3.3. Chemical Data
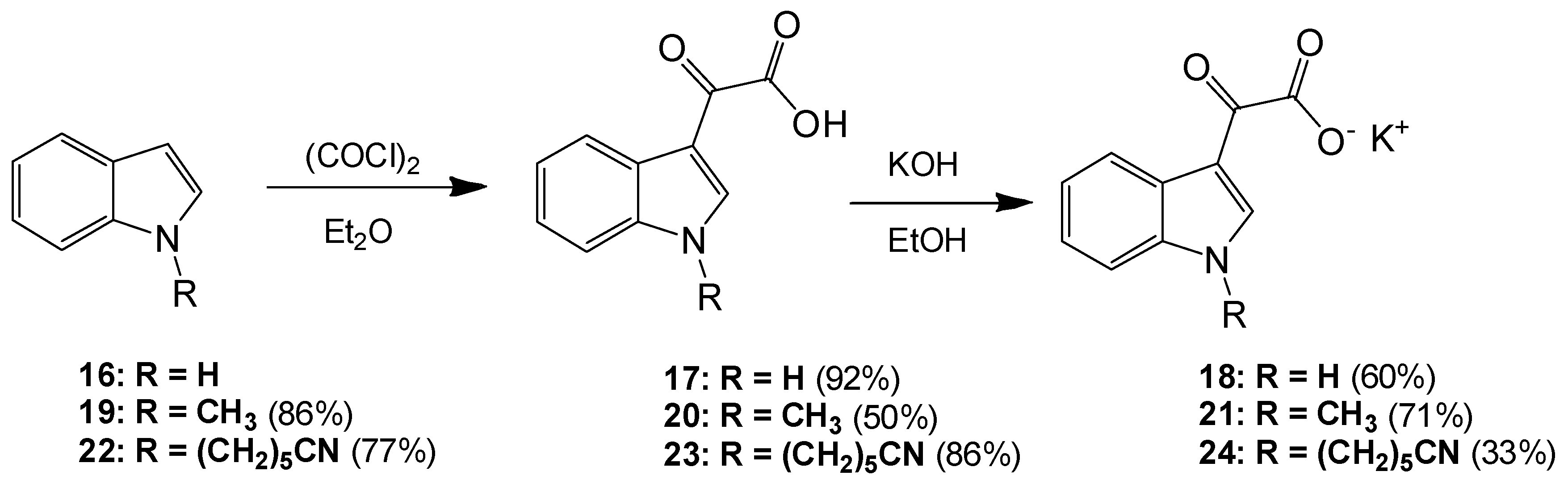
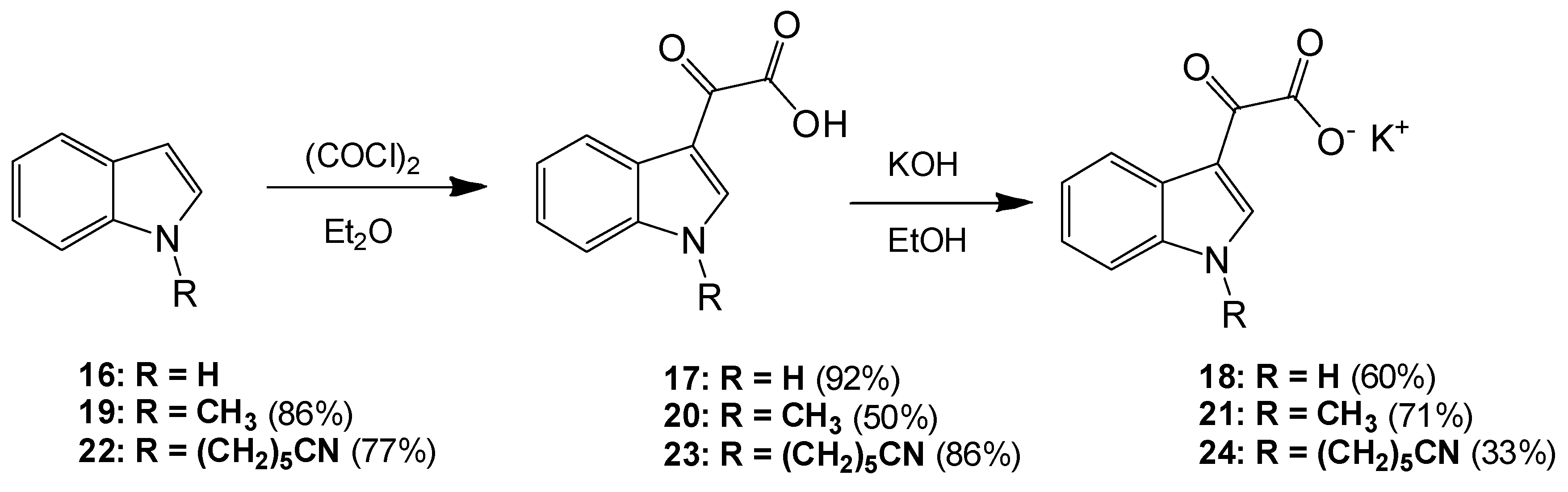
6-(1H-Indol-1-yl)hexanenitrile (22). To a solution of indole (2.51 g, 21.4 mmol) in dry DMF (60 mL) at 0 °C was added sodium hydride (1.31 g, 32.75 mmol) in a portion-wise manner. The resultant mixture was allowed to stir at room temperature for 30 min after which time 6-bromohexanitrile (4.25 mL, 1.328 g/mL, 32 mmol) was added with care. The reaction mixture was then allowed to warm to room temperature and stirred overnight. The reaction mixture was subsequently and carefully poured into ice-cold water, and this resulting mixture was extracted with ethyl acetate (6× 50 mL). Combined organic layers were then washed with water (5× 50 mL) and brine (3× 50 mL) before being dried over anhydrous magnesium sulphate and concentrated under reduced pressure to yield a brown oil, which was subject to flash column chromatography (65:35, hexane/ethyl acetate) to yield a viscous yellow oil, which was used without further purification (3.49 g, 16.4 mmol, 77%): νmax/cm−1 (NaCl) 3053, 2937, 2866, 2244, 1611; δH (300 MHz, CDCl3) 1.43 (m, 2H, CH2(CH2)2CN), 1.61 (m, 2H, CH2CH2CN), 1.84 (m, 2H, CH2(CH2)3CN), 2.25 (t, 2H, J = 7.1 Hz, CH2-CN), 4.11 (t, 2H, J = 6.9 Hz, N-CH2), 6.48 (dd, 1H, J = 3.2, 0.86 Hz, C-H3), 7.05 (d, 1H, J = 3.1 Hz, C-H2) 7.09 (overlapping ddd, 1H, J = 0.9, 7.1, 7.9 Hz, C-H5), 7.19 (m, 1H, J = 1.1, 7.1 Hz, C-H6), 7.30 (dd, 1H, J = 8.3, 0.8 Hz, C-H7), 7.62 (dt, 1H, J = 7.9, 0.9 Hz, C-H4); δC (75 MHz, CDCl3) 17.1 (CH2, CH2), 25.1 (CH2, CH2), 26.2 (CH2, CH2), 29.5 (CH2, CH2), 46.0 (CH2, NCH2), 101.3 (CH, aromatic CH), 109.3 (CH, aromatic CH), 119.4 (C, CN), 119.5 (CH, aromatic CH), 121.1 (CH, aromatic CH), 121.5 (CH, aromatic CH), 127.7 (CH, aromatic CH), 128.7 (C, aromatic C), 135.9 (C, aromatic C); m/z (ES+) 213.4 [M + H]+ (100%); HRMS (ES+): exact mass calculated for C14H17N2 213.1392. Found 213.1386.
2-(1-(5-Cyanopentyl)-1H-indol-3-yl)-2-oxoacetic acid (23). To a solution of 6-(1H-indol-1-yl)hexanenitrile 6 (3.49 g, 16.4 mmol) in diethyl ether (100 mL) at 0 °C was added oxalyl chloride (2.20 mL, 1.48 g/mL, 24.64 mmol) dropwise over a 20 min. The resultant deep orange slurry was allowed to stir at room temperature for 30 min. After this time saturated, aqueous sodium bicarbonate solution was added very carefully (15 mL). The resulting biphasic mixture was left to stir overnight. Following this, the reaction mixture was vacuum filtered, and the product cake was washed with ice-cold diethyl ether (10 mL) and ice-cold water (5 mL) before being allowed to dry to yield the oxoacetic acid as a pink solid (4.03 g, 14.2 mmol, 86%), m.p. 133–135 °C: νmax/cm−1 (KBr) 3428, 2952, 2241, 1752, 1628, 1520, 1148; δH (400 MHz, DMSO-d6) 1.38 (m, 2H, CH2(CH2)2CN), 1.60 (m, 2H, CH2CH2CN), 1.83 (m, 2H, CH2(CH2)3CN), 2.48 (t, 2H, J = 2.4 Hz, CH2-CN), 4.34 (t, 2H, J = 7.2 Hz, N-CH2), 7.33 (m, 2H, C-H5, C-H6), 7.68 (d, 1H, J = 8.1 Hz, C-H7), 8.22 (d, 1H, J = 8.0 Hz, C-H4), 8.52 (s, 1H, C-H2); δC (100 MHz, DMSO-d6) 16.0 (CH2, CH2), 24.3 (CH2, CH2), 25.2 (CH2, CH2), 28.6 (CH2, CH2), 46.2 (CH2, NCH2), 111.3 (CH, aromatic CH), 111.4 (C, aromatic C), 120.6 (C, CN), 121.4 (CH, aromatic CH), 123.0 (CH, aromatic CH), 123.7 (CH, aromatic CH), 126.2 (C, aromatic C), 136.6 (C, aromatic C), 140.3 (CH, aromatic CH), 165.2 (C, aromatic C=O), 180.4 (C, C=O); m/z (ES+) 285.3 [M + H]+ (100%); HRMS (ES+): exact mass calculated for C16H17N2O3: 285.1239. Found 285.1236.
Potassium 2-(1-(5-Cyanopentyl)-1H-indol-3-yl)-2-oxoacetate (24). To a solution of 2-(1-(5-cyanopentyl)-1H-indol-3-yl)-2-oxoacetic acid 7 (4.03 g, 14.17 mmol) in ethanol (200 mL) was added potassium hydroxide (0.83 g, 14.8 mmol). The reaction mixture was allowed to stir for 4 h before being isolated by vacuum filtration. The resulting product cake was allowed to dry to yield a beige solid (1.48 g, 4.6 mmol, 33 %), m.p. 167–170 °C: νmax/cm−1 (KBr) 3406, 2944, 2245, 1884, 1765, 1632 (broad), 1523, 1149; δH (400 MHz, DMSO-d6) 1.35 (m, 2H, CH2(CH2)2CN), 1.58 (m, 2H, CH2CH2CN), 1.79 (m, 2H, CH2(CH2)3CN), 2.47 (t, 2H, J = 7.12 Hz, CH2-CN), 4.25 (t, 2H, J = 7.26, N-CH2), 7.19 (m, 2H, C-H5, C-H6), 7.55 (d, 1H, J = 8.09 Hz, C-H7), 8.18 (s, 1H, C-H2), 8.20 (d, 1H, J = 7.58 Hz, C-H4); δC (100 MHz, DMSO-d6) 16.0 (CH2, CH2), 24.4 (CH2, CH2), 25.3 (CH2, CH2), 28.8 (CH2, CH2), 45.7 (NCH2), 110.5 (CH, aromatic CH), 113.1 (C, CN), 120.6 (C, aromatic C), 121.4 (CH, aromatic CH), 121.5 (CH, aromatic CH), 122.3 (CH, aromatic CH), 126.5 (C, aromatic C), 136.2 (C, aromatic C), 138.3 (CH, aromatic CH), 169.6 (C, C=O), 192.9 (C, C=O); m/z (ES−) 283.4 [M−H]− (100%); HRMS (ES+): exact mass calculated for C16H17N2O3: 285.1239. Found 285.1232.
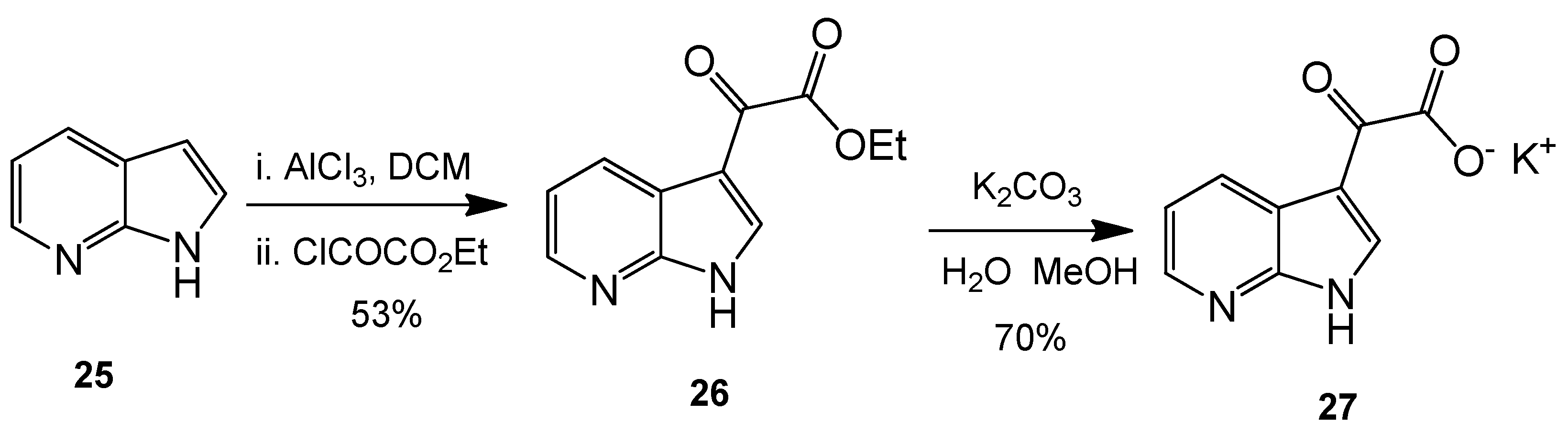
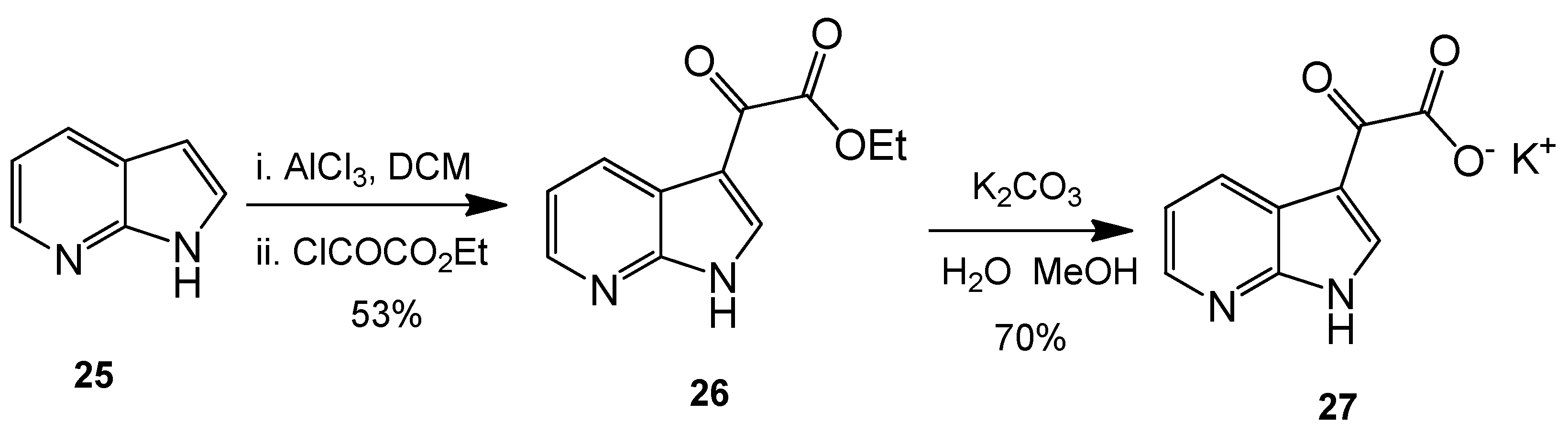
Ethyl 2-oxo-2-(1H-pyrrolo[2,3-b]pyridin-3-yl)acetate (26). 7-Azaindole (5.00 g, 42 mmol) was added to a suspension of aluminium trichloride (30.00 g, 225 mmol) in DCM (500 mL), and the resulting mixture was allowed to stir for 30 min at room temperature. After this time, ethyl chlorooxoacetate (24 mL, 215 mmol) was added dropwise over 20 min with the resulting mixture left to stir vigorously at room temperature overnight. The reaction mixture was then poured over ice cold water, and the layers were separated. The aqueous layer was washed with DCM (4× 50 mL). Combined organic layers were then washed very carefully with ice-cold, saturated aqueous sodium bicarbonate solution (3× 20 mL) and brine (1× 100 mL) before being dried over anhydrous magnesium sulphate and concentrated under reduced pressure to yield a brown oil. Purification by flash column chromatography (60:40, hexane/ethyl acetate) afforded the oxoacetate as a fluffy, light yellow solid (4.83 g, 22.1 mmol, 53%), m.p. 157–158 °C; νmax/cm−1 (KBr) 3418, 1872, 1723, 1611, 1174; δH (400 MHz, DMSO-d6) 1.35 (t, 3H, J = 7.1 Hz, OCH2CH3), 4.38 (q, 2H, J = 14.2, 7.1 Hz, OCH2CH3), 7.34 (dd, 1H, J = 7.9, 4.7 Hz, C-H5), 8.40 (dd, 1H, J = 4.7, 1.6 Hz, C-H6), 8.47 (dd, 1H, J = 7.9, 1.6 Hz, C-H4), 8.57 (s, 1H, C-H2); m/z (ES+) 219.3 [M + H]+ 100%.
Potassium 2-oxo-2-(1H-pyrrolo[2,3-b]pyridin-3-yl)acetate (27). To a suspension of ethyl 2-oxo-2-(1H-pyrrolo[2,3-b]pyridin-3-yl)acetate 9 (4.83 g, 22.1 mmol) in a 1:1 solution of water/MeOH was added potassium carbonate (5.67 g, 41.03 mmol). The resulting mixture was allowed to stir for 5 h before the product was isolated by vacuum filtration and the cake dried overnight to yield the glyoxylate salt as a brilliant white powder (3.53 g, 15.5 mmol, 70%), m.p. > 250 °C; νmax/cm−1 (KBr) 3244, 3061, 2530, 1980, 1860, 1780, 1634, 1455; δH (400 MHz, DMSO-d6) 7.18 (q, 1H, J = 7.9, 4.5 Hz, C-H5), 8.13 (s, 1H, C-H2), 8.27 (dd, 1H, J = 4.7, 1.6 Hz, C-H6), 8.42 (dd, 1H, J = 7.8, 1.6 Hz, C-H4); m/z (ES−) 189.3 [M−H]− 100%.
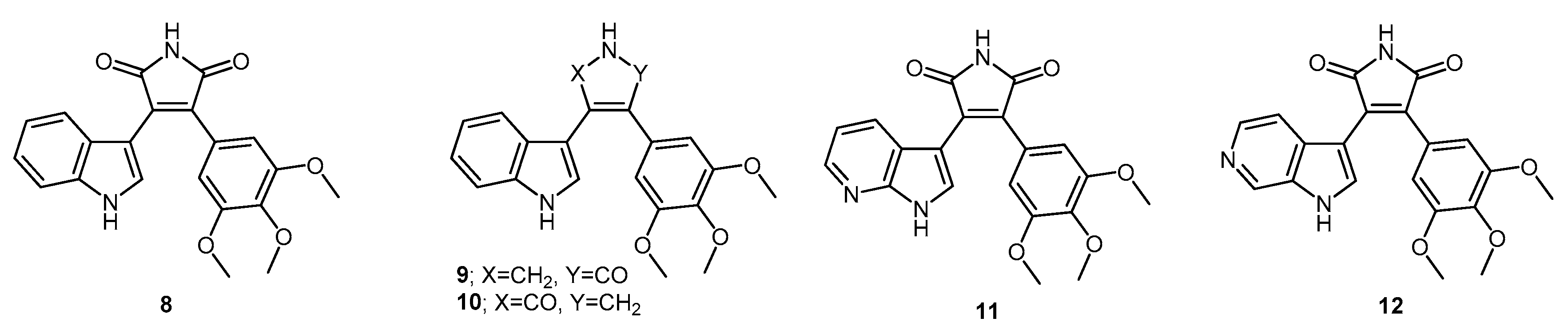
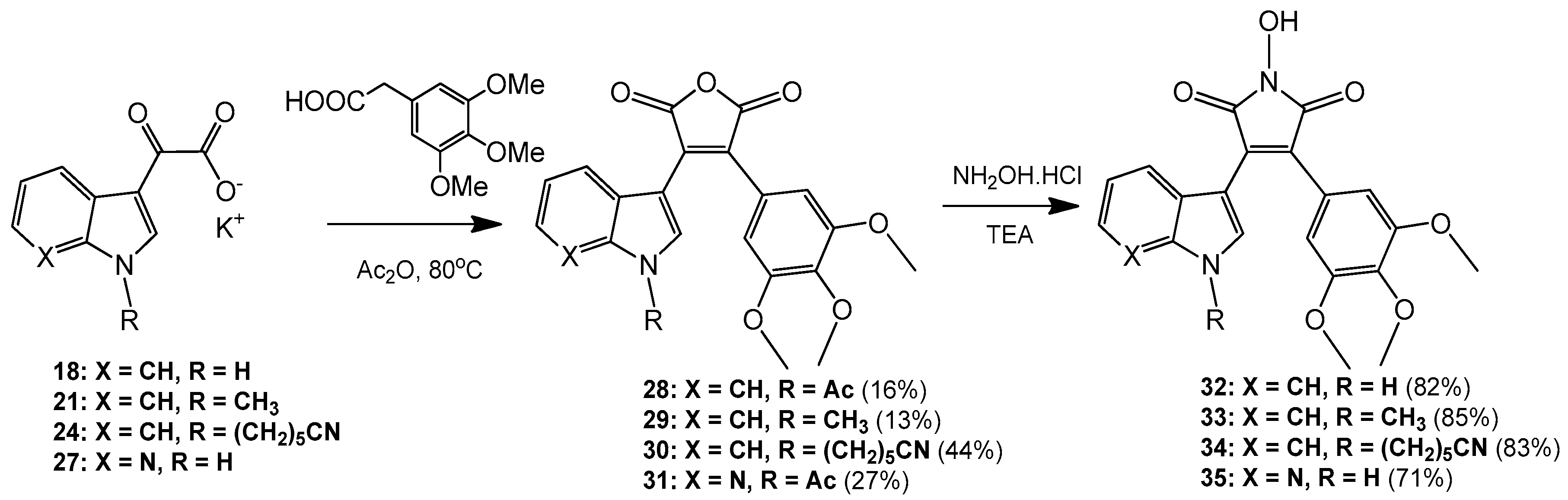
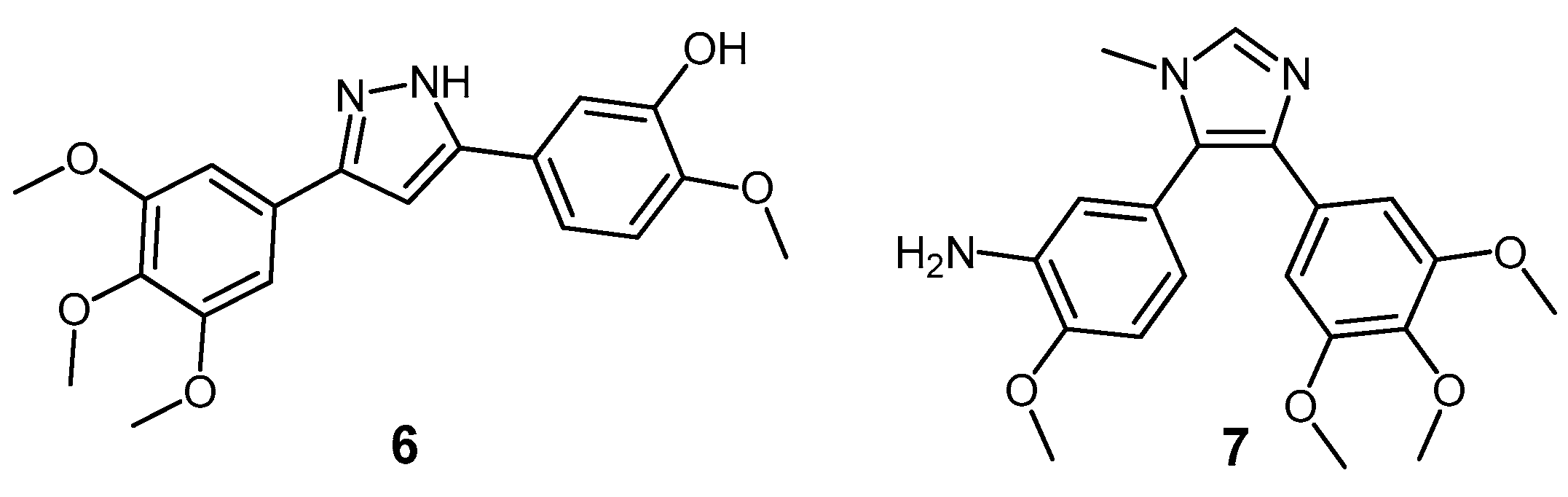
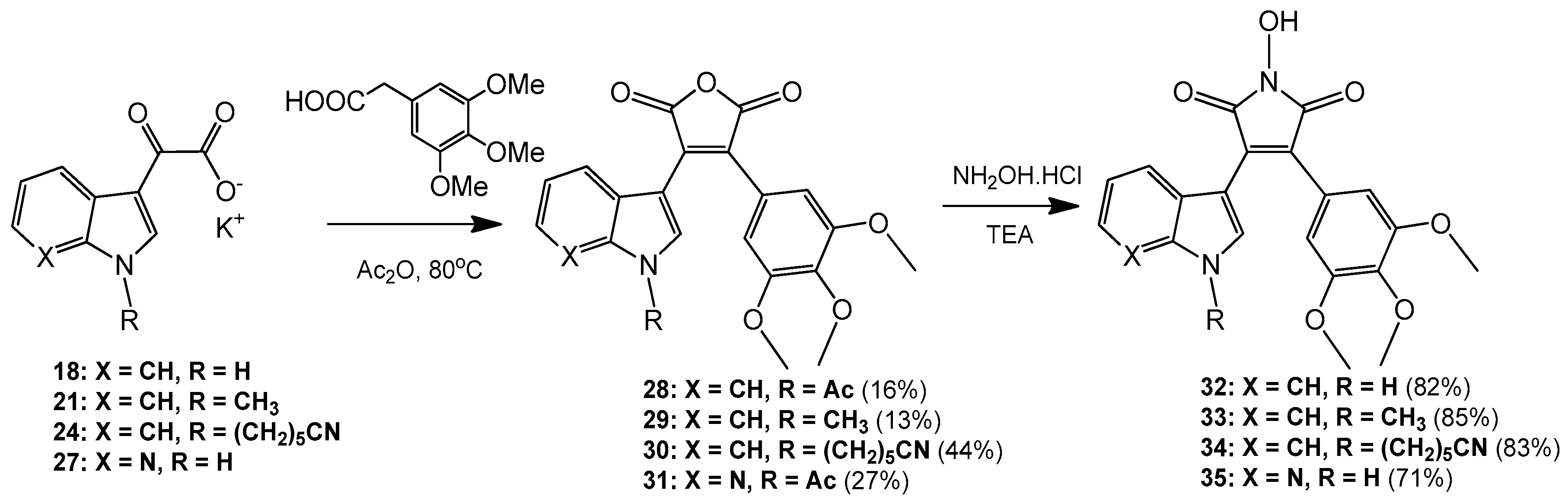
3-(1-Acetyl-1H-indol-3-yl)-4-(3,4,5-trimethoxyphenyl)furan-2,5-dione (28). Potassium 2-(1H-indol-3-yl)-2-oxoacetate 2 (703 mg, 3.09 mmol) was added to a round-bottomed flask along with 3,4,5-trimethoxyphenylacetic acid (700 mg, 3.09 mmol). Acetic anhydride was added (25 mL) and the reaction mixture was heated for 24 h at 80 °C. Following this, the acetic anhydride was removed under reduced pressure, and to the crude residue was added saturated aqueous sodium bicarbonate solution. This mixture was extracted with ethyl acetate (5× 50 mL). The ethyl acetate was then washed with saturated aqueous sodium bicarbonate solution (4× 50 mL), water (4× 50 mL) and brine (2× 100 mL) before being dried over anhydrous magnesium sulphate and concentrated under reduced pressure to yield a brown residue. Purification by flash chromatography (65:35, hexane/ethyl acetate) yielded a bright yellow solid (216 mg, 0.5 mmol, 16%), m.p. 153–155 °C; νmax/cm−1 (KBr) 3417, 2942, 1824, 1763, 1718, 1676, 1477; δH (400 MHz, CDCl3) 2.77 (s, 3H, acyl CH3), 3.50 (s, 6H, 2× m-OCH3), 3.89 (s, 3H, p-OCH3), 6.50 (d, 1H, J = 8.1 Hz, C-H7), 6.84 (s, 2H, Ar-CH2′ and Ar-CH6′), 7.03 (overlapping ddd, 1H, J = 8.1, 7.6, 0.9 Hz, C-H5), 7.34 (overlapping ddd, 1H, J = 8.4, 7.3, 1.0 Hz, C-H6), 8.23 (s, 1H, C-H2), 8.49 (d, 1H, J = 8.4 Hz, C-H4); δC (100 MHz, CDCl3) 24.0 (CH3, Acyl-CH3), 56.0 (2× CH3, 2× m-OCH3), 61.0 (CH3, p-OCH3), 108.2 (2× CH, 2× aromatic CH), 110.2 (C, aromatic C), 116.9 (CH, aromatic CH), 122.3 (CH, aromatic CH), 122.7 (C, aromatic C), 124.0 (CH, aromatic CH), 125.4 (C, aromatic C), 126.3 (CH, aromatic CH), 130.0 (C, aromatic C), 130.5 (CH, aromatic CH), 134.5 (C, aromatic C), 135.9 (C, aromatic C), 141.0 (C, aromatic C), 153.0 (2× C, 2× aromatic C), 165.0 (C, C=O), 165.3 (C, C=O), 168.6 (C, C=O); m/z (ES+) 422.2 [M + H]+ (20%); HRMS (ES+): exact mass calculated for C23H20NO7 422.1240. Found 422.1227.
3-(1-Methyl-1H-indol-3-yl)-4-(3,4,5-trimethoxyphenyl)furan-2,5-dione (29). Potassium 2-(1-methyl-1H-indol-3-yl)-2-oxoacetate 5 (1.43 g, 5.93 mmol) and 3,4,5-trimethoxyphenylacetic acid (1.31 g, 5.8 mmol) were placed in a round-bottomed flask. Acetic anhydride (55 mL) was added, and the resulting mixture was heated for 24 h at 80 °C. Following this, the excess acetic anhydride was removed under reduced pressure, and to the solid residue was added saturated aqueous sodium bicarbonate solution. The crude organic material was extracted with ethyl acetate (4× 50 mL). The combined ethyl acetate washings were then washed with saturated aqueous sodium bicarbonate solution (3× 100 mL), water (3× 100 mL) and brine (2× 100 mL) before being dried over anhydrous magnesium sulphate and concentrated under reduced pressure to yield a sticky red solid. Purification by flash chromatography (70:30, hexane/ethyl acetate) yielded the desired maleic anhydride as a red solid (293 mg, 0.75 mmol, 13 %), m.p. 175–177 °C; νmax/cm−1 (KBr) 2940, 1987, 1846, 1814, 1748, 1601, 1247, 1124; δH (400 MHz, CDCl3) 3.51 (s, 6H, 2× m-OCH3), 3.89 (s, 3H, p-OCH3), 3.92 (s, 3H, N-CH3), 6.42 (d, 1H, J = 8.2 Hz, C-H7), 6.81 (s, 2H, Ar-CH2′ and Ar-CH6′), 6.90 (overlapping ddd, 1H, J = 8.1, 7.2, 0.9 Hz, C-H5), 7.21 (overlapping ddd, 1H, J = 8.0, 7.1, 0.9 Hz, C-H6), 7.35 (d, 1H, J = 8.3 Hz, C-H4), 8.05 (d, 1H, J = 1.3 Hz, C-H2); δC (75 MHz, CDCl3) 33.7 (CH3, N-CH3), 55.9 (2× CH3, 2× m-OCH3), 61.0 (CH3, p-OCH3), 104.2 (C, aromatic C), 107.7 (2× CH, 2× aromatic CH), 110.1 (CH, aromatic CH), 121.3 (CH, aromatic CH), 123.2 (CH, aromatic CH), 123.3 (CH, aromatic CH), 124.1 (C, aromatic C), 124.2 (C, aromatic C), 128.1 (C, aromatic C), 132.5 (C, aromatic C), 136.0 (CH, aromatic CH), 137.5 (C, aromatic C), 139.7 (C, aromatic C), 152.8 (2× C, 2× aromatic C), 165.9 (C, C=O), 166.4 (C, C=O); m/z (ES+) 394.4 [M + H]+ (40%); HRMS (ES+): exact mass calculated for C22H20NO6 394.1291. Found 394.1283.
6-(3-(2,5-Dioxo-4-(3,4,5-trimethoxyphenyl)-2,5-dihydrofuran-3-yl)-1H-indol-1-yl)hexanenitrile (30). Potassium 2-(1-(5-cyanopentyl)-1H-indol-3-yl)-2-oxoacetate 8 (511 mg, 1.58 mmol) and 3,4,5-trimethoxyphenylacetic acid (365 mg, 1.62 mmol) were added to a round-bottomed flask. Acetic anhydride (15 mL) was added, and the mixture was heated at 80 °C for 24 h. Following this, the excess acetic anhydride was removed under reduced pressure. Saturated aqueous sodium bicarbonate solution was added to the residue to remove any residual acetic anhydride. Crude organic material was then extracted with ethyl acetate (4× 50 mL). The combined ethyl acetate extracts were then washed with saturated aqueous sodium bicarbonate solution (3× 50 mL), water (3× 50 mL) and brine (3× 50 mL) before being dried over anhydrous magnesium sulphate and concentrated under reduced pressure to yield a red residue. Purification by flash chromatography (50:50, hexane/ethyl acetate) yielded the maleic anhydride as a red solid (333 mg, 0.7 mmol, 44%), m.p. 134–136 °C; νmax/cm−1 (KBr) 3056, 2247, 1815, 1748, 1464; δH (400 MHz, CDCl3) 1.56 (m, 2H, CH2(CH2)2CN), 1.73 (m, 2H, CH2CH2CN), 1.99 (m, 2H, CH2(CH2)3CN), 2.37 (t, 2H, J = 7 Hz, CH2CN), 3.52 (s, 6H, 2× m-OCH3), 3.89 (s, 3H, p-OCH3), 4.26 ppm (t, 2H, J = 7.1 Hz, N-CH2), 6.46 (d, 1H, J = 8.12 Hz, C-H7), 6.81 (s, 2H, Ar-CH2′ and Ar-CH6′), 6.92 (dt, 1H, J = 8.0, 0.9 Hz, C-H5), 7.23 (dt, 1H, J = 8.0, 0.9 Hz, C-H6), 7.37 (d, 1H, J = 8.3 Hz, C-H4), 8.07 (d, 1H, J = 1.46 Hz, C-H2); δC (100 MHz, CDCl3) 17.1 (CH2, CH2), 24.9 (CH2, CH2), 26.0 (CH2, CH2), 29.2 (CH2, CH2), 46.8 (CH2, N-CH2), 55.9 (2× CH3, 2× m-OCH3), 61.0 (CH3, p-OCH3), 104.5 (C, aromatic C), 107.8 (2× CH, 2× aromatic CH), 110.1 (CH, aromatic CH), 119.2 (C, CN), 121.4 (CH, aromatic CH), 123.4 (2× CH, 2× aromatic CH), 124.0 (C, aromatic C), 124.3 (C, aromatic C), 128.4 (C, aromatic C), 132.4 (C, aromatic C), 134.7 (CH, aromatic CH), 136.7 (C, aromatic C), 139.8 (C, aromatic C), 152.9 (2× C, 2× aromatic C), 165.8 (C, C=O), 166.4 (C, C=O); m/z (ES+) 475.2 [M + H]+ (100%); HRMS (ES+): exact mass calculated for C27H27N2O6 475.1869. Found 475.1863
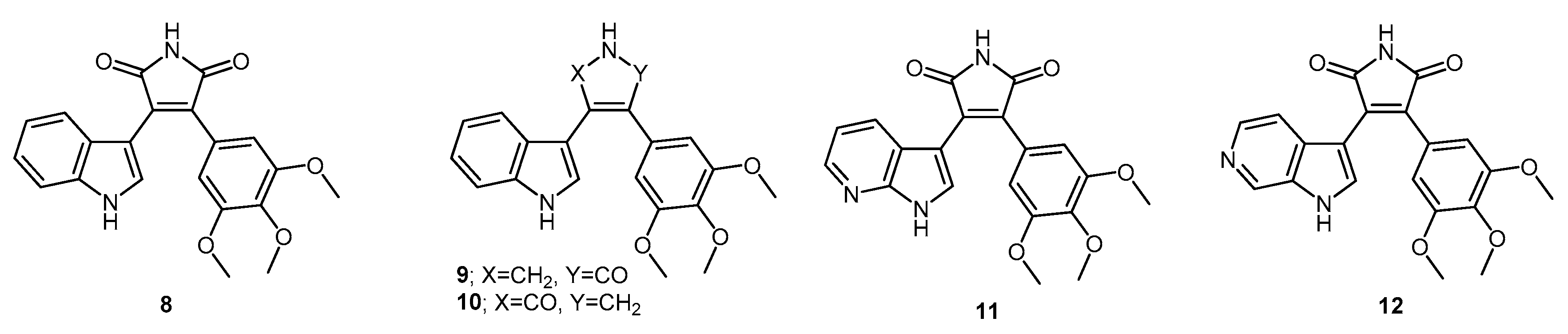
3-(1-Acetyl-1H-pyrrolo[2,3-b]pyridin-3-yl)-4-(3,4,5-trimethoxyphenyl)furan-2,5-dione (31). Potassium 2-oxo-2-(1H-pyrrolo[2,3-b]pyridin-3-yl)acetate 10 (410 mg, 1.8 mmol) and 3,4,5-trimethoxyphenylacetic acid (407 mg, 1.8 mmol) were added to a round-bottomed flask. Acetic anhydride (20 mL) was added and the mixture was heated for 24 h at 80 °C. Following this, the excess acetic anhydride was removed under reduced pressure. Saturated aqueous sodium bicarbonate solution was added to the residue in order to remove any excess acetic anhydride. Crude organic material was then extracted with ethyl acetate (4× 50 mL). The combined ethyl acetate extracts were then washed with saturated aqueous sodium bicarbonate solution (3× 50 mL), water (3× 50 mL) and brine (3× 50 mL) before being dried over anhydrous magnesium sulphate and concentrated under reduced pressure to yield a dull golden residue. Purification by flash chromatography (65:35, hexane/ethyl acetate) yielded the maleic anhydride as a light orange solid (208 mg, 0.49 mmol, 27%), m.p. 103–105 °C; νmax/cm−1 (KBr) 3311, 1824, 1757, 1625, 1580; δH (400 MHz, CDCl3) 3.12 (s, 3H, acyl CH3), 3.58 (s, 6H, 2× m-OCH3), 3.90 (s, 3H, p-OCH3), 6.81 (s, 2H, Ar-CH2′ and Ar-CH6′), 6.88 (dd, 1H, J = 8.05, 1.38 Hz, C-H4), 7.00 (m, 1H, J = 8.0, 4.7 Hz, C-H5), 8.38 (dd, 1H, J = 4.7, 1.6 Hz, C-H6), 8.76 (s, 1H, C-H2); δC (100 MHz, CDCl3) 26.1 (CH3, Acyl CH3), 56.1 (2× CH3, 2× m-OCH3), 61.1 (CH3, p-OCH3), 107.0 (C, aromatic C), 107.9 (2× CH, 2× aromatic CH), 118.9 (CH, aromatic CH), 119.3 (C, aromatic C), 122.7 (C, aromatic C), 130.4 (C, aromatic C), 130.7 (CH, aromatic CH), 131.0 (CH, aromatic CH), 135.0 (C, aromatic C), 141.0 (C, aromatic C), 145.2 (CH, aromatic CH), 147.6 (C, aromatic C), 153.2 (2× C, 2× aromatic C), 164.5 (C, C=O), 164.8 (C, C=O), 168.4 (C, C=O); m/z (ES+) 423.2 [M + H]+ (40%); HRMS (ES+): exact mass calculated for C22H19N2O7 423.1192. Found 423.1189.
Synthesis of aryl hydroxymaleimides: General Procedure (i): To a solution of the maleic anhydride (1 eq.) in acetonitrile (or DMF) was added hydroxylamine hydrochloride (5 eq.) and triethylamine (5 eq.). The mixture was heated for 24 h at 80 °C. Following this, 1 M aqueous HCl was added to quench the reaction. Organic components were extracted with ethyl acetate, and the combined extracts were washed with saturated aqueous sodium bicarbonate solution, water, brine, dried over anhydrous magnesium sulphate and concentrated under reduced pressure to yield the corresponding hydroxymaleimide.
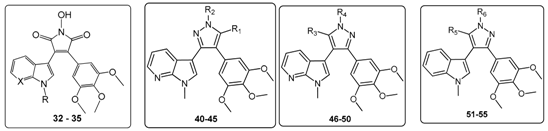
1-Hydroxy-3-(1H-indol-3-yl)-4-(3,4,5-trimethoxyphenyl)-1H-pyrrole-2,5-dione (32). Applying General Procedure (i) starting from maleic anhydride 11 (125 mg, 0.33 mmol), hydroxylamine HCl (115 mg, 1.65 mmol) and triethylamine (0.23 mL, 0.726 g/mL, 1.65 mmol) in acetonitrile (15 mL), hydroxymaleimide 15 was formed as a deep red solid (107.3 mg, 0.27 mmol, 82%), m.p. 123–125 °C; νmax/cm−1 (KBr) 3262, 2943, 1767, 1711 (broad), 1601, 1562, 1124; δH (400 MHz, DMSO-d6) 3.40 (s, 6H, 2× m-OCH3), 3.69 (s, 3H, p-OCH3), 6.37 (d, 1H, J = 8.05 Hz, C-H7), 6.75 (s, 2H, Ar-CH2′ and Ar-CH6′), 6.79 (t, 1H, J = 7.7 Hz, C-H5), 7.11 (t, 1H, J = 7.6 Hz, C-H6), 7.48 (d, 1H, J = 8.17 Hz, C-H4), 8.05 (s, 1H, C-H2), 10.54 (brs, 1H, N-H), 11.98 (brs, 1H, N-OH); δC (100 MHz, DMSO-d6) 55.5 (2× CH3, 2× m-OCH3), 60.1 (CH3, p-OCH3), 104.1 (C, aromatic C), 107.6 (2× CH, 2× aromatic CH), 112.2 (CH, aromatic CH), 119.8 (CH, aromatic CH), 121.6 (CH, aromatic CH), 122.2 (CH, aromatic CH), 123.6 (C, aromatic C), 124.6 (C, aromatic C), 125.2 (C, aromatic C), 128.8 (C, aromatic C), 131.6 (CH, aromatic CH), 136.5 (C, aromatic C), 138.2 (C, aromatic C), 152.3 (2× C, 2× aromatic C), 167.6 (C, C=O), 167.9 (C, C=O); m/z (ES+) 395.6 [M + H]+ (10%); HRMS (ES+): exact mass calculated for C21H19N2O6 395.1243. Found 395.1226.
1-Hydroxy-3-(1-methyl-1H-indol-3-yl)-4-(3,4,5-trimethoxyphenyl)-1H-pyrrole-2,5-dione (33). Applying General Procedure (i) starting from maleic anhydride 12 (257 mg, 0.65 mmol), hydroxylamine HCl (248 mg, 3.25 mmol) and triethylamine (0.45 mL, 0.726 g/mL, 3.25 mmol) in dry DMF (30 mL), hydroxymaleimide 16 was formed as a red solid (226 mg, 0.55 mmol, 85%), m.p. > 250 °C; νmax/cm−1 (KBr) 3262, 3125, 2942, 1769, 1715 (broad), 1597, 1467, 1125; δH (400 MHz, DMSO-d6) 3.40 (s, 6H, 2× m-OCH3), 3.68 (s, 3H, p-OCH3), 3.93 (s, 3H, N-CH3), 6.32 (d, 1H, J = 7.98 Hz, C-H7), 6.73 (s, 2H, Ar-CH2′ and Ar-CH6′), 6.82 (overlapping ddd, 1H, J = 8.1, 7.7, 1.0 Hz, C-H5), 7.18 (overlapping ddd, 1H, J = 8.2, 7.5, 0.9 Hz, C-H6), 7.53 (d, 1H, J = 8.25 Hz, C-H4), 8.11 (s, 1H, C-H2), 10.56 (brs, 1H, N-OH); δC (100 MHz, DMSO-d6) 33.1 (CH3, N-CH3), 55.5 (2× CH3, 2× m-OCH3), 59.7 (CH3, p-OCH3), 103.2 (C, aromatic C), 107.7 (2× CH, 2× aromatic CH), 110.6 (CH, aromatic CH), 120.1 (CH, aromatic CH), 121.8 (CH, aromatic CH), 122.3 (CH, aromatic CH), 124.0 (C, aromatic C), 124.3 (C, aromatic C), 125.2 (C, aromatic C), 128.3 (C, aromatic C), 135.2 (CH, aromatic CH), 137.0 (C, aromatic C), 138.2 (C, aromatic C), 152.3 (2× C, 2× aromatic C), 167.6 (C, C=O), 167.8 (C, C=O); m/z (ES+) 409.2 [M + H]+ (30%); HRMS (ES+): exact mass calculated for C22H21N2O6 409.1400. Found 409.1394.
6-(3-(1-Hydroxy-2,5-dioxo-4-(3,4,5-trimethoxyphenyl)-2,5-dihydro-1H-pyrrol-3-yl)-1H-indol-1-yl)hexanenitrile (34). Applying general procedure (i) starting from maleic anhydride 13 (257 mg, 0.54 mmol), hydroxylamine HCl (188 mg, 2.7 mmol) and triethylamine (0.38 mL, 0.726 g/mL, 2.7 mmol) in dry DMF (30 mL), hydroxymaleimide 17 was formed as a light red solid (219 mg, 0.45 mmol, 83%), m.p. 182–184 °C; νmax/cm−1 (KBr) 3467, 3213, 3000, 2942, 2260, 1720 (broad), 1626, 1580; δH (400 MHz, DMSO-d6) 1.34 (m, 2H, CH2(CH2)2CN), 1.58 (m, 2H, CH2CH2CN), 1.83 (m, 2H, CH2(CH2)3CN), 2.46 (t, 2H, J = 7.03 Hz, CH2-CN), 3.34 (s, 6H, 2× m-OCH3), 3.68 (s, 3H, p-OCH3), 4.34 (t, 2H, J = 6.81 , N-CH2), 6.40 (d, 1H, J = 8.10 Hz, CH7), 6.75 (s, 2H, Ar-CH2′ and Ar-CH6′), 6.82 (t, 1H, J = 7.74 Hz, C-H5), 7.17 (t, 1H, J = 7.62 Hz, C-H6), 7.59 (d, 1H, J = 8.21 Hz, C-H4), 8.09 (s, 1H, C-H2), 10.54 (brs, 1H, N-OH); δC (100 MHz, DMSO-d6) 16.0 (CH2, CH2), 24.2 (CH2, CH2), 25.1 (CH2, CH2), 28.7 (CH2, CH2), 45.6 (CH2, N-CH2), 55.4 (2× CH3, 2× m-OCH3), 60.1 (CH3, p-OCH3), 103.4 (C, aromatic C), 107.5 (2× CH, 2× aromatic CH), 110.7 (CH, aromatic CH), 120.0 (CH, aromatic CH), 120.5 (C, CN), 121.8 (CH, aromatic CH), 122.3 (CH, aromatic CH), 124.2 (C, aromatic C), 124.7 (C, aromatic C), 125.1 (C, aromatic C), 128.3 (C, aromatic C), 134.1 (CH, aromatic CH), 136.3 (C, aromatic C), 138.2 (C, aromatic C), 152.3 (2× C, 2× aromatic C), 167.5 (C, C=O), 167.8 (C, C=O); m/z (ES+) 490.2 [M + H]+ (70%); HRMS (ES+): exact mass calculated for C27H28N3O6 490.1978. Found 490.1992.
1-Hydroxy-3-(1H-pyrrolo[2,3-b]pyridin-3-yl)-4-(3,4,5-trimethoxyphenyl)-1H-pyrrole-2,5-dione (35). Applying General Procedure (i) starting from maleic anhydride 14 (147 mg, 0.38 mmol), hydroxylamine HCl (136 mg, 2 mmol) and triethylamine (0.27 mL, 0.726 g/mL, 1.9 mmol) in acetonitrile (15 mL), hydroxymaleimide 35 was formed as an orange solid (106 mg, 0.27 mmol, 71%), m.p. > 250 °C; νmax/cm−1 (KBr) 3157, 3098, 2937, 1712 (broad), 1616, 1500; δH (400 MHz, DMSO-d6) 3.45 (s, 6H, 2× m-OCH3), 3.70 (s, 3H, p-OCH3), 6.68 (dd, 1H, J = 8.05, 1.54 Hz, C-H4), 6.74 (s, 2H, Ar-CH2′ and Ar-CH6′), 6.88 (m, 1H, J = 8.01, 4.69 Hz, C-H5), 8.14 (s, 1H, C-H2), 8.23 (dd, 1H, J = 4.69, 1.53 Hz, C-H6), 10.61 (brs, 1H, N-OH), 12.50 (brs, 1H, N-H); δC (100 MHz, DMSO-d6) 55.6 (2× CH3, 2× m-OCH3), 60.2 (CH3, p-OCH3), 102.9 (C, aromatic C), 107.6 (2× CH, 2× aromatic CH), 116.1 (CH, aromatic CH), 116.2 (C, aromatic C), 124.9 (C, aromatic C), 126.1 (C, aromatic C), 128.3 (C, aromatic C), 129.5 (CH, aromatic CH), 131.7 (CH, aromatic CH), 138.5 (C, aromatic C), 143.7 (CH, aromatic CH), 148.8 (C, aromatic C), 152.5 (2× C, 2× aromatic C), 167.3 (C, C=O), 167.5 (C, C=O); m/z (ES+) 396.2 [M + H]+ (50%); HRMS (ES+): exact mass calculated for C20H18N3O6 396.1196. Found 396.1181.
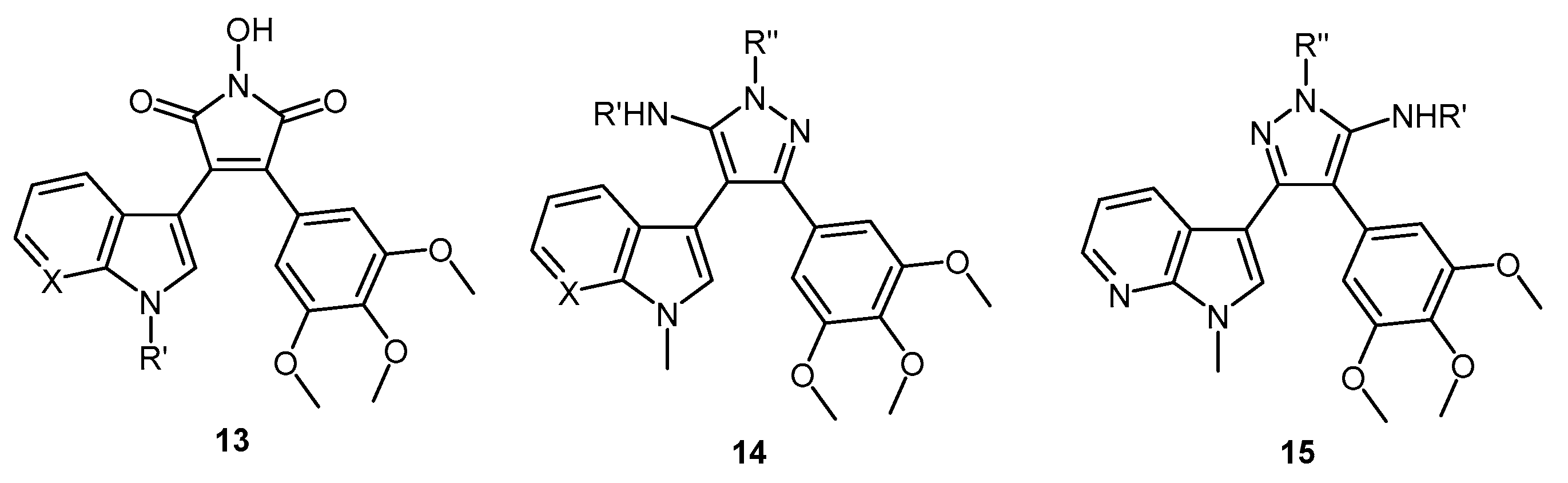
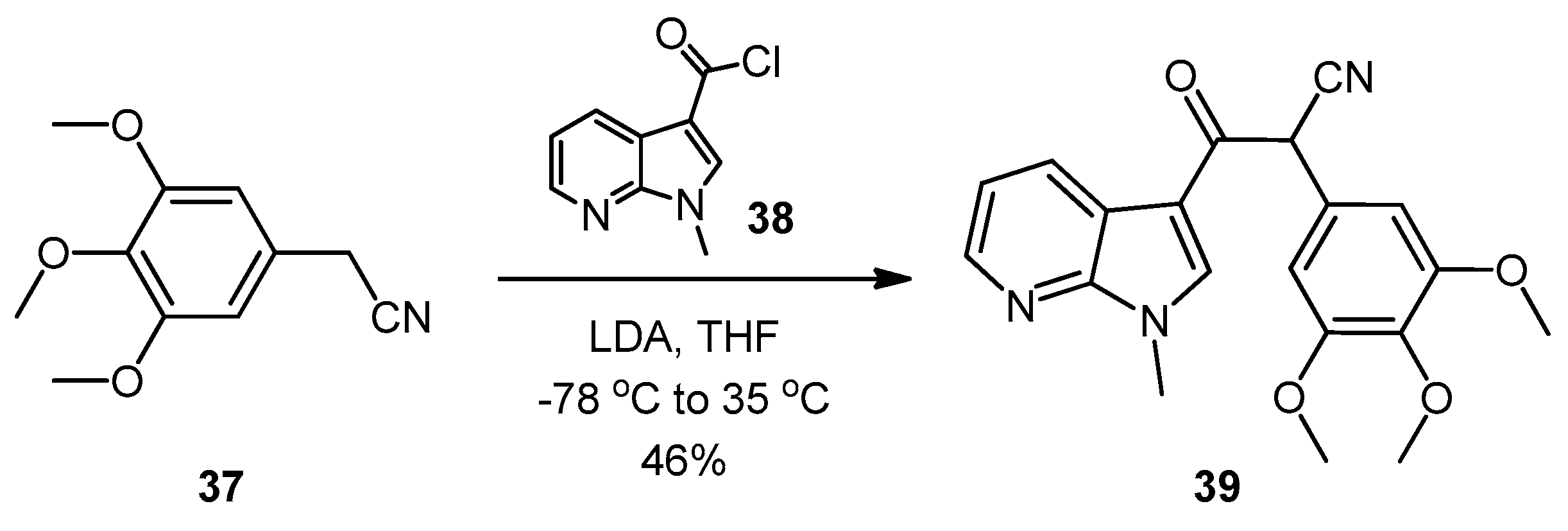
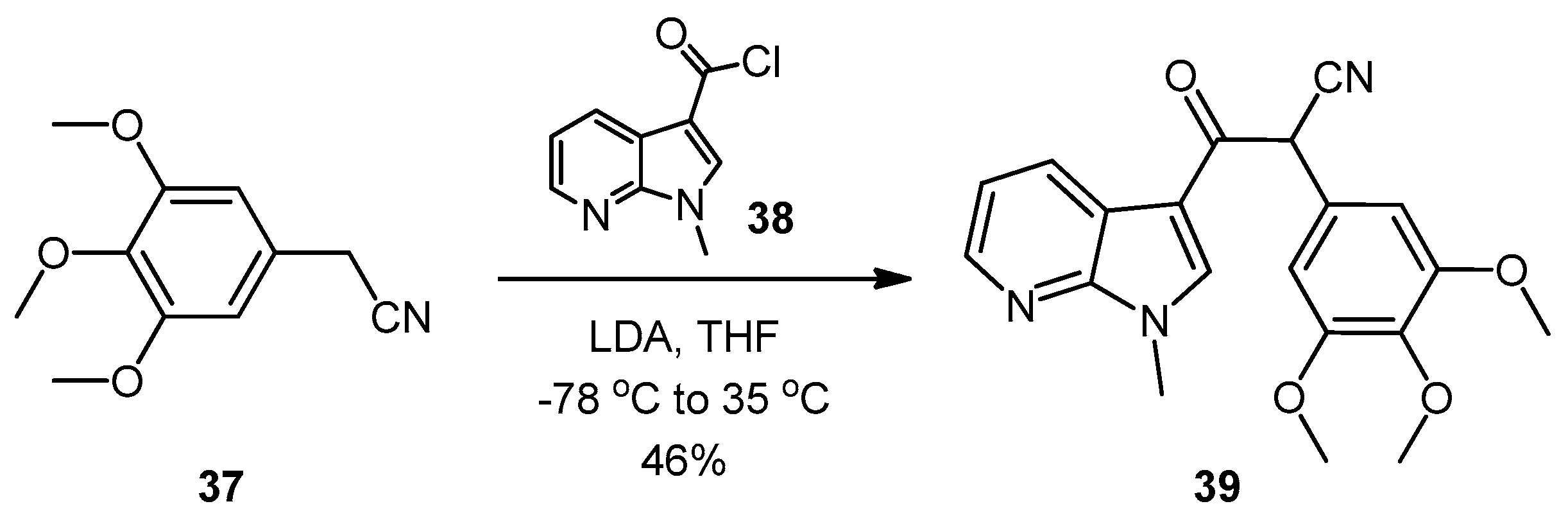
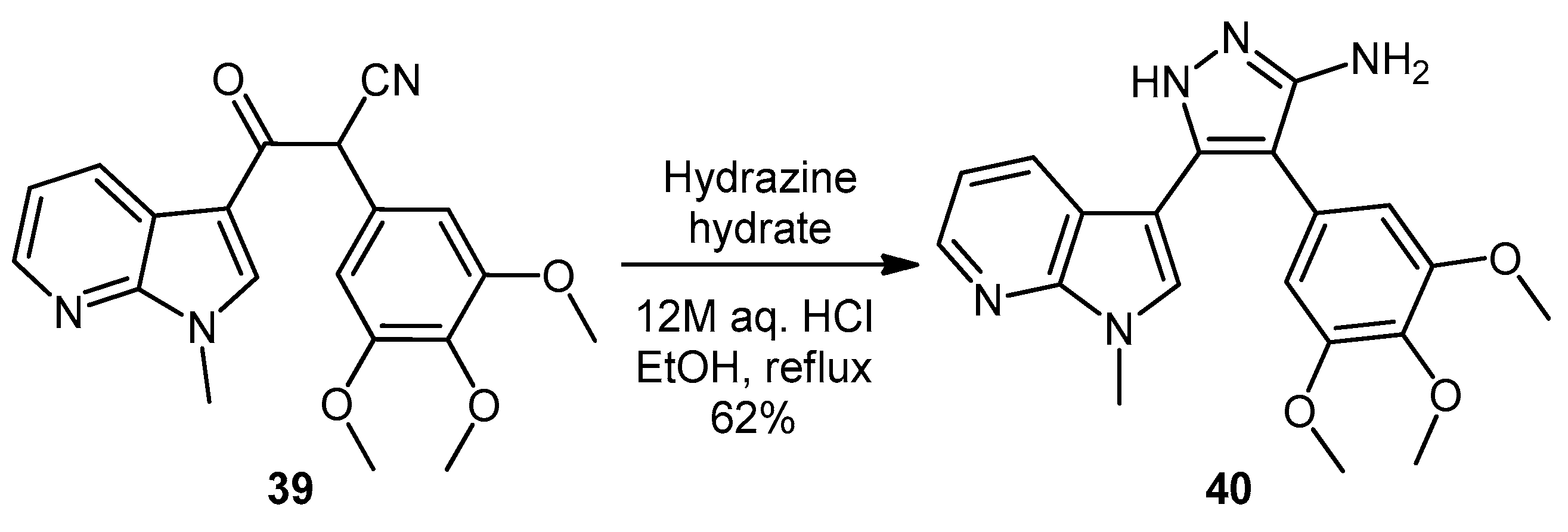
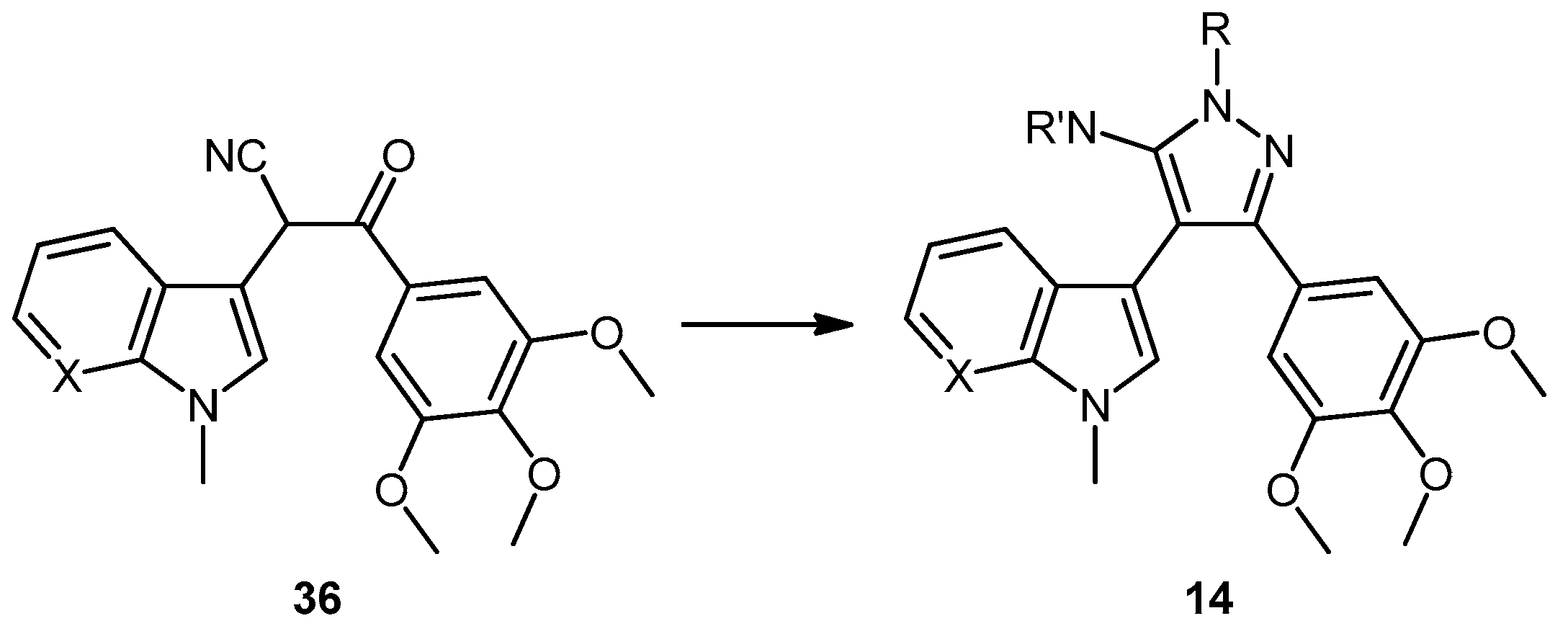
3-(1-Methyl-1H-pyrrolo[2,3-b]pyridin-3-yl)-3-oxo-2-(3,4,5-trimethoxyphenyl)propanenitrile (39). A solution of Lithium diisopropylamide (LDA) (1.8 M, 9.8 mL, 17.7 mmol) in THF (10 mL) was then cooled to −78 °C under a nitrogen atmosphere. A second solution of 3,4,5-trimethoxyphenyl acetonitrile (37, 1.36 g, 6.5 mmol) in THF (5 mL) was then added in a dropwise manner, with the resulting mixture stirred for 1 h at −78 °C. A suspension of 1-methyl-1H-pyrrolo[2,3-b]pyridine-3-carbonyl chloride (38, 1.40 g, 7.2 mmol) in THF (25 mL) was slowly added to the reaction mixture over a 20-min period. Once the addition was complete, the reaction mixture was warmed to 35 °C and allowed to stir at this temperature, still under an inert atmosphere, for 12 h. The solvent was evaporated under reduced pressure, and the resultant residue was dissolved in ethyl acetate (40 mL). The organic phase was washed with a saturated aqueous sodium bicarbonate solution (50 mL), water (50 mL) and then brine (50 mL), before being dried over magnesium sulphate. The solvent was removed under reduced pressure, and the crude residue was subjected to flash column chromatography (hexane/ethyl acetate, 30:70) to yield oxopropanenitrile 39 as a yellow solid (1.09 g, 46%): m.p. 137–140 °C; vmax/cm−1 (KBr) 2933, 2243, 1728, 1643, 1595, 1450, 1130; δH (400 MHz, CDCl3) 3.83 [3H, s, p-OCH3], 3.87 [6H, s, 2× m-OCH3], 3.94 [3H, s, NCH3], 5.23 [1H, s, CHα], 6.70 [2H, s, C-H2′6′], 7.28 [1H, q, J 7.9, 4.8, C-H5], 7.97 [1H, s, C-H2], 8.43 [1H, dd, J 4.8, 1.6, C-H6], 8.60 [1H, dd, J 7.9, 1.6, C-H4]; δC (75 MHz, CDCl3) 32.3 (CH3, NCH3), 47.8 (CH, CHCN), 56.4 (2CH3, 2× m-OCH3), 60.9 (CH3, p-OCH3), 105.1 (2CH, 2× aromatic CH), 111.4 (C, aromatic C), 117.3 (C, aromatic C), 119.3 (C, C≡N), 119.4 (CH, aromatic CH), 126.5 (C, aromatic C), 131.2 (CH, aromatic CH), 136.0 (CH, aromatic CH), 138.5 (C, aromatic C), 145.4 (CH, aromatic CH), 148.2 (C, aromatic C), 154.0 (2C, 2× aromatic C), 182.6 (C, C=O); m/z (ES+) 364.1 [M−H]− (100%); HRMS (ES+): exact mass calculated for C20H20N3O4 366.1454. Found 366.1472.
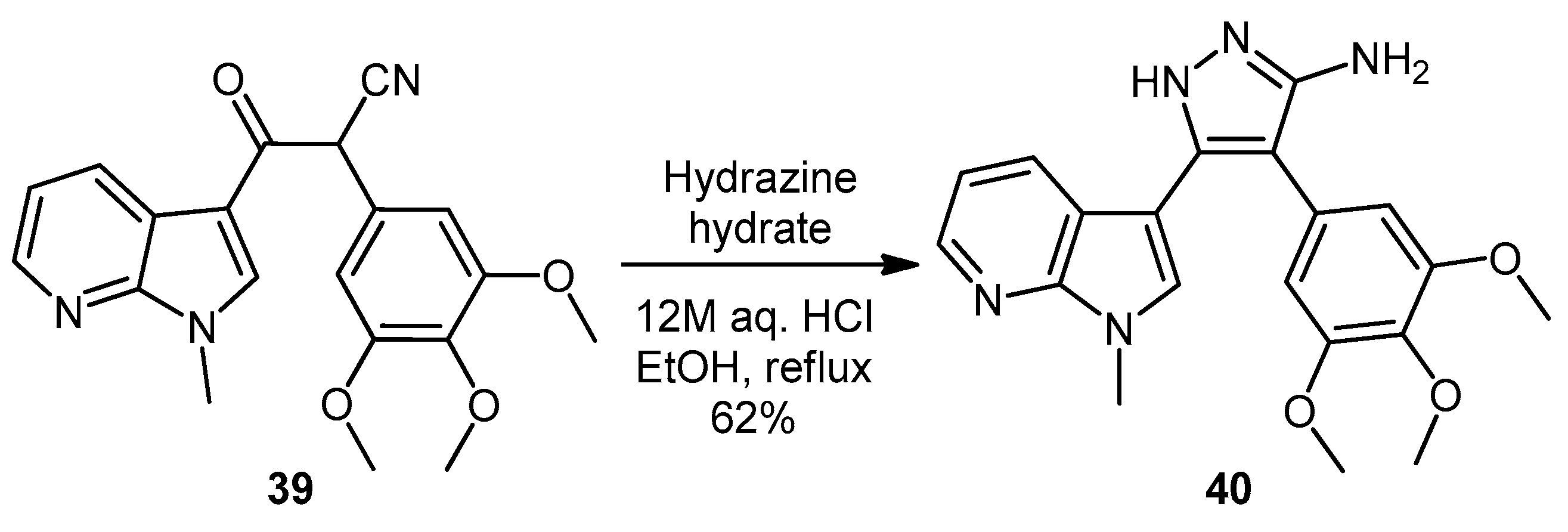
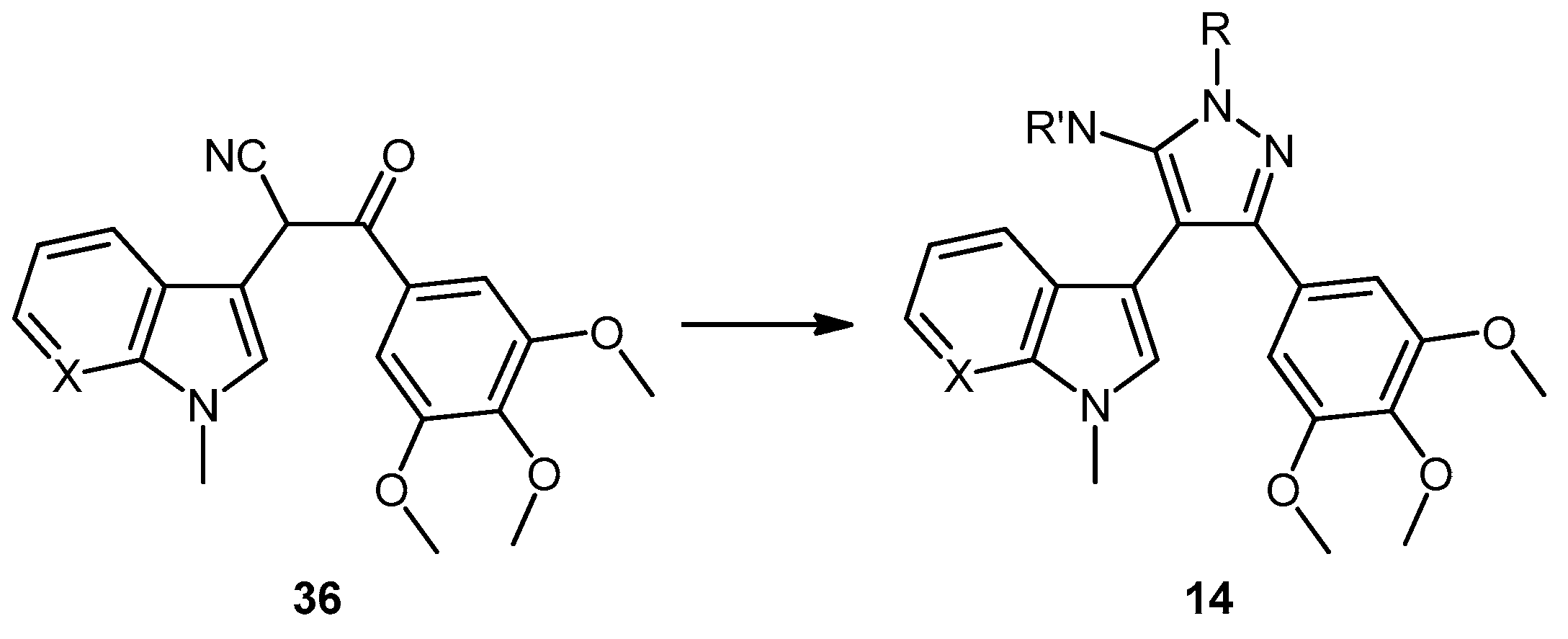
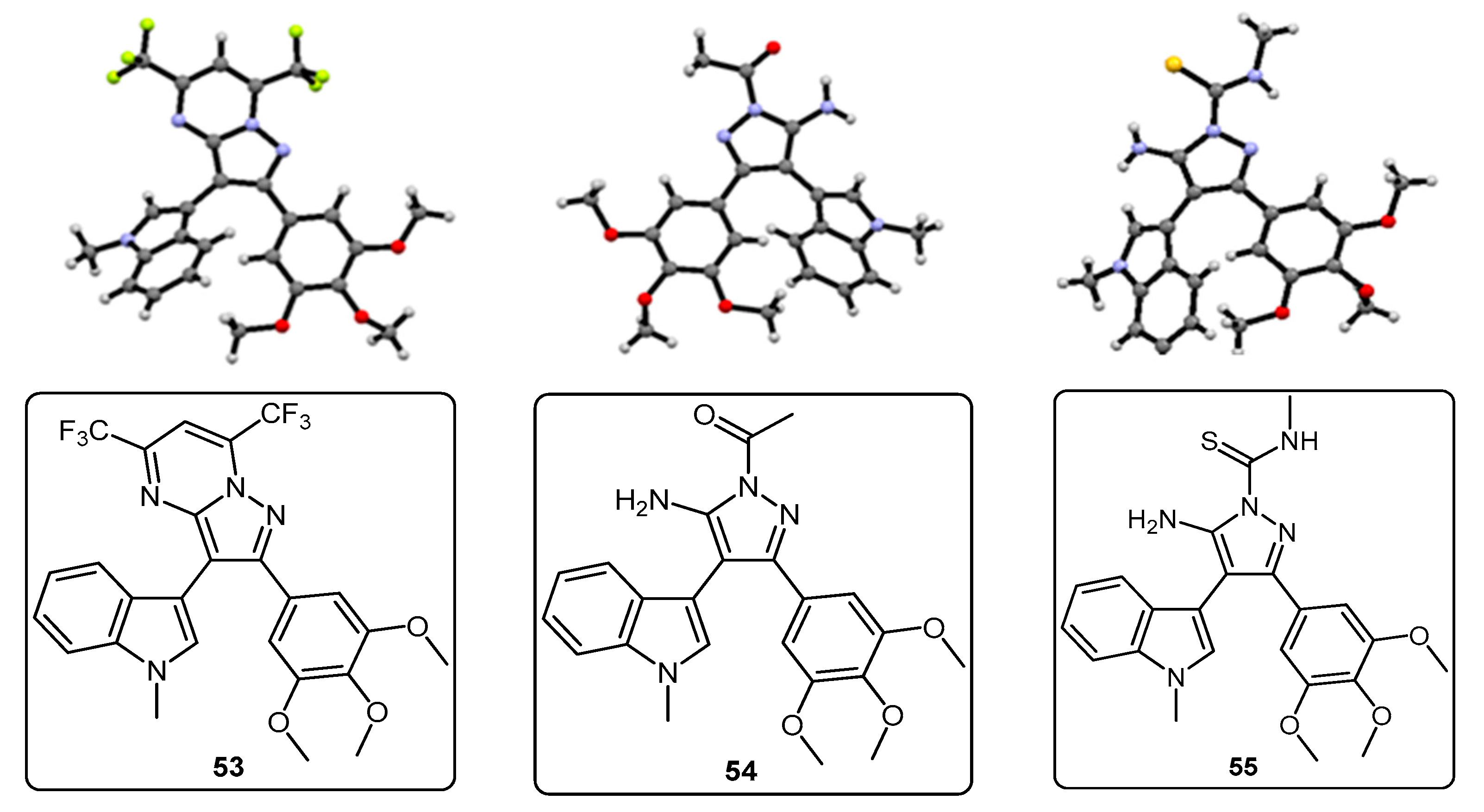
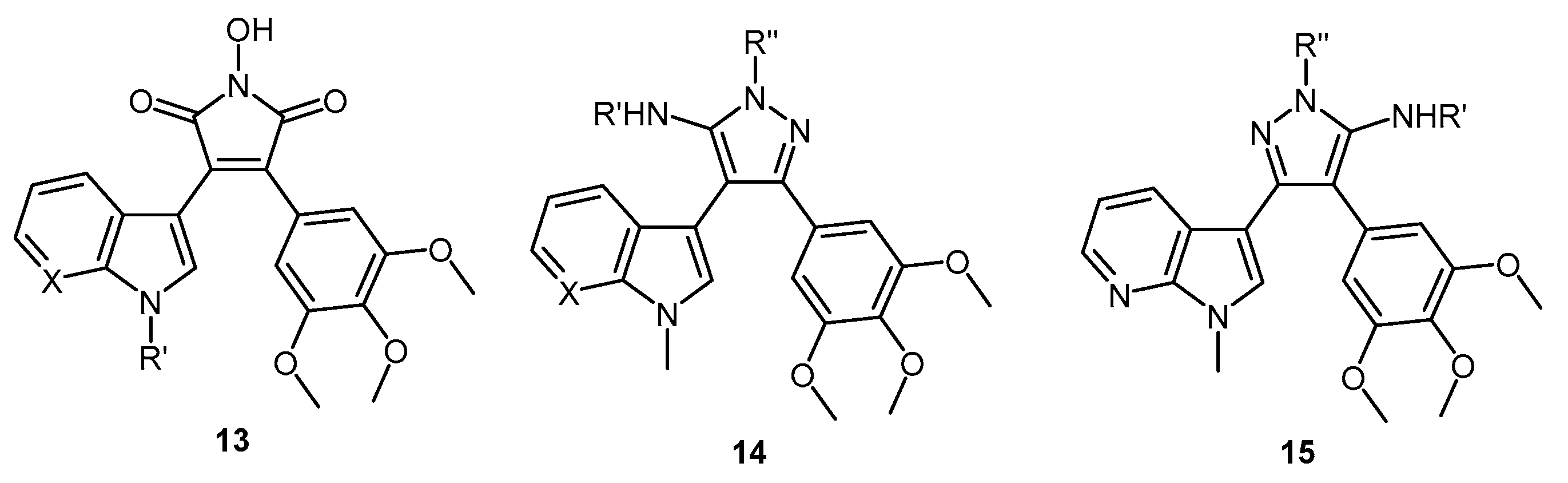
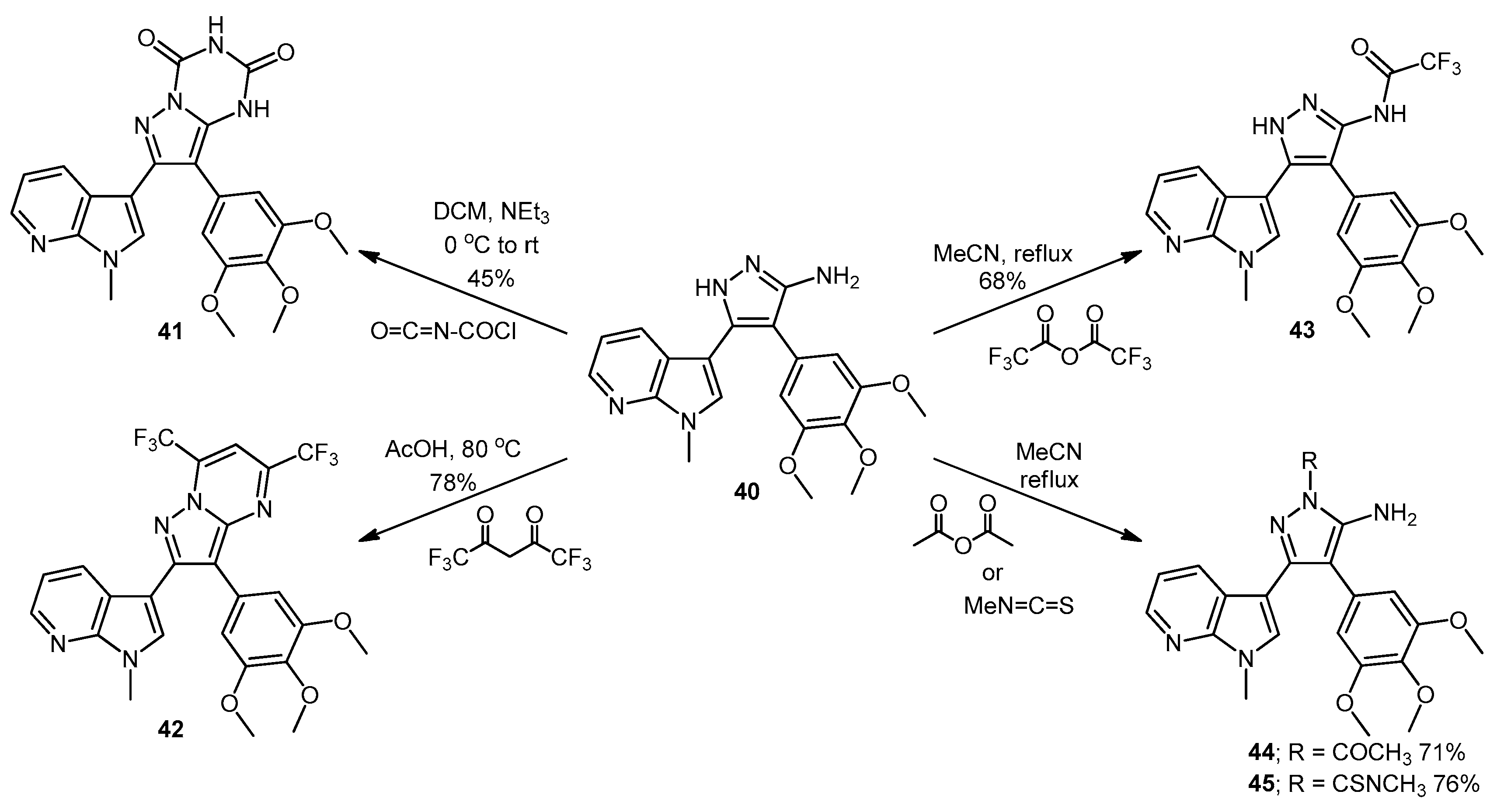
5-(1-Methyl-1H-pyrrolo[2,3-b]pyridin-3-yl)-4-(3,4,5-trimethoxyphenyl)-1H-pyrazol-3-amine (40). To a solution of 3-(1-methyl-1H-pyrrolo[2,3-b]pyridin-3-yl)-3-oxo-2-(3,4,5-trimethoxyphenyl)propanenitrile (39, 1.02 g, 2.8 mmol) in absolute alcohol (30 mL) was added 12 M aqueous HCl (1 mL) in a dropwise manner. Hydrazine hydrate (50% aqueous solution, 0.87 mL, 13.9 mmol) was then added, and the resulting mixture was heated to reflux for 26 h. Once cooled to room temperature, the solvent was evaporated under reduced pressure. The residue was dissolved in ethyl acetate (50 mL), then washed successively with saturated aqueous sodium bicarbonate solution (40 mL), water (40 mL) and then brine (40 mL), before being dried over magnesium sulphate. The solvent was removed under reduced pressure, and the crude brown residue was subjected to flash column chromatography (ethyl acetate/methanol, 90:10) to yield pure aminopyrazole 40 as a light brown solid (0.66 g, 62%): m.p. 135–137 °C; vmax/cm−1 (KBr) 3199, 2931, 1602, 1518, 1470, 1351, 1115; δH (300 MHz, DMSO-d6) 3.53 [6H, s, 2× m-OCH3], 3.64 [3H, s, p-OCH3], 3.82 [3H, s, NCH3], 4.68 [2H, bs, NH2], 6.53 [2H, s, C-H2′6′], 6.98 [1H, q, J 7.9, 4.6, C-H5], 7.55 [2H, m, C-H2,4], 8.24 [1H, dd, J 4.6, 1.3, C-H6], 11.73 [1H, bs, NH]; δC (75 MHz, DMSO-d6) 30.8 (C, NCH3), 55.5 (2C, 2× m-OCH3), 60.0 (C, p-OCH3), 94.9 (C, broad aromatic C), 106.1 (2CH, 2× aromatic CH), 115.5 (CH, aromatic CH), 117.9 (C, aromatic C), 125.4 (C, broad aromatic C), 128.2 (CH, aromatic CH), 128.8 (CH, aromatic CH), 129.4 (C, aromatic C), 135.4 (C, aromatic C), 141.9 (C, broad aromatic C), 142.7 (CH, aromatic CH), 147.2 (C, aromatic C), 151.8 (C, aromatic C), 152.7 (2C, 2× aromatic C); m/z (ES+) 380.2 [M + H]+ (100%); HRMS (ES+): exact mass calculated for C20H22N5O3 380.1723. Found 380.1724.
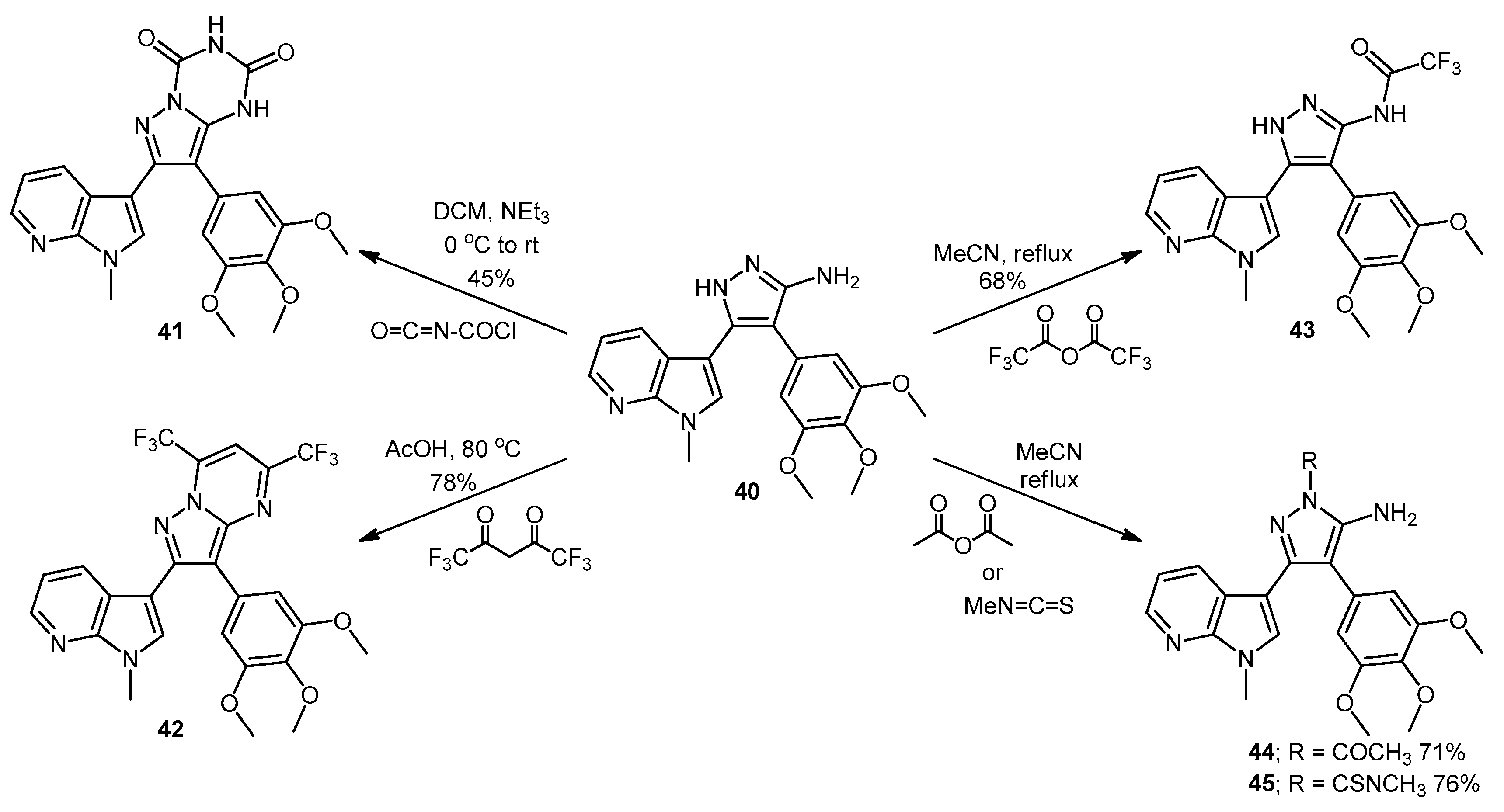
7-(1-Methyl-1H-pyrrolo[2,3-b]pyridin-3-yl)-8-(3,4,5-trimethoxyphenyl)pyrazolo[1,5-a][1,3,5]triazine-2,4(1H,3H)-dione (41). 5-(1-Methyl-1H-pyrrolo[2,3-b]pyridin-3-yl)-4-(3,4,5-trimethoxyphenyl)-1H-pyrazol-3-amine (40, 100 mg, 0.26 mmol) was dissolved in DCM (5 mL) containing three drops of triethylamine, and the resulting solution was cooled to 0 °C in an ice bath. This solution was then treated, while stirring, with N-chlorocarbonyl isocyanate (0.03 mL, 0.29 mmol). The reaction mixture was subsequently allowed to warm to room temperature and was stirred for 15 h. After careful addition of water (3 mL), a solid precipitated, which was filtered off and then washed with water (4× 10 mL), followed by ether (2× 10 mL). The precipitate was desiccated at 50 °C overnight to give the desired pyrazolotriazinedione 41 as a white solid (51 mg, 45%): m.p. 284–285 °C; vmax/cm−1 (KBr) 3215, 3067, 1761, 1713, 1647, 1586, 1411, 1127; δH (300MHz, DMSO-d6) 3.72 [6H, s, 2× m-OCH3], 3.74 [3H, s, p-OCH3], 3.78 [3H, s, NCH3], 6.64 [2H, s, C-H2′,6′], 7.18 [1H, q, J 7.8, 4.9, C-H5], 7.31 [1H, s, C-H2], 8.32-8.34 [2H, m, C-H4,6], 11.59 [1H, bs, C=C-NHCO], 11.74 [1H, bs, CONHCO]; δC (75 MHz, DMSO-d6) 31.0 (CH3, NCH3), 55.8 (2CH3, 2× m-OCH3), 60.1 (CH3, p-OCH3), 102.0 (C, aromatic C), 104.8 (C, aromatic C), 107.9 (2CH, 2× aromatic CH), 116.5 (CH, aromatic CH), 118.0 (C, aromatic C), 124.9 (C, aromatic C), 129.5 (CH, aromatic CH), 130.0 (CH, aromatic CH), 137.2 (C, aromatic C), 138.1 (C, aromatic C), 143.3 (CH, aromatic CH), 144.1 (C, aromatic C), 147.2 (C, aromatic C), 148.7 (C, C=O), 149.7 (C, C=O), 153.0 (2C, 2× aromatic C); m/z (ES+) 447.2 [M−H]− (100%); HRMS (ES+): exact mass calculated for C22H21N6O5 449.1573. Found 449.1587.
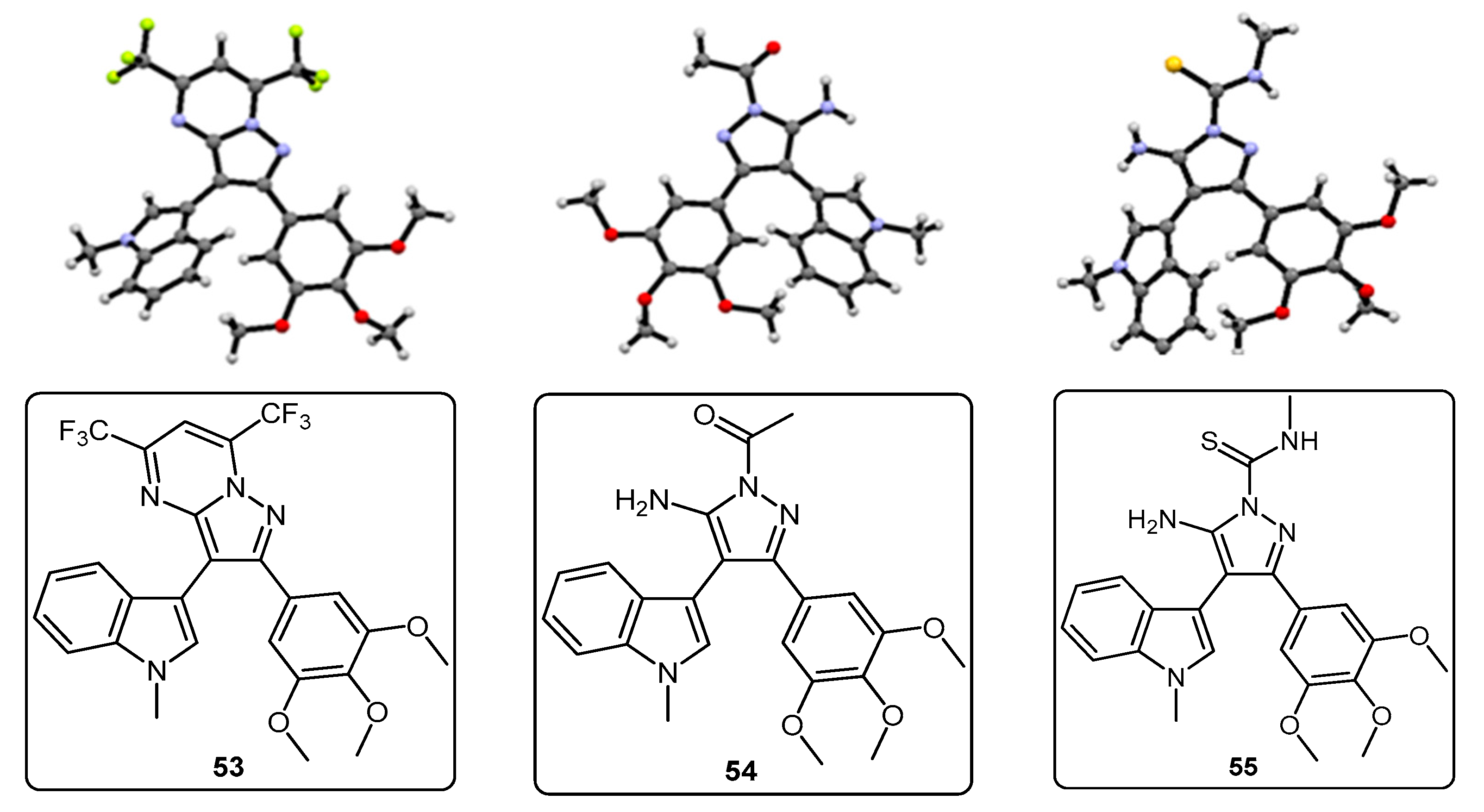
2-(1-Methyl-1H-pyrrolo[2,3-b]pyridin-3-yl)-5,7-bis(trifluoromethyl)-3-(3,4,5-trimethoxyphenyl)pyrazolo[1,5-a]pyrimidine (42). To a stirred solution of 5-(1-methyl-1H-pyrrolo[2,3-b]pyridin-3-yl)-4-(3,4,5-trimethoxyphenyl)-1H-pyrazol-3-amine (40, 60 mg, 0.16 mmol) in acetic acid (5 mL), was added hexafluoroacetylacetone (0.44 mL, 3.2 mmol). The mixture was then heated to 80 °C for 22 h. Once cooled to room temperature, the excess solvent was removed under reduced pressure. The residue was dissolved in ethyl acetate (20 mL), then washed with water (20 mL) and brine (20 mL), before being dried over magnesium sulphate. The solvent was removed under reduced pressure, and the crude residue was subjected to flash column chromatography (hexane/ethyl acetate, 60:40) to yield 42 as an orange solid (69 mg, 78%): m.p. 176–178 °C; vmax/cm−1 (KBr) 2923, 2841, 1586, 1560, 1509, 1279, 1131; δH (300MHz, CDCl3) 3.71 [6H, s, 2× m-OCH3], 3.81 [3H, s, p-OCH3], 3.88 [3H, s, NCH3], 6.82 [2H, s, C-H2′6′], 7.11 [1H, q, J 8.0, 4.8, C-H5], 7.33 [1H, s, C-H2], 7.44 [1H, s, CHCCF3], 8.33-8.38 [2H, m, C-H4,6]; δC (75 MHz, CDCl3) 30.6 (CH3, NCH3), 55.1 (2CH3, 2× m-OCH3), 60.0 (CH3, p-OCH3), 100.4 (CH, aromatic CH), 104.3 (C, aromatic C), 106.2 (2CH, 2× aromatic CH), 110.0 (C, aromatic C), 116.2 (CH, aromatic CH), 112.8-123.7 (C, q, JF-C 274.9, CF3), 113.8-124.8 (C, q, JF-C 274.6, CF3), 117.8 (C, aromatic C), 124.5 (C, aromatic C), 129.1 (CH, aromatic CH), 129.7 (CH, aromatic CH), 132.8-134.4 (C, q, JF-C 39.0, aromatic C), 137.0 (C, aromatic C), 143.0 (CH, aromatic CH), 143.6-145.1 (C, q, JF-C 38.2, aromatic C), 145.3 (C, aromatic C), 147.0 (C, aromatic C), 151.8 (C, aromatic C), 152.6 (2C, 2× aromatic C); m/z (ES+) 552.2 [M + H]+ (100%); HRMS (ES+): exact mass calculated for C25H20N5O3F6 552.1470. Found 552.1493.
2,2,2-Trifluoro-N-(5-(1-methyl-1H-pyrrolo[2,3-b]pyridin-3-yl)-4-(3,4,5-trimethoxyphenyl)-1H-pyrazol-3-yl)acetamide (43). To a solution of 5-(1-methyl-1H-pyrrolo[2,3-b]pyridin-3-yl)-4-(3,4,5-trimethoxyphenyl)-1H-pyrazol-3-amine (40, 60 mg, 0.16 mmol) in acetonitrile (10 mL) was added trifluoroacetic anhydride (0.03 mL, 0.19 mmol). The resulting mixture was heated to reflux for 28 h. Once cooled to room temperature, the solvent was evaporated under reduced pressure, and the crude brown residue was subjected to flash column chromatography (hexane/ethyl acetate, 30:70), yielding trifluoroacetamide 43 as an-off white solid (52 mg, 68%): m.p. 129–131 °C; vmax/cm−1 (KBr) 3215, 3067, 1713, 1603, 1586, 1411; δH (300 MHz, DMSO-d6) 3.49 [6H, s, 2× m-OCH3], 3.66 [3H, s, p-OCH3], 3.91 [3H, s, NCH3], 6.53 [2H, s, C-H2′,6′], 7.03 [1H, q, J 7.9, 4.6, C-H5], 7.32 [1H, d, J 6.5, C-H4], 7.86 [1H, s, C-H2], 8.31 [1H, d, J 3.5, C-H6], 11.37 [1H, bs, NHCOCF3], 13.30 [1H, bs, C=N-NH-C]; δC (75 MHz, DMSO-d6) 31.0 (CH3, NCH3), 55.4 (2CH3, 2× m-OCH3), 60.0 (CH3, p-OCH3), 102.0 (C, aromatic C), 105.8 (2CH, 2× aromatic CH), 110.2-121.6 (C, q, JF-C 288.0, CF3), 113.9 (C, aromatic C), 115.9 (CH, aromatic CH), 117.3 (C, aromatic C), 127.1 (C, aromatic C), 128.2 (CH, aromatic CH), 129.4 (CH, aromatic CH), 136.2 (C, aromatic C), 143.1 (CH, aromatic CH), 147.2 (C, aromatic C), 152.7 (2C, 2× aromatic C), 152.9 (C, aromatic C), 155.8-157.2 (C, q, JF-C 36.2, C=O); m/z (ES+) 474.3 [M−H]− (100%); HRMS (ES+): exact mass calculated for C22H21N5O4F3 476.1546. Found 476.1532.
1-(5-Amino-3-(1-methyl-1H-pyrrolo[2,3-b]pyridin-3-yl)-4-(3,4,5-trimethoxyphenyl)-1H-pyrazol-1-yl)ethanone (44). To a solution of 5-(1-methyl-1H-pyrrolo[2,3-b]pyridin-3-yl)-4-(3,4,5-trimethoxyphenyl)-1H-pyrazol-3-amine (40, 60 mg, 0.16 mmol) in acetonitrile (10 mL) was added acetic anhydride (0.016 mL, 0.17 mmol). The resulting mixture was then heated to reflux for 20 h. Once cooled to room temperature, the solvent was removed under reduced pressure. The crude residue was purified by flash column chromatography (hexane/ethyl acetate, 50:50) to yield acetylated product 44 as an off-white solid (48 mg, 71%): m.p. 171–172 °C; vmax/cm−1 (KBr) 3454, 3358, 2927, 1704, 1624, 1509, 1404, 1124; δH (300 MHz, CDCl3) 2.80 [3H, s, NCOCH3], 3.79 [3H, s, p-OCH3], 3.79 [6H, s, 2× m-OCH3], 3.92 [3H, s, NCH3], 5.56 [2H, bs, NH2], 6.59 [2H, s, C-H2′,6′], 7.05 [1H, s, C-H2], 7.14 [1H, q, J 7.9, 4.7, C-H5], 8.36 [1H, dd, J 4.7, 1.5, C-H6], 8.47 [1H, dd, J 7.9, 1.6, C-H4]; δC (75 MHz, CDCl3) 23.3 (CH3, NCOCH3), 31.4 (CH3, NCH3), 56.2 (CH3, 2× m-OCH3), 61.0 (CH3, p-OCH3), 101.9 (C, aromatic C), 105.9 (C, aromatic C), 107.1 (CH, 2× aromatic CH), 116.6 (CH, aromatic CH), 118.8 (C, aromatic C), 127.4 (C, aromatic C), 129.3 (CH, aromatic CH), 130.7 (CH, aromatic CH), 137.5 (C, aromatic C), 143.7 (CH, aromatic CH), 147.0 (C, aromatic C), 147.9 (C, aromatic C), 148.5 (C, aromatic C), 153.9 (C, 2× aromatic C), 173.7 (C, C=O); m/z (ES+) 422.3 [M + H]+ (100%); HRMS (ES+): exact mass calculated for C22H24N5O4 422.1828. Found 422.1813.
5-Amino-N-methyl-3-(1-methyl-1H-pyrrolo[2,3-b]pyridin-3-yl)-4-(3,4,5-trimethoxyphenyl)-1H-pyrazole-1-carbothioamide (45). To a solution of 5-(1-methyl-1H-pyrrolo[2,3-b]pyridin-3-yl)-4-(3,4,5-trimethoxyphenyl)-1H-pyrazol-3-amine (40, 60 mg, 0.16 mmol) in acetonitrile (10 mL) was added methyl isothiocyanate (14 mg, 0.19 mmol). The mixture was heated to reflux for 22 h, before then being cooled to room temperature. The solvent was evaporated under reduced pressure, and the crude residue was subjected to flash column chromatography (hexane/ethyl acetate, 50:50), yielding pure thiourea 45 as a white solid (55 mg, 76%): m.p. 162–164 °C; vmax/cm−1 (KBr) 3378, 2931, 1601, 1523, 1406, 1362, 1129; δH (300 MHz, CDCl3) 3.33 [3H, d, J 5.0, NHCH3], 3.78 [6H, s, 2× m-OCH3], 3.80 [3H, s, p-OCH3], 3.92 [3H, s, NCH3], 6.47 [2H, bs, NH2], 6.59 [2H, s, C-H2′,6′], 7.08 [1H, s, C-H2], 7.12 [1H, q, J 7.9, 4.7, C-H5], 8.27 [1H, dd, J 7.9, 1.6, C-H4], 8.36 [1H, dd, J 4.7, 1.5, C-H6], 9.24 [1H, bd, J 4.7, NHCH3]; δC (75 MHz, CDCl3) 30.9 (CH3, NHCH3), 31.4 (CH3, NCH3), 56.2 (2CH3, 2× m-OCH3), 61.0 (CH3, p-OCH3), 102.5 (C, aromatic C), 105.4 (C, aromatic C), 107.2 (2CH, 2× aromatic CH), 116.5 (CH, aromatic CH), 118.6 (C, aromatic C), 127.6 (C, aromatic C), 129.7 (CH, aromatic CH), 130.2 (CH, aromatic CH), 137.5 (C, aromatic C), 143.7 (CH, aromatic CH), 145.7 (C, aromatic C), 147.8 (C, aromatic C), 148.1 (C, aromatic C), 153.9 (2C, 2× aromatic C), 176.8 (C, C=S); m/z (ES+) 453.2 [M + H]+ (100%); HRMS (ES+): exact mass calculated for C22H25N6O3S 453.1709. Found 453.1718.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}