Martin, J.L.; Halayko, A.J.; Gerthoffer, W.T. Overexpression of human Hsp27 inhibits serum-induced proliferation in airway smooth muscle myocytes and confers resistance to hydrogen peroxide cytotoxicity. Am. J. Physiol. Lung Cell. Mol. Physiol. 2007, 293, 1194–1207. [Google Scholar] [CrossRef] [PubMed]
- Lazaar, A.L.; Panettieri, R.A. Airway smooth muscle as a regulator of immune responses and bronchomotor tone. Clin. Chest Med. 2006, 27, 53–69. [Google Scholar] [CrossRef] [PubMed]
- Chiappara, G.; Gagliardo, R.; Siena, A.; Bonsignore, M.R.; Bousquet, J.; Bonsignore, G.; Vignola, A.M. Airway remodelling in the pathogenesis of asthma. Curr. Opin. Allergy Clin. Immunol. 2001, 1, 85–93. [Google Scholar] [CrossRef] [PubMed]
- Feigin, V. Global, regional, and national life expectancy, all-cause mortality, and cause-specific mortality for 249 causes of death, 1980–2015: A systematic analysis for the Global Burden of Disease Study 2015. Lancet 2016, 388, 1459–1544. [Google Scholar]
- Vos, T.; Allen, C.; Arora, M.; Barber, R.M.; Bhutta, Z.A.; Brown, A.; Carter, A.; Casey, D.C.; Charlson, F.J.; Chen, A.Z.; et al. Global, regional, and national incidence, prevalence, and years lived with disability for 310 diseases and injuries, 1990–2015: A systematic analysis for the Global Burden of Disease Study 2015. Lancet 2016, 388, 1545–1602. [Google Scholar] [CrossRef]
- Wolff, P.T.; Arison, L.; Rahajamiakatra, A.; Raserijaona, F.; Niggemann, B. High asthma prevalence and associated factors in urban malagasy schoolchildren. J. Asthma 2012, 49, 575–580. [Google Scholar] [CrossRef] [PubMed]
- Adeloye, D.; Chan, K.Y.; Rudan, I.; Campbell, H. An estimate of asthma prevalence in Africa: A systematic analysis. Croatian Med. J. 2013, 54, 519–531. [Google Scholar] [CrossRef]
- Bertorelli, G.; Bocchino, V.; Zhuo, X.; Chetta, A.; Del Donno, M.; Foresi, A.; Testi, R.; Olivieri, D. Heat shock protein 70 upregulation is related to HLA-DR expression in bronchial asthma. Effects of inhaled glucocorticoids. Clin. Exp. Allergy 1998, 28, 551–560. [Google Scholar] [CrossRef] [PubMed]
- Changchun, H.; Haijin, Z.; Wenjun, L.; Zhenyu, L.; Dan, Z.; Laiyu, L.; Wancheng, T.; Shao-Xi, C.; Fei, Z. Increased heat shock protein 70 levels in induced sputum and plasma correlate with severity of asthma patients. Cell Stress Chaperones 2011, 16, 663–671. [Google Scholar] [CrossRef] [PubMed]
- Vignola, A.M.; Chanez, P.; Polla, B.S.; Vic, P.; Godard, P.; Bousquet, J. Increased expression of heat shock protein 70 on airway cells in asthma and chronic bronchitis. Am. J. Respir. Cell. Mol. Biol. 1995, 13, 683–691. [Google Scholar] [CrossRef] [PubMed]
- Polla, B.S.; Bachelet, M.; Dall’Ava, J.; Vignola, A.M. Heat shock proteins in inflammation and asthma: Dr Jekyll or Mr Hyde? Clin. Exp. Allergy 1998, 28, 527–529. [Google Scholar] [CrossRef] [PubMed]
- Aron, Y.; Busson, M.; Polla, B.S.; Dusser, D.; Lockhart, A.; Swierczewski, E.; Favatier, F. Analysis of HSP70 gene polymorphism in allergic asthma. Allergy 1999, 54, 165–170. [Google Scholar] [CrossRef] [PubMed]
- Wong, H.R.; Wispe, J.R. The stress response and the lung. Am. J. Physiol. Lung Cell. Mol. Physiol. 1997, 273, L1–L9. [Google Scholar]
- Shingai, R.; Maeda, T.; Onishi, S.; Yamamoto, Y. Autonatibody against 70 kD heat shock protein in patients with autoimmune liver diseases. J. Hepatol. 1995, 23, 382–390. [Google Scholar] [CrossRef]
- Xu, Q.; Kiechl, S.; Mayr, M.; Metzler, B.; Egger, G.; Oberhollenzer, F.; Willeit, J.; Wick, G. Association of serum antibodies to heat-shock protein 65 with carotid atherosclerosis. Circulation 1999, 100, 1169–1174. [Google Scholar] [CrossRef] [PubMed]
- Salman, A.N. Serum Levels Evaluation of Heat Shock Protein70 during Gestation and Fetal Birth Weight in Asthmatic Women of Thi-QAR Province, Iraq. J. Al-Nahrain Univ. 2015, 18, 117–122. [Google Scholar] [CrossRef]
- Schatz, M. Is maternal asthma a life or death issue for the baby? Thorax 2009, 64, 93–95. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Wu, T.; Cheng, L.; Wang, F.; Wei, Q.; Tanguay, R.M. Plasma antibodies against heat shock protein 70 correlate with the incidence and severity of asthma in a Chinese population. Respir. Res. 2005, 6. [Google Scholar] [CrossRef] [PubMed]
- PeRIšIć, T.; Srećković, M.; Matić, G. Changes of antioxidant enzyme activity and heat shock protein content in lymphocytes of children with asthma. Arch. Biol. Sci. 2007, 59, 257–266. [Google Scholar] [CrossRef]
- Tong, W.; Luo, W. Heat shock proteins’ mRNA expression in asthma. Respirology 2000, 5, 227–230. [Google Scholar] [CrossRef] [PubMed]
- Karin, M.; Greten, F.R. NF-[kappa] B: Linking inflammation and immunity to cancer development and progression. Nat. Rev. Immunol. 2005, 5, 749–759. [Google Scholar] [CrossRef] [PubMed]
- Torre, L.A.; Bray, F.; Siegel, R.L.; Ferlay, J.; Lortet-Tieulent, J.; Jermal, A. Global cancer statistics 2012. CA Cancer J. Clin. 2015, 65, 87–108. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef]
- Calderwood, S.K.; Stevenson, M.A.; Murshid, A. Heat shock proteins, autoimmunity, and cancer treatment. Autoimmun. Dis. 2012, 2012, 486069. [Google Scholar] [CrossRef] [PubMed]
- Kapoor, C.; Vaidya, S. Heat shock protein (HSP) and cancer: An overview. Am. J. Med. Dent. Sci. 2013, 1, 31–34. [Google Scholar]
- Beere, H.M.; Green, D.R. Stress management–heat shock protein-70 and the regulation of apoptosis. Trends Cell Biol. 2001, 11, 6–10. [Google Scholar] [CrossRef]
- Wu, J.; Liu, T.; Rios, Z.; Mei, Q.; Lin, X.; Cao, S. Heat shock proteins and cancer. Trends Pharmacol. Sci. 2017, 38, 226–256. [Google Scholar] [CrossRef] [PubMed]
- Vergara, D.; Simeone, P.; del Boccio, P.; Toto, C.; Pieragostino, D.; Tinelli, A.; Acierno, R.; Alberti, S.; Salzet, M.; Giannelli, G.; et al. Comparative proteome profiling of breast tumor cell lines by gel electrophoresis and mass spectrometry reveals an epithelial mesenchymal transition associated protein signature. Mol. BioSyst. 2013, 9, 1127–1138. [Google Scholar] [CrossRef] [PubMed]
- Shiota, M.; Bishop, J.L.; Nip, K.M.; Zardan, A.; Takeuchi, A.; Cordonnier, T.; Beraldi, E.; Bazov, J.; Fazli, L.; Chi, K.; et al. Hsp27 regulates epithelial mesenchymal transition, metastasis, and circulating tumor cells in prostate cancer. Cancer Res. 2013, 73, 3109–3119. [Google Scholar] [CrossRef] [PubMed]
- Lianos, G.D.; Alexiou, G.A.; Mangano, A.; Mangano, A.; Rausei, S.; Boni, L.; Dionigi, G.; Roukos, D.H. The role of heat shock proteins in cancer. Cancer Lett. 2015, 360, 114–118. [Google Scholar] [CrossRef] [PubMed]
- Jego, G.; Hazoumé, A.; Seigneuric, R.; Garrido, C. Targeting heat shock proteins in cancer. Cancer Lett. 2013, 332, 275–285. [Google Scholar] [CrossRef] [PubMed]
- Chaiwatanasirikul, K.A.; Sala, A. The tumour-suppressive function of CLU is explained by its localisation and interaction with HSP60. Cell Death Dis. 2011, 2, e219. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, J.C.; Siegelin, M.D.; Dohi, T.; Altieri, D.C. Heat shock protein 60 regulation of the mitochondrial permeability transition pore in tumor cells. Cancer Res. 2010, 70, 8988–8993. [Google Scholar] [CrossRef] [PubMed]
- Chalmin, F.; Ladoire, S.; Mignot, G.; Vincent, J.; Bruchard, M.; Remy-Martin, J.P.; Boireau, W.; Rouleau, A.; Simon, B.; Lanneau, D.; et al. Membrane-associated Hsp72 from tumor-derived exosomes mediates STAT3-dependent immunosuppressive function of mouse and human myeloid-derived suppressor cells. J. Clin. Investig. 2010, 120, 457–471. [Google Scholar] [CrossRef] [PubMed]
- Trinh, D.L.; Elwi, A.N.; Kim, S.W. Direct interaction between p53 and Tid1 proteins affects p53 mitochondrial localization and apoptosis. Oncotarget 2010, 1, 396–404. [Google Scholar] [CrossRef] [PubMed]
- Spooner, R.; Yilmaz, Ö. The role of reactive-oxygen-species in microbial persistence and inflammation. Int. J. Mol. Sci. 2011, 12, 334–352. [Google Scholar] [CrossRef] [PubMed]
- Simon, H.U.; Haj-Yehia, A.; Levi-Schaffer, F. Role of reactive oxygen species (ROS) in apoptosis induction. Apoptosis 2000, 5, 415–418. [Google Scholar] [CrossRef] [PubMed]
- Fischer, R.; Maier, O. Interrelation of oxidative stress and inflammation in neurodegenerative disease: Role of TNF. Oxid. Med. Cell. Longev. 2015, 2015, 610813. [Google Scholar] [CrossRef] [PubMed]
- Nowsheen, S.; Yang, E.S. The intersection between DNA damage response and cell death pathways. Exp. Oncol. 2012, 34, 243–254. [Google Scholar] [PubMed]
- Nita, M.; Grzybowski, A. The role of the reactive oxygen species and oxidative stress in the pathomechanism of the age-related ocular diseases and other pathologies of the anterior and posterior eye segments in adults. Oxid. Med. Cell. Longev. 2016, 2016, 3164734. [Google Scholar] [CrossRef] [PubMed]
- Barnes, P.J. Reactive oxygen species and airway inflammation. Free Radic. Biol. Med. 1990, 9, 235–243. [Google Scholar] [CrossRef]
- Henricks, P.A.; Nijkamp, F.P. Reactive oxygen species as mediators in asthma. Pulm. Pharmacol. Ther. 2001, 14, 409–421. [Google Scholar] [CrossRef] [PubMed]
- Winrow, V.R.; McLean, L.; Morris, C.J.; Blake, D.R. The heat shock protein response and its role in inflammatory disease. Ann. Rheum. Dis. 1990, 49, 128–132. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Voegeli, T.S.; Liu, P.P.; Noble, E.G.; Currie, R.W. Heat shock paradox and a new role of heat shock proteins and their receptors as anti-inflammation targets. Inflamm. Allergy Drug Targets 2007, 6, 91–100. [Google Scholar] [CrossRef] [PubMed]
- Hauet-Broere, F.; Wieten, L.; Guichelaar, T.; Berlo, S.; Van der Zee, R.; Van Eden, W. Heat shock proteins induce T cell regulation of chronic inflammation. Ann. Rheum. Dis. 2006, 65, 65–68. [Google Scholar] [CrossRef] [PubMed]
- Tóth, M.E.; Gombos, I.; Sántha, M. Heat shock proteins and their role in human diseases. Acta Biol. Szeged. 2015, 59, 121–141. [Google Scholar]
- Arnal, M.E.; Lallès, J.P. Gut epithelial inducible heat-shock proteins and their modulation by diet and the microbiota. Nutr. Rev. 2016, 74, 181–197. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, M.G.; Li, Z. Heat-shock proteins in infection-mediated inflammation-induced tumorigenesis. J. Hematol. Oncol. 2009, 2. [Google Scholar] [CrossRef] [PubMed]
- Hatfield, P.D.; Lovas, S. Role of Hsp70 in cancer growth and survival. Protein Pept. Lett. 2012, 19, 616–624. [Google Scholar] [CrossRef] [PubMed]
- Chauhan, D.; Li, G.; Hideshima, T.; Podar, K.; Mitsiades, C.; Mitsiades, N.; Catley, L.; Tai, Y.T.; Hayashi, T.; Shringarpure, R.; et al. Hsp27 inhibits release of mitochondrial protein Smac in multiple myeloma cells and confers dexamethasone resistance. Blood 2003, 102, 3379–3386. [Google Scholar] [CrossRef] [PubMed]
- Nylandsted, J.; Rohde, M.; Brand, K.; Bastholm, L.; Elling, F.; Jäättelä, M. Selective depletion of heat shock protein 70 (HSP70) activates a tumor-specific death program that is independent of caspases and bypasses Bcl-2. Proc. Natl. Acad. Sci. USA 2000, 97, 7871–7876. [Google Scholar] [CrossRef] [PubMed]
- Zininga, T.; Shonhai, A. Are heat shock proteins druggable candidates? Am. J. Biochem. Biotech. 2014, 8, 427–432. [Google Scholar] [CrossRef]
- Henstridge, D.C.; Febbraio, M.A.; Hargreaves, M. Heat shock proteins and exercise adaptations. Our knowledge thus far and the road still ahead. J. Appl. Physiol. 2016, 120, 683–691. [Google Scholar] [CrossRef] [PubMed]
- Noble, E.G.; Shen, G.X. Impact of exercise and metabolic disorders on heat shock proteins and vascular inflammation. Autoimmun. Dis. 2012, 2012, 836519. [Google Scholar] [CrossRef] [PubMed]
- Naughton, L.; Lovell, R.; Madden, L. Heat shock proteins in exercise: A review. J. Exerc. Sci. Physiother. 2006, 2, 13–26. [Google Scholar]


{kind=link}
{kind=link}
| Classification | Location | Cellular Function | Reference |
|---|---|---|---|
| Hsp10 | Mitochondria | Serves as biomarker in endometrial cancer and helps protein folding | [19,20] |
| Hsp27 | Cytosol, endoplasmic reticulum & nucleus | Facilitates refolding of denatured proteins (chaperoning activity) and serves as a biomarker in many cellular diseases such as cancer | [21] |
| Hsp40 | cytosol | Assists HSP70 in protein folding (co-chaperoning with HSP70) | [22] |
| HSP60 | Cytoplasm & mitochondria | Assists in protein folding, prevents protein aggregation and assembling of unfolding proteins via the formation of the hetero-oligomeric complex | [23] |
| Hsp70 | Cytoplasm & nucleus | Aids protein assembling, protein folding, degradation of improperly folded peptides and translocation of organelles | [5] |
| Hsp90 | Cytoplasm | Assists in protein folding, refolding and degradation. It also facilitates signal transduction and important roles in cancer and sarcomere formation as well as in myosin folding | [5,24] |
| Hsp100 | Cytoplasm | Complexes with other HSPs to refold aggregated or misfolded proteins | [25] |
| Hsp110 | Cytosol & nucleus | Helps immune response and complexes with HSP70 to promote protein refolding and cell survival under stress | [26] |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ikwegbue, P.C.; Masamba, P.; Oyinloye, B.E.; Kappo, A.P. Roles of Heat Shock Proteins in Apoptosis, Oxidative Stress, Human Inflammatory Diseases, and Cancer. Pharmaceuticals 2018, 11, 2. https://doi.org/10.3390/ph11010002
Ikwegbue PC, Masamba P, Oyinloye BE, Kappo AP. Roles of Heat Shock Proteins in Apoptosis, Oxidative Stress, Human Inflammatory Diseases, and Cancer. Pharmaceuticals. 2018; 11(1):2. https://doi.org/10.3390/ph11010002
Chicago/Turabian StyleIkwegbue, Paul Chukwudi, Priscilla Masamba, Babatunji Emmanuel Oyinloye, and Abidemi Paul Kappo. 2018. "Roles of Heat Shock Proteins in Apoptosis, Oxidative Stress, Human Inflammatory Diseases, and Cancer" Pharmaceuticals 11, no. 1: 2. https://doi.org/10.3390/ph11010002
APA StyleIkwegbue, P. C., Masamba, P., Oyinloye, B. E., & Kappo, A. P. (2018). Roles of Heat Shock Proteins in Apoptosis, Oxidative Stress, Human Inflammatory Diseases, and Cancer. Pharmaceuticals, 11(1), 2. https://doi.org/10.3390/ph11010002