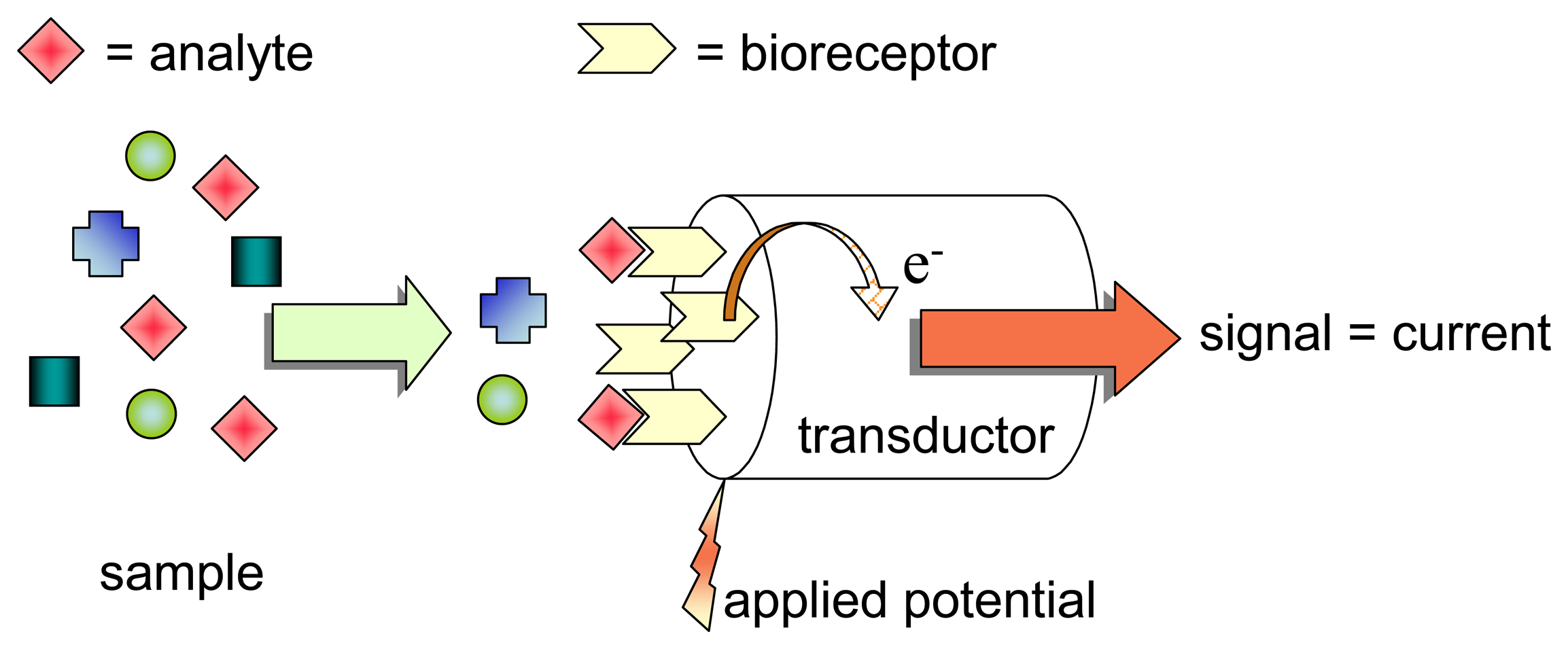
Assembling Amperometric Biosensors for Clinical Diagnostics

Abstract
:1. Introduction
2. Selectivity-Specificity
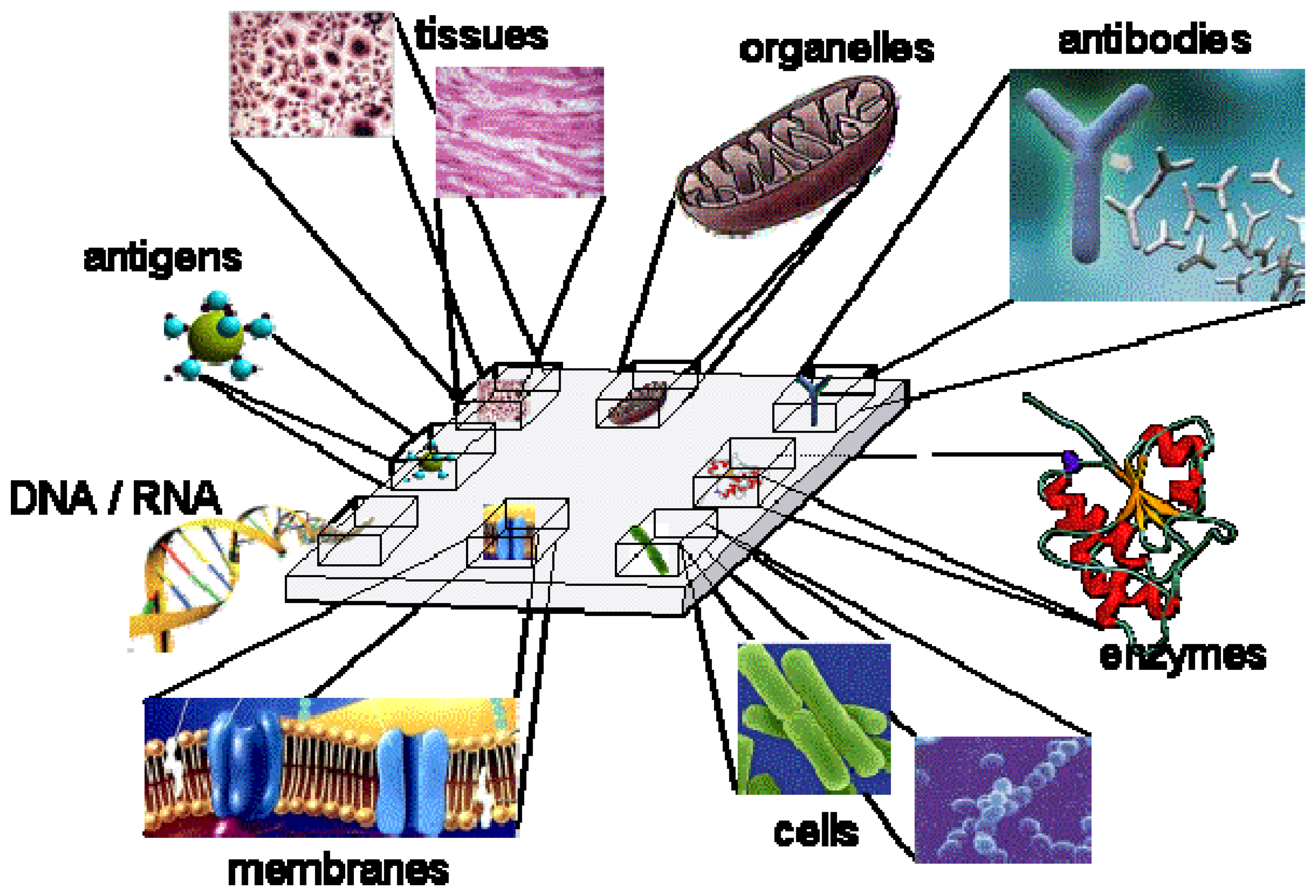
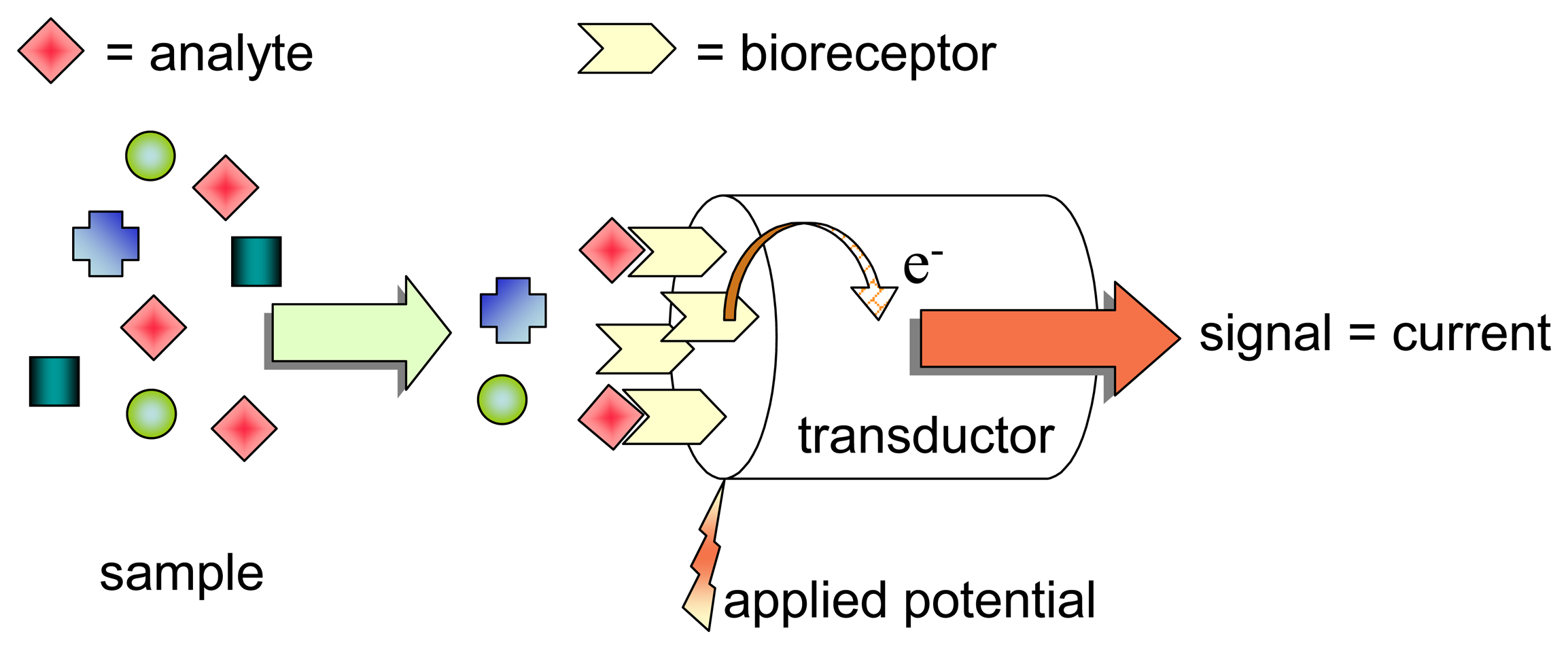
2.1. Choosing the bioreceptor system
2.1.1. Enzymes



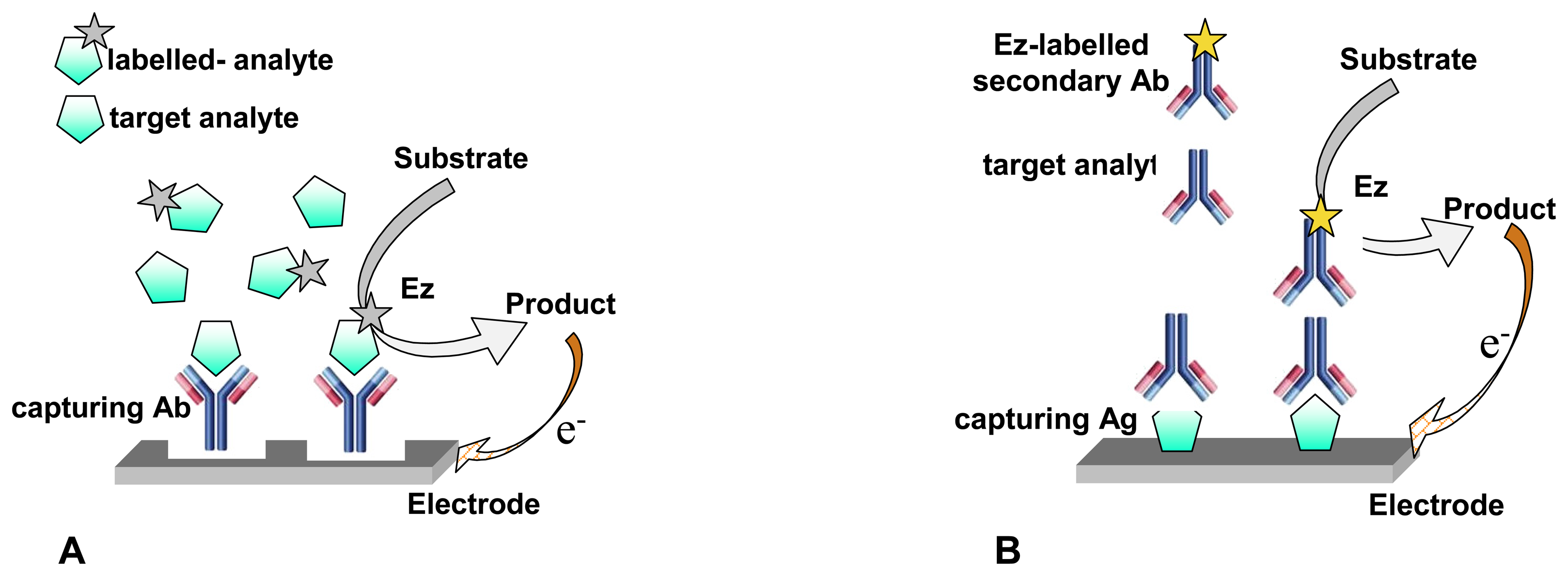
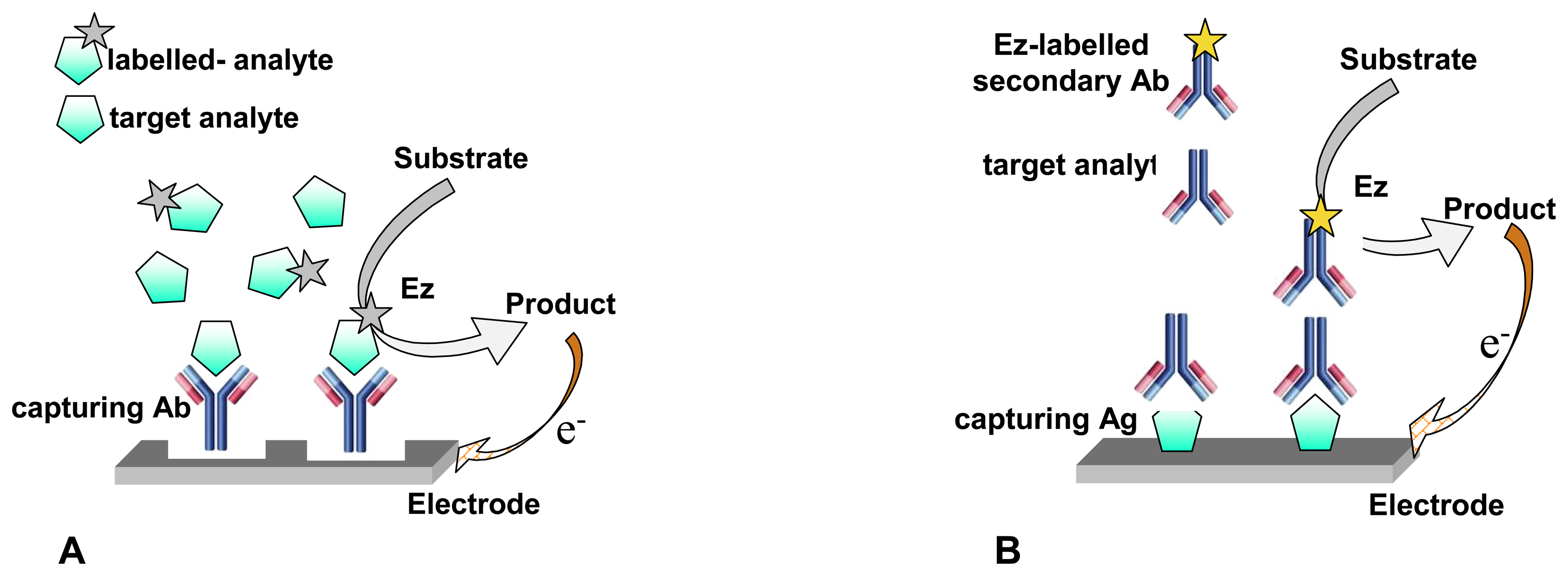
2.1.2. Antibodies, antigens & aptamers
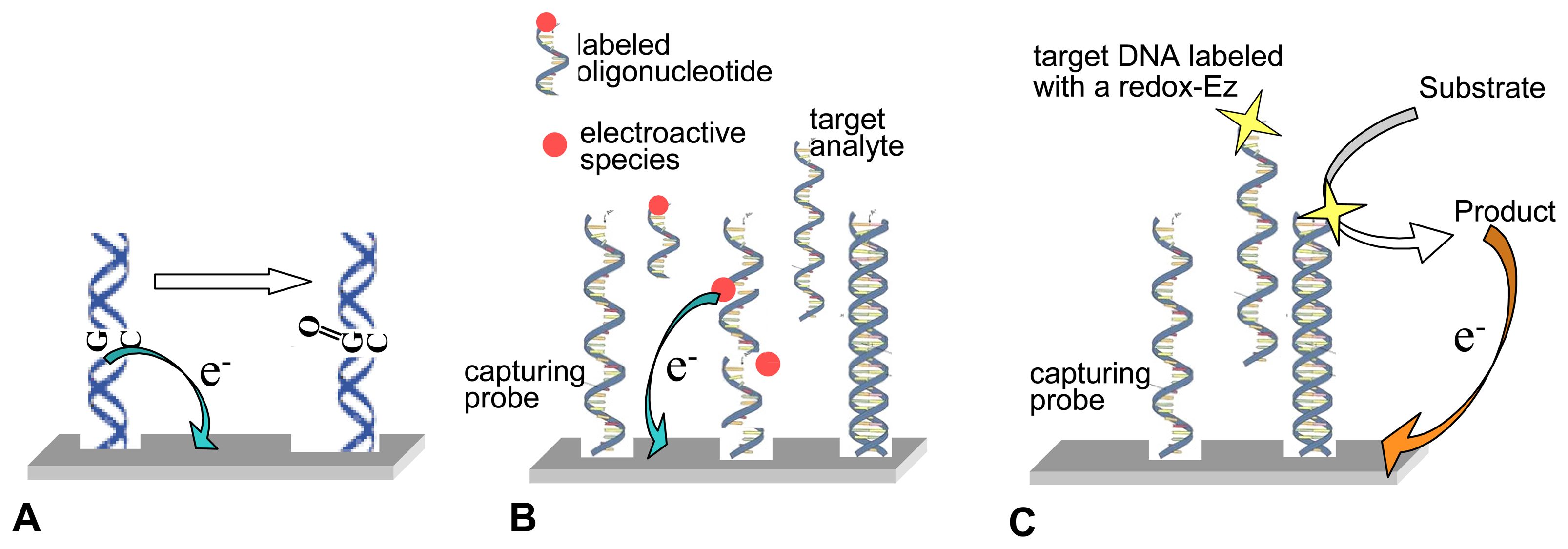
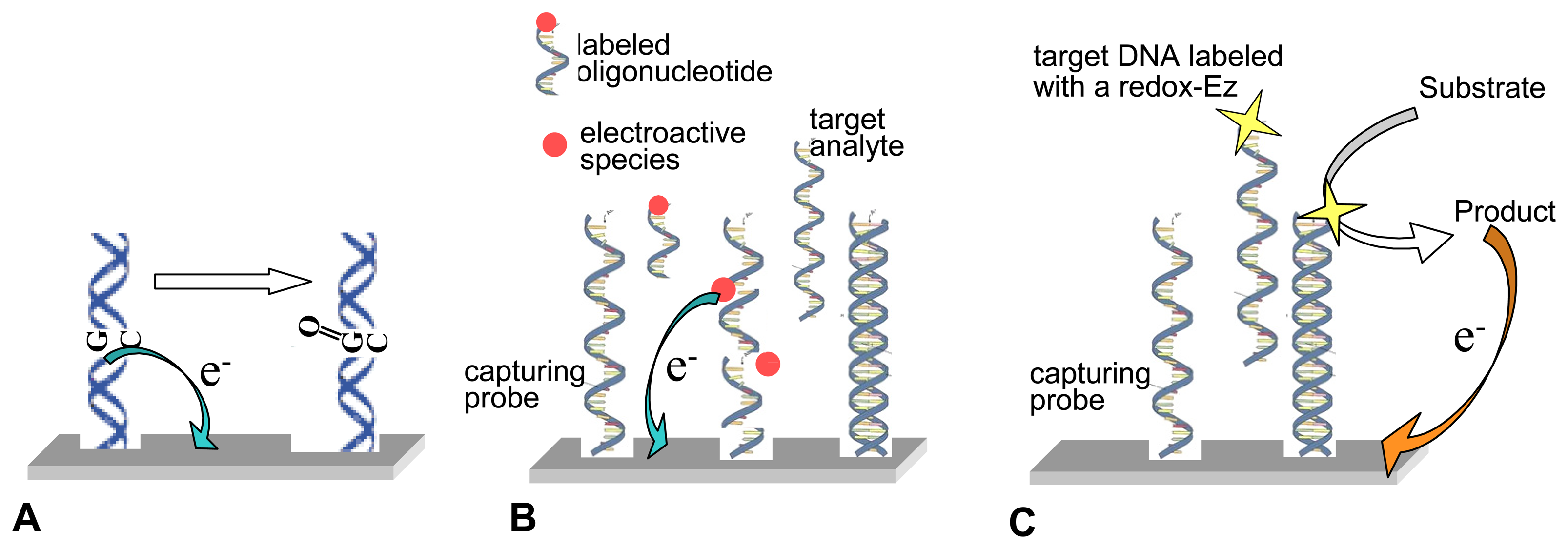
2.1.3. Nucleic acids
2.2. Avoiding interfering reactions
2.3. Preserving the bioreceptor layer
3. Sensitivity
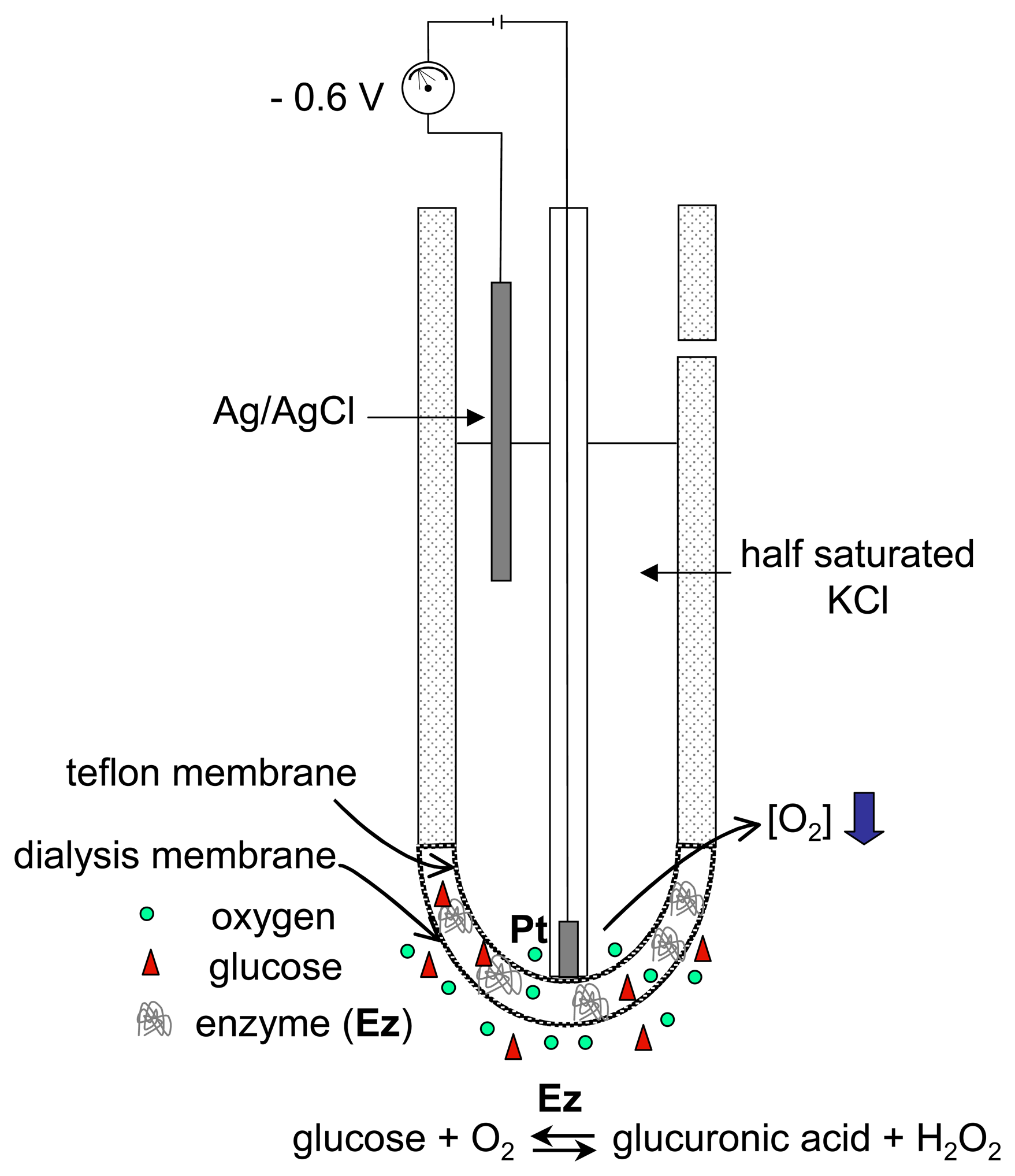
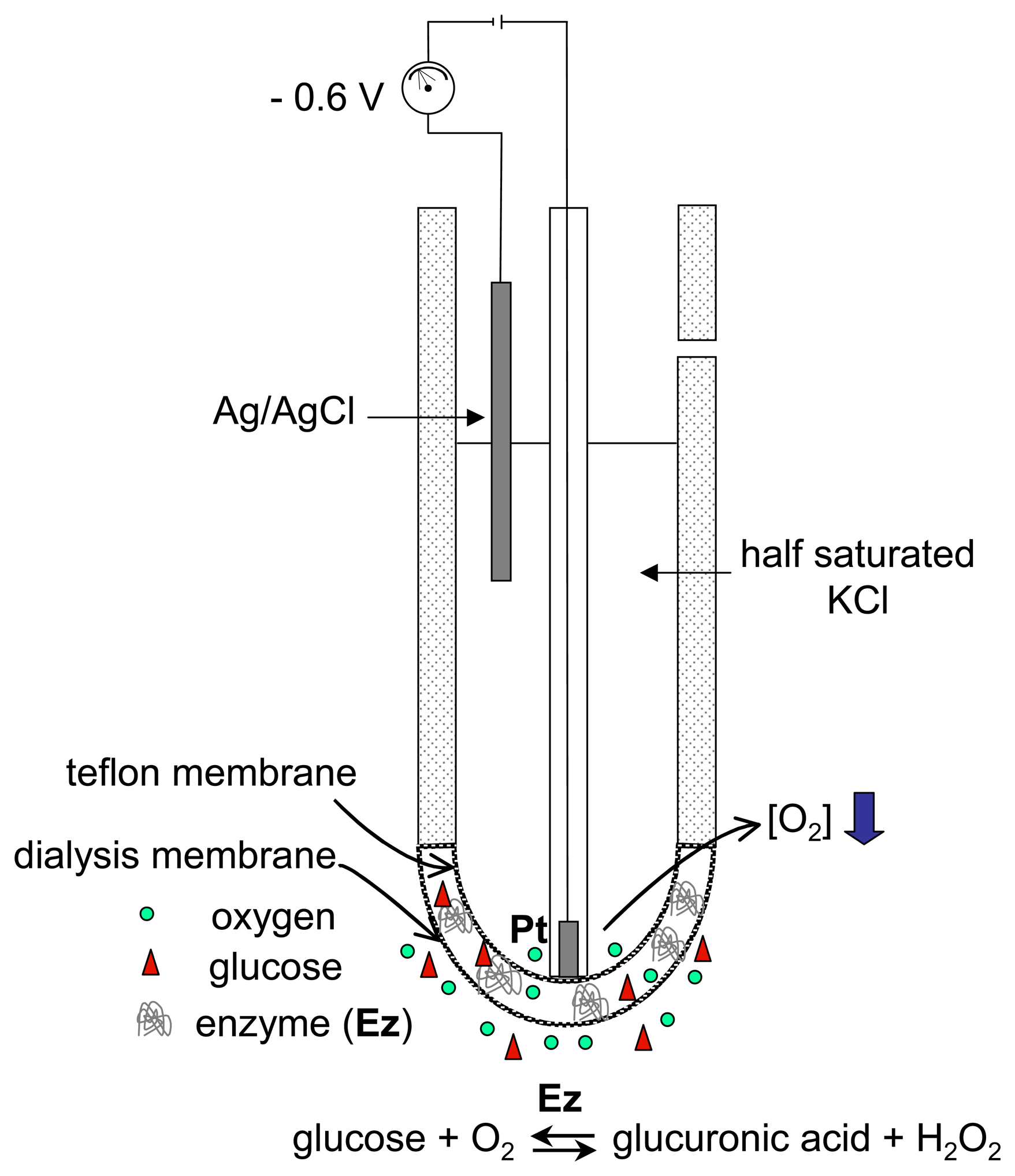
3.1. Magnifying the electron-transfer process
- (i)
- Recycling the electrochemically-transformed compound. This has already been described above in item 2.2 when considering catalytic enzymatic cycles involving the regeneration of the active-site of the redox enzyme using a charge transfer mediator that is then electro-transformed at the electrode surface (see Fig. 7). Usually, in these cases, the enzyme substrate should be present at excess concentration to keep the enzymatic cycle. The signal that evidences the occurrence of the enzymatic cycle is generally related to the presence (or absence) of the analyte in the sample when performing an indirect (or competitive) immunoassay with electrochemical detection, as described previously in point 2.1.2.
- (ii)
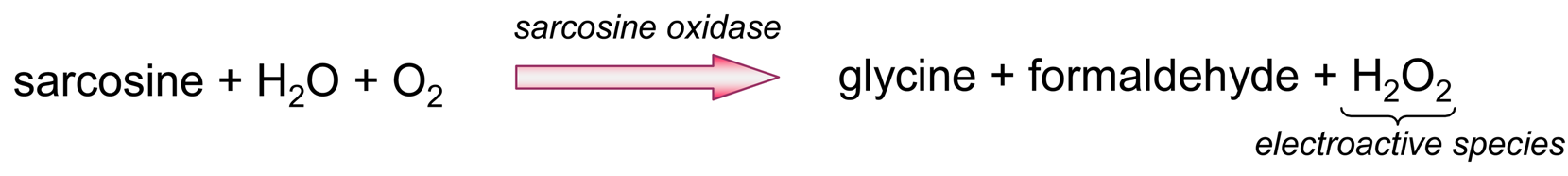
- Amplifying the electron exchange process at the electrode surface using coupled enzymatic systems. In this approach, the analyte (or one analyte-related species), is the substrate of the first enzyme (Ez-1). The analyte and the Ez-1 co-substrate, which is the electro-active species, react with Ez1 to render the products, one of which is the Ez-2 substrate (Fig. 8). Ez-2 reacts with its substrate to regenerate the analyte (or the analyte-related species), thus enabling its recycling to keep consuming Ez-1 co-substrate, this latter one being electrochemically detected. Therefore, even tiny amounts of the analyte can be detected because the consumption of the electroactive species is supported by the cyclic reaction of both enzymes.
- (iii)
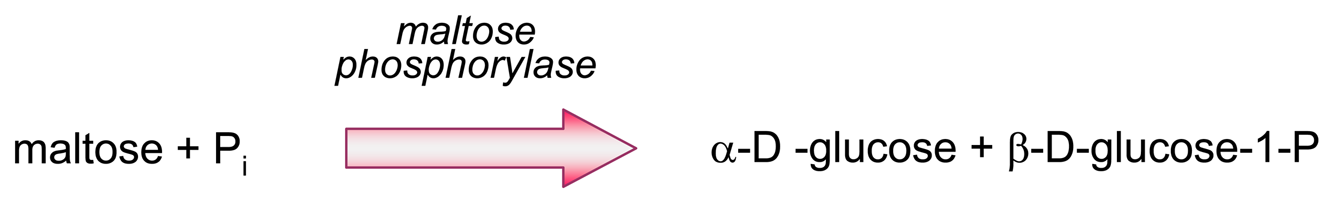
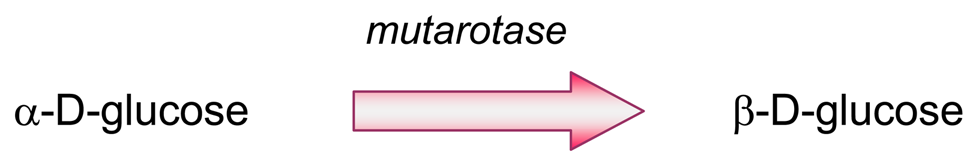
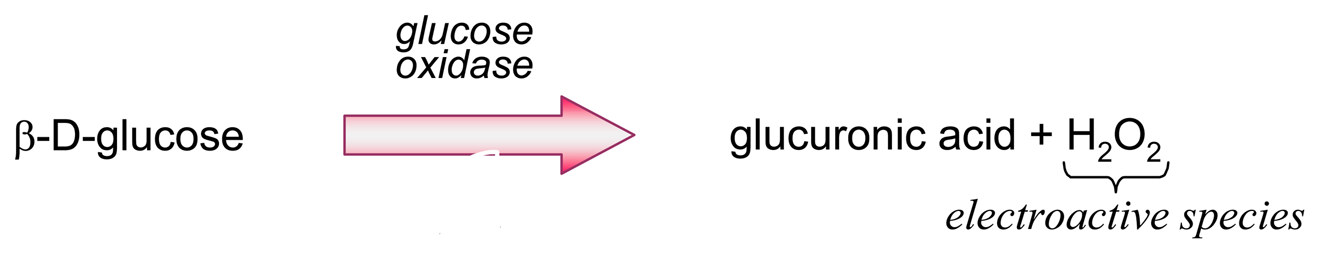
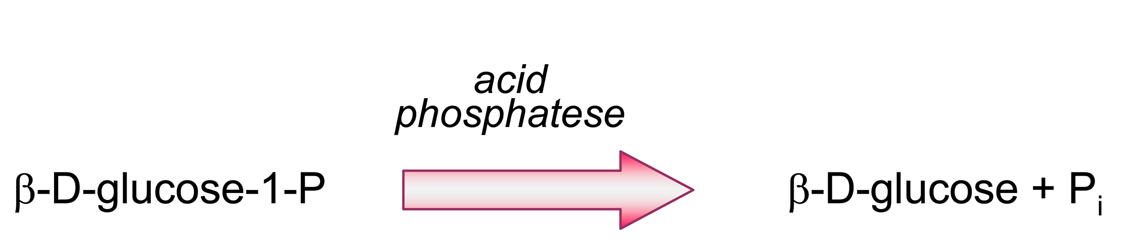
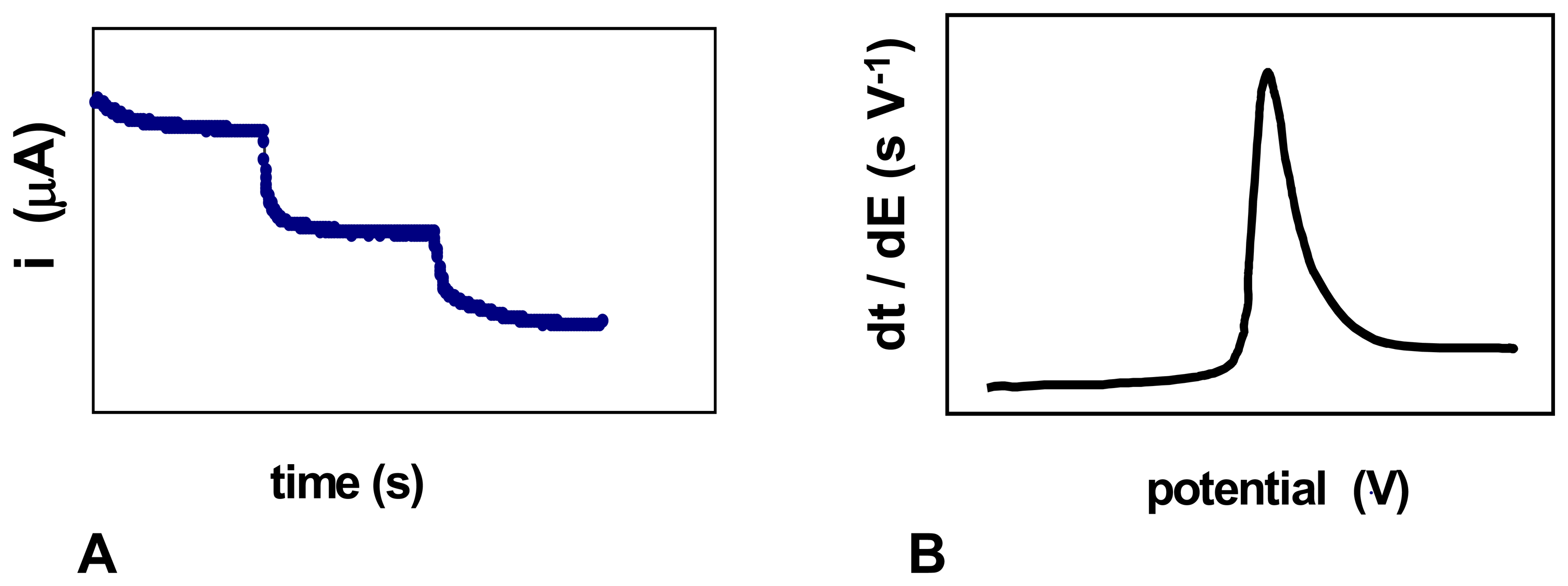
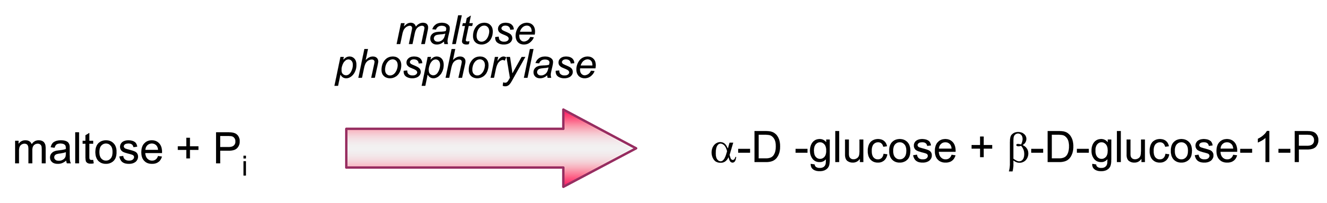
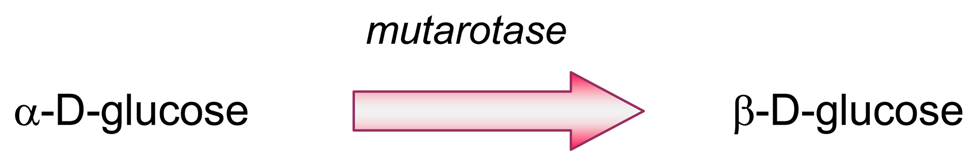
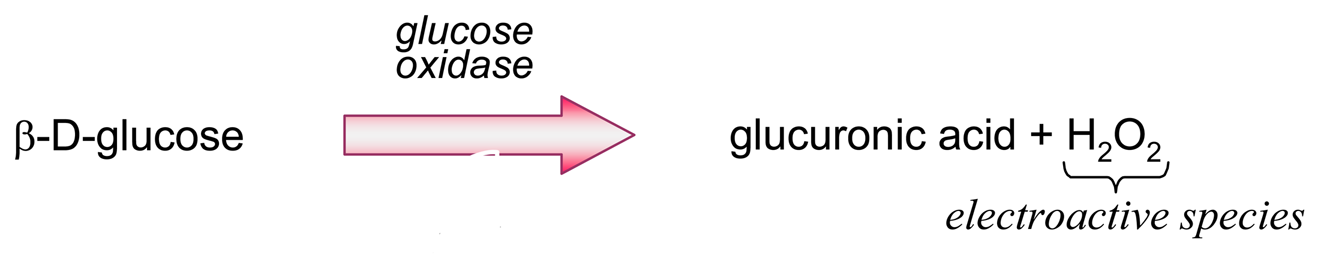
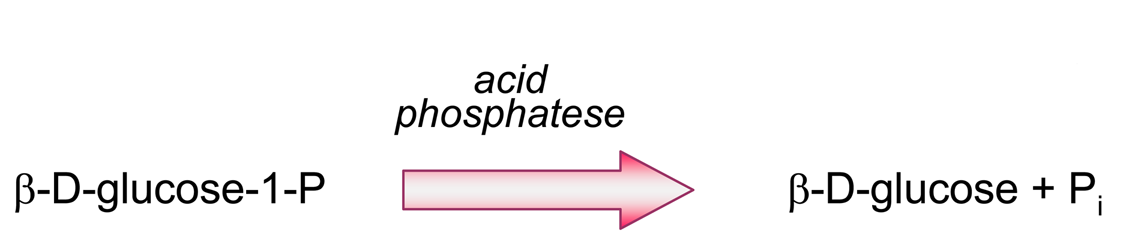
- Producing additional equivalents of electroactive species, generated by a second enzyme that uses a “side product” of the first enzymatic reaction, which had rendered the first equivalent of electroactive species. An amperometric biosensor using this model was presented by Conrath et al. to detect inorganic phosphate [179]. In the clinical practice, phosphate levels in both blood and urine are determined as indicative of osteopathies, and of parathyroid function [180]. In Conrath's proposal, the first enzyme, maltose phosphorylase, catalyses the reaction between the analyte and maltose, to render β-D-glucose-1-phosphate and α-D-glucose (4). This latter species is further transformed by mutarotase into to β-D-glucose (5). β-D-glucose acts as substrate of glucose oxidase to render glucuronic acid and hydrogen peroxide (6), which is the electrochemically detected species. Another enzyme, acid phosphatase, hydrolyses the “side product”, glucose-1-phosphate, generating an extra β-D-glucose (7), which will be also further oxidised according to (6), thus releasing the analyte, phosphate, which can be recycled again.
3.2. Increasing the number of biorecognition species
4. Simplicity of use and packaging
5. Future directions
Acknowledgments
References and Notes
- Luppa, P. B.; Sokoll, L. J.; Chan, D. W. Immunosensors: principles and applications to clinical chemistry. Clin. Chim. Acta 2001, 314, 1–26. [Google Scholar]
- Clark, L. C., Jr; Lyons, C. Electrode systems for continuous monitoring in cardiovascular surgery. Ann. NY. Acad. Sci. 1962, 102, 29–45. [Google Scholar]
- Updike, S. J.; Hicks, G. P. The enzyme electrode. Nature 1967, 214, 986–988. [Google Scholar]
- Anon: News and Market Update. Biosens. Bioelectron. 1998, 13, I–II.Newman, J.; Turner, A. P. F. Home blood glucose biosensors: a commercial perspective. Biosens. Bioelectron. 2005, 20, 2435–2453. [Google Scholar]
- Tsuchida, T.; Yoda, K. Multi-enzyme membrane electrodes for determination of creatinine and creatine in serum. Clin. Chem. 1983, 29, 51–55. [Google Scholar]
- Senda, M.; Yamamoto, Y. Urea biosensor based on amperometric ammonium ion electrode. 1993, 5, 775–779. [Google Scholar]
- Kost, G. J.; Nguyen, T. H.; Tang, Z. Whole blood glucose and lactate: trilayer biosensors, drug interference, metabolism and practice guidelines. Arch. Pathol. Lab. Med. 2000, 124, 1128–1134. [Google Scholar]
- Aduen, J.; Bernstein, W. K.; Khastgir, T.; Miller, J.; Kerzner, R.; Bhatiani, A.; Lustgarten, J.; Bassin, A. S.; Davison, L.; Chernow, B. The use and clinical importance of a substrate specific electrode for rapid determination of blood lactate concentrations. Jama-J. Am. Med. Assoc. 1994, 272, 1678–1685. [Google Scholar]
- Durliat, H.; Causserand, C.; Comtat, M. Bienzyme amperometric lactate-specific electrode. Anal. Chim. Acta 1990, 231, 309–311. [Google Scholar]
- Gilis, M.; Durliat, H.; Comtat, M. Amperometric biosensors for L-alanine and pyruvate assays in biological fluids. Anal. Chim. Acta 1997, 355, 235–240. [Google Scholar]
- Rasmussen, S. K.; Rasmussen, L. K.; Weilguny, D.; Tolstrup, A. B. Manufacture of recombinant polyclonal antibodies. Biotechnol. Lett. 2007, 9, 845–852. [Google Scholar]
- de StGroth, S. F.; Scheidegger, D. Production of monoclonal antibodies: strategy and tactics. J. Immunol. Methods 1980, 35, 1–21. [Google Scholar]
- Morgan, C. L.; Newman, D. J.; Price, C. P. Immunosensors: technology and opportunities in laboratory medicine. Clin. Chem. 1996, 42, 192–209. [Google Scholar]
- Sarkar, P.; Pal, P.; Ghosh, D.; Setford, S.; Tothill, I. Amperometric biosensors for detection of the prostate cancer marker (PSA). Int. J. Pharm. 2002, 238, 1–9. [Google Scholar]
- Okuno, J.; Maehashi, K.; Kerman, K.; Takamura, Y.; Matsumoto, K.; Tamiya, E. Label-free immunosensor for prostate-specific antigen based on single-walled carbon nanotube array-modified microelectrodes. Biosens Bioelectron. 2006, 22, 2377–2381. [Google Scholar]
- Tang, H.; Chen, J.; Nie, L.; Kuang, Y.; Yao, S. A label-free electrochemical immunoassay for carcinoembryonic antigen (CEA) based on gold nanoparticles (AuNPs) and nonconductive polymer film. Biosens Bioelectron. 2007, 22, 1061–1067. [Google Scholar]
- Aizawa, M.; Morioka, A.; Suzuki, S.; Nagamura, Y. Enzyme immunosensor III. Amperometric determination of human choriogonadotropin by membrane bound antibody. Anal. Biochem. 1979, 94, 22–28. [Google Scholar]
- Pemberton, R. M.; Hart, J. P.; Mottran, T. T. An electrochemical immunosensor for milk progesterone using continuous flow system. Biosens. Bioelectron. 2001, 16, 715–723. [Google Scholar]
- Gilmartin, M. A. T.; Hart, J. P. Fabrication and characterization of a screen printed, disposable amperometric cholesterol biosensor. Analyst 1994, 119, 2331–2336. [Google Scholar]
- Benkert, A.; Scheller, F. W.; Schoessler, W.; Micheel, B.; Warsinke, Axel. Size Exclusion Redox-Labeled Immunoassay (SERI): A New Format for Homogeneous Amperometric Creatinine Determination. 2000, 12, 1318–1321. [Google Scholar]
- Lee, S. H.; Lee, H.; Park, J. S.; Choi, H.; Han, K. Y.; Seo, H. S.; Ahn, K. Y.; Han, S. S.; Cho, Y.; Lee, K. H.; Lee, J. A novel approach to ultrasensitive diagnosis using supramolecular protein nanoparticles. FASEB J. 2007, 21, 1324–1334. [Google Scholar]
- Luppa, P. B.; Sokoll, L. J.; Chan, D. W. Immunosensors-principles and applications to clinical chemistry. Clin. Chim. Acta 2001, 314, 1–26. [Google Scholar]
- Wang, J. Electrochemical biosensors: Towards point-of-care cancer diagnostics. Biosen. Bioelctron. 2006, 21, 1887–1892. [Google Scholar]
- Ribone, M. E.; Belluzo, M. S.; Pagani, D.; Macipar, I. S.; Lagier, C. M. Amperometric bioelectrode for specific human immunoglobulin G determination: optimization of the method to diagnose American trypanosomiasis. Anal. Biochem. 2006, 350, 61–70. [Google Scholar]
- Ferreira, A. A. P.; Colli, W.; Da Costa, P. I.; Yamanaka, H. Immunosensor for the diagnosis of Chagas' disease. Biosens. Bioelectron. 2005, 21, 175–181. [Google Scholar]
- Lei, C.-X.; Gong, F.-C.; Shen, G.-L.; Yu, R.-Q. Amperometric immunosensor for Schistosoma japonicum antigen using antibodies loaded on a nano-Au monolayer modified chitosan-entrapped carbon paste electrode. Sens. Actuat. B-Chem 2003, 96, 582–588. [Google Scholar]
- Zhou, Y.-M.; Wu, Z.-Y.; Shen, G.-L.; Yu, R.-Q. An amperometric immunosensor based on Nafion-modified electrode for the determination of Schistosoma japonicum antibody. Sens. Actuat. B-Chem. 2003, 89, 292–298. [Google Scholar]
- Krishnan, R.; Ghindilis, A. L.; Atanasov, P.; Wilkins, E.; Montoya, J.; Koster, F. T. Fast amperometric immunoassay for hantavirus infection. 1996, 8, 1131–1134. [Google Scholar]
- Aguirre, S.; Silber, A. M.; Brito, M. E. F.; Ribone, M. E.; Lagier, C. M.; Mancipar, I. S. Design, Construction and Evaluation of a Specific Chimeric Antigen to Diagnose Chagasic Infection. J. Clin. Microbiol. 2006, 44, 1043–1046. [Google Scholar]
- Marcipar, I. S.; Roodveldt, C; Corradi, G.; Cabeza, M. L.; Brito, M. E. F.; Floeter Winter, L. M.; Marcipar, A. J.; Silber, A. M. Use of full-length recombinant calflagin and its c fragment for improvement of diagnosis of Trypanosoma cruzi infection. J. Clin. Microbiol. 2005, 43, 5498–5503. [Google Scholar]
- Belluzo, M. S.; Ribone, M. E.; Caussone, C.; Marcipar, I. S; Lagier, C. M. Oriented Protein Attachment Onto Gold Electrodes To Perform A Highly Sensitive And Selective Immunoassay With Amperometric Detection. 11th Int. Conf. Electroanal. ESEAC 2006, P1–021. [Google Scholar]
- Ellington, A. D.; Szostak, J. W. In vitro selection of RNA molecules that bind specific ligands. Nature 1990, 346, 818–822. [Google Scholar]
- Tombelli, S; Minunni, M; Mascini, M. Analytical applications of aptamers. Biosens Bioelectron. 2005, 20, 2424–34. [Google Scholar]
- Willner, I.; Zayats, M. Electronic Aptamer-Based Sensors. Angew. Chem. Int. Edit. 2007, 46, 6408–6418. [Google Scholar]
- Jayasena, S. D. Aptamers: An Emerging Class of Molecules That Rival Antibodies in Diagnostics. Clin. Chem. 1999, 45, 1628–1650. [Google Scholar]
- Luzi, E.; Minunni, M.; Tombelli, S.; Mascini, M. New trends in affinity sensing: Aptamers for ligand binding. Trends Anal. Chem. 2003, 22, 810–818. [Google Scholar]
- Hesselberth, J.; Robertson, M. P.; Jhaveri, S.; Ellington, A. D. In vitro selection of nucleic acids for diagnostic applications. J Biotechnol. 2000, 74, 15–25. [Google Scholar]
- O'Suvillan, C. K. Aptasensors: the future of biosensing? Anal. Bioanal. Chem. 2002, 372, 44–48. [Google Scholar]
- McCauley, T. G.; Hamaguchi, N.; Stanton, M. Aptamer-based biosensor arrays for detection and quantification of biological macromolecules. Anal. Biochem. 2003, 319, 244–250. [Google Scholar]
- Xu, D.; Xu, D.; Yu, X.; Liu, Z.; He, W.; Ma, Z. Label-Free Electrochemical Detection for Aptamer-Based Array Electrodes. Anal. Chem. 2005, 77, 5107–5113. [Google Scholar]
- Ikebukuro, K.; Kiyohara, C.; Sode, K. Novel electrochemical sensor system for protein using the aptamers in sandwich manner. Biosens. Bioelectron. 2005, 20, 2168–2172. [Google Scholar]
- Tombelli, S.; Minunni, M.; Mascini, M. Aptamers-based assays for diagnostics, environmental and food analysis. Biomol. Eng. 2007, 24, 191–200. [Google Scholar]
- Jiang, T.; Minunni, M.; Mascini, M. Towards fast and inexpensive molecular diagnostic: the case of TP53. Clin. Chim. Acta. 2004, 343, 45–60. [Google Scholar]
- Kim, Y. S.; Jung, H. S.; Matsuura, T.; Lee, H. Y.; Kawai, T.; Gu, M. B. Electrochemical detection of 17β-estradiol using DNA aptamer immobilized gold electrode chip. Biosens. Bioelectron. 2007, 22, 2525–2531. [Google Scholar]
- Baker, B. R.; Lai, R. Y.; Wood, M. C. S.; Doctor, E. H.; Heeger, A. J.; Plaxco, K. W. An Electronic Aptamer-Based Small-Molecule Sensor for the Rapid, Label-Free Detection of Cocaine in Adulterated Samples and Biological Fluids. J. Am. Chem. Soc. 2006, 128, 3138–3139. [Google Scholar]
- Radi, A. E.; Acero Sanchez, J. L.; Baldrich, E.; O'Sullivan, C. K. Reagentless, Reusable, Ultrasensitive Electrochemical Molecular Beacon Aptasensor. J. Am. Chem. Soc. 2006, 128, 117–124. [Google Scholar]
- Mir, M.; Vreeke, M.; Katakis, I. Different Strategies To Develop an Electrochemical Thrombin Aptasensor. Electrochem. Commun. 2006, 8, 505–511. [Google Scholar]
- Bini, A.; Minunni, M.; Tombelli, S.; Centi, S.; Mascini, M. Analytical performances of aptamer-based sensing for thrombin detection. Anal. Chem. 2007, 79, 3016–3019. [Google Scholar]
- Goldsack, N. R.; Chambers, R. C.; Dabbagh, K.; Laurent, G. J. Molecules in focus Thrombin. Int. J. Biochem. Cell. B. 1998, 30, 641–646. [Google Scholar]Wallerstein, R. O., Jr. Laboratory evaluation of a bleeding patient. Western J. Med. 1989, 150, 51–58. [Google Scholar]
- Hansen, J. A.; Wang, J.; Kawde, A. –N.; Xiang, Y.; Gothelf, K. V.; Collins, G. Quantum-Dot/Aptamer-Based Ultrasensitive Multi-Analyte Electrochemical biosensor. J. Am. Chem. Soc. 2006, 128, 2228–2229. [Google Scholar]
- Gasior-Chrzan, B. The biological role of lysozyme and its usefulness in clinical practice. Wiad Lek. 1987, 40, 1693–1696. [Google Scholar]
- Navani, N. K.; Li, Y. Nucleic acid aptamers and enzymes as sensors. Curr. Opin. Chem. Biol. 2006, 10, 272–281. [Google Scholar]
- Wang, J.; Rivas, G.; Cai, X. Screen-Printed Electrochemical Hybridization DNA Biosensor for the E. Coli Pathogen. 1997, 9, 395–398. [Google Scholar]
- Millan, K. M.; Mikkelsen, S. R. Sequence-Selective Biosensor for DNA Based on Electroactive Hybridization Indicators. Anal. Chem. 1993, 65, 2317–2323. [Google Scholar]
- Hashimoto, K.; Ito, K.; Ishimori, Y. Sequence-Specific Gene Detection with a Gold Electrode Modified with DNA Probes and an Electrochemically Active Dye. Anal. Chem. 1994, 66, 3830–3833. [Google Scholar]
- Thorp, H. H. Cutting out the middleman: DNA biosensors based on electrochemical oxidation. Trends Biotechnol. 1998, 16, 117–121. [Google Scholar]
- Palecek, E. Oscillographic polarography of highly polymerized deoxyribonucleic acid. Nature 1960, 188, 656–657. [Google Scholar]
- Palecek, E. Polarographic techniques in nucleic acid research. Prog. Nucleic Acid Res. Mol. Biol. 1969, 9, 31–73. [Google Scholar]
- Palecek, E. Adsorptive transfer stripping voltametry: determination of nanogram quantities of DNA immobilized at the electrode surface. Anal. Biochem. 1988, 170, 421–431. [Google Scholar]
- Jelen, F.; Yosypchuk, B.; Kourilova, A.; Novotny, L.; Palecek, E. Label-free determination of picogram quantities of DNA by stripping voltammetry with solid copper amalgam or mercury electrodes in the presence of copper. Anal. Chem. 2002, 74, 4788–4793. [Google Scholar]
- Palecek, E.; Fojta, M. Detecting DNA Hybridization and Damage. Anal. Chem. 2001, 73, 75A–83A. [Google Scholar]
- Palecek, E.; Fojta, M.; Jelen, F. New approaches in the development of DNA sensors: hybridization and electrochemical detection of DNA and RNA at two different surfaces. Bioelectrochemistry 2002, 56, 85–90. [Google Scholar]
- Gooding, J. J. Electrochemical DNA Hybridization Biosensors. Electroanalysis 2002, 14, 1149–1156. [Google Scholar]
- Wang, J.; Rivas, G.; Fernandes, J. R.; Lopez Paz, J. L.; Jiang, M.; Waymire, R. Indicator-free electrochemical DNA hybridization biosensor. Anal. Chim. Acta 1998, 375, 197–203. [Google Scholar]
- Domínguez, E.; Rincón, O.; Narváez, A. Electrochemical DNA sensors based on enzyme dendritic architectures: an approach for enhanced sensitivity. Anal. Chem. 2004, 76, 3132–3138. [Google Scholar]
- Popovich, N. D.; Eckhardt, A. E.; Mikulecky, J. C.; Napier, M. E.; Thomas, R. S. Electrochemical sensor for detection of unmodified nucleic acids. Talanta 2002, 56, 821–828. [Google Scholar]
- Cai, H.; Cao, X. N.; Jiang, Y.; He, P. G.; Fang, Y. Z. Carbon nanotube-enhanced electrochemical DNA biosensor for DNA hybridization detection. Anal. Bioanal. Chem. 2003, 375, 287–293. [Google Scholar]
- Wang, J.; Rivas, G.; Cai, X.; Chicharro, M.; Dontha, N.; Luo, D.; Palecek, E.; Nielsen, P. E. Adsorption and detection of peptide nucleic acids at carbon paste electrodes. Electroanalysis 2005, 9, 120–124. [Google Scholar]
- Liao, J. C.; Mastali, M.; Gau, V.; Suchard, M. A.; Møller, A. K.; Bruckner, D. A.; Babbitt, J. T.; Li, Y.; Gornbein, J.; Landaw, E. M.; McCabe, E. R. B.; Churchill, B. M.; Haake, D. A. Use of Electrochemical DNA Biosensors for Rapid Molecular Identification of Uropathogens in Clinical Urine Specimens. J Clin Microbiol. 2006, 44, 561–570. [Google Scholar]
- Abad-Valle, P.; Fernández-Abedul, M. T.; Costa-García, A. Genosensor on gold films with enzymatic electrochemical detection of a SARS virus sequence. Biosens. Bioelectron. 2005, 20, 2251–2260. [Google Scholar]
- Lin, X.-H.; Wu, P.; Chen, W.; Zhang, Y.-F.; Xia, X.-H. Electrochemical DNA biosensor for the detection of short DNA species of Chronic Myelogenous Leukemia by using methylene blue. Talanta 2007, 72, 468–471. [Google Scholar]
- Lin, X. H.; Wan, H. Y.; Zhang, Y. F.; Chen, J. H. Studies of the interaction between Aloe-emodin and DNA and preparation of DNA biosensor for detection of PML-RARα fusion gene in acute promyelocytic leukemia. Talanta 2008, 74, 944–950. [Google Scholar]
- Palecek, E.; Jelen, F. Electrochemistry of nucleic acids and development of DNA sensors. Crit. Rev. Anal. Chem. 2002, 3, 261–270. [Google Scholar]
- Wang, J. Electrochemical nucleic acid biosensors. Anal. Chim. Acta 2002, 469, 63–71. [Google Scholar]
- Drummond, T. G.; Hill, M. G.; Barton, J. K. Electrochemical DNA sensors. Nat. Biotechnol. 2003, 21, 1192–1199. [Google Scholar]
- Ferapontova, E. E. Electrochemistry of guanine and 8-oxoguanine at gold electrodes. Electrochim. Acta 2004, 49, 1751–1759. [Google Scholar]
- Brooks, S. C.; Richter, M. M. Determination of DNA Bases Using Electrochemistry: A Discovery-Based Experiment. The Chemical Educator 2002, 7, 284–287. [Google Scholar]
- Oliveira Brett, A. M.; Piedade, J. A. P.; Serrano, S. H. P. Electrochemical Oxidation of 8-Oxoguanine. Electroanalysis 2000, 12, 969–973. [Google Scholar]
- Palecek, E. From polarography of DNA to microanalysis with nucleic acid-modifield electrodes. Electroanalysis 1996, 8, 7–14. [Google Scholar]
- Singhal, P.; Kuhr, W. G. Ultrasensitive Voltammetric Detection of Underivatized Oligonucleotides and DNA. Anal. Chem. 1997, 69, 4828–4832. [Google Scholar]
- Casegreen, S.; Southern, E. Studies on the base pairing properties of deoxyinosine by solid phase hybridisation to oligonucleotides. Nucleic acids Res. 1994, 22, 131–136. [Google Scholar]
- Wang, J.; Cai, X.; Rivas, G.; Shiraishi, H.; Dontha, N. Nucleic-acid immobilization, recognition and detection at chronopotentiometric DNA chips. Biosens. Bioelectron. 1997, 12, 587–599. [Google Scholar]
- Wang, J.; Rivas, G.; Parrado, C.; Cai, X.; Flair, M. N. Electrochemical biosensor for detecting DNA sequences from the pathogenic protozoan. Cryptosporidium parvum. Talanta 1997, 44, 2003–2010. [Google Scholar]
- Erdem, A.; Kerman, K.; Meric, B.; Akarca, U. S.; Ozsoz, M. DNA electrochemical biosensor for the detection of short DNA sequences related to the hepatitis B virus. Electroanalysis 1999, 11, 586–588. [Google Scholar]
- Wang, J.; Cai, X.; Rivas, G.; Shiraishi, H.; Farias, P. A. M.; Dontha, N. DNA electrochemical biosensor for the detection of short DNA sequences related to the human immunodeficiency virus. Anal. Chem. 1996, 68, 2629–2634. [Google Scholar]
- Pan, D.; Zuo, X.; Wan, Y.; Wang, L.; Zhang, J.; Song, S.; Fan, C. Electrochemical Interrogation of Interactions between Surface-Confined DNA and Methylene Blue. Sensors 2007, 7, 2671–2680. [Google Scholar]
- Mascini, M.; Palchetti, I.; Marrazza, G. DNA electrochemical biosensors. Fresenius J Anal Chem. 2001, 369, 15–22. [Google Scholar]
- Liepold, P.; Wieder, H.; Hillebrandt, H.; Friebel, A.; Hartwich, G. DNA-arrays with electrical detection: A label-free low cost technology for routine use in life sciences and diagnostics. Bioelectrochemistry 2005, 67, 143–150. [Google Scholar]
- Kerman, K.; Kobayashi, M.; Tamiya, E. Recent trends in electrochemical DNA biosensor technology. Meas. Sci. Technol. 2004, 15, R1–R11. [Google Scholar]
- Caruana, D. J.; Heller, A. Enzyme-amplified amperometric detection of hybridization and of a single base pair mutation in an 18-base oligonucleotide on a 7 μm diameter microelectrode. J. Am. Chem. Soc. 1999, 121, 769–774. [Google Scholar]
- Xie, H.; Zhang, C.; Gao, Z. Amperometric detection of nucleic acid at femtomolar levels with a nucleic acid/electrochemical activator bilayer on gold electrode. Anal. Chem. 2004, 76, 1611–1617. [Google Scholar]
- Suye, S.; Matsuura, T.; Kimura, T.; Zheng, H.; Hori, T.; Amano, Y.; Katayama, H. Amperometric DNA sensor using gold electrode modified with polymerized mediator by layer-by-layer adsorption. Microelectron. Eng. 2005, 81, 441–447. [Google Scholar]
- Zhao, W.; Xu, J. –J.; Chen, H. -Y. Electrochemical Biosensors Based on Layer-by-Layer Assemblies. Electroanalysis 2006, 18, 1737–1748. [Google Scholar]
- Xie, H.; Yu, Y. H.; Mao, P. –L; Gao, Z. Highly sensitive amperometric detection of genomic DNA in animal tissues. Nucleic Acids Res. 2004, 32, 1–7. [Google Scholar]
- Fojta, M.; Havran, L.; Billova, S.; Kostecka, P.; Masarik, M.; Kizek, R. Two-Surface Strategy in Electrochemical DNA Hybridization Assays: Detection of Osmium-Labeled Target DNA at Carbon Electrodes. Electroanalysis 2003, 15, 431–440. [Google Scholar]
- Mikkelsen, S. R. Electrochemical Biosensors for DNA Sequence Detection. Electroanalysis 1996, 8, 15–19. [Google Scholar]
- Pividori, M. I.; Merkoci, A.; Alegret, S. Electrochemical genosensor design: immobilisation of oligonucleotides onto transducer surfaces and detection methods. Biosens. Bioelectron. 2000, 15, 291–303. [Google Scholar]
- Lucarelli, F.; Marrazza, G.; Turner, A. P. F.; Mascini, M. Carbon and gold electrodes as electrochemical transducers for DNA hybridisation sensors. Biosens. Bioelectron. 2004, 19, 515–530. [Google Scholar]
- Siest, G.; Appel, W.; Blijenberg, G. B.; Capolaghi, B.; Galteau, M. M.; Heusghem, C.; Hjelm, M.; Lauer, K. L.; Le Perron, B.; Loppinet, V.; Love, C.; Royer, R. J.; Tognoni, C.; Wilding, P. C.; Wilding, P. Drug interference in clinical chemistry: studies on ascorbic acid. J. Clin. Chem. Clin. Biochem. 1978, 16, 103–110. [Google Scholar]
- Palleschi, G.; Rahni, M. A.; Lubrano, G. J.; Nwainbi, Ngeh; Guilbault, G. G. A study of interferences in glucose measurement in blood by hydrogen peroxide based glucose probes. Anal. Biochem. 1986, 159, 114–121. [Google Scholar]
- Hills, L.; Azurin, G.; Wang, X.; et al. Glutathione as an interferent in near patient whole blood glucose devices. In Proc. 17th Int. Symp. Int. Fed. Clin. Chem.; Omnipress: Madison, Wis, 1998; pp. 207–219. [Google Scholar]
- D'Orazio, P.; Parker, B. Interference by the oxidizable pharmaceuticals acetaminophen and dopamine at electrochemical biosensors for blood glucose. Clin. Chem. 1995, 41, S156. [Google Scholar]
- D'Orazio, P. Interference by thiocyanate on electrochemical biosensors for blood glucose. Clin. Chem. 1996, 42, 1124–1126. [Google Scholar]
- Hasebe, Y.; Nawa, K.; Ujita, S.; Uchiyama, S. Highly sensitive flow detection of uric acid based on intermediate regeneration of uricase. Analyst 1998, 123, 1775–1780. [Google Scholar]
- Zhang, Y.; Hu, Y.; Wilson, G. S.; Moatti-Sirat, D.; Poitout, V.; Reach, G. Elimination of the acetaminophen interference in an implantable glucose sensor. Anal. Chem. 1994, 66, 1183–1188. [Google Scholar]
- Lobel, E.; Rishpon, J. Enzyme electrode for the determination of glucose. Anal. Chem. 1981, 53, 51–53. [Google Scholar]
- Palmisano, F.; Rizzi, R.; Centonze, D.; Zambonin, P. G. Simultaneous monitoring of glucose and lactate by an interference and cross-talk free dual electrode amperometric biosensor based on electropolymerized thin films. Biosens. Bioelectron. 2000, 15, 531–539. [Google Scholar]
- Mizutani, F.; Yabuki, S.; Iijima, S. Use of Polydimethylsiloxane for Constructing Amperometric Glucose-Sensing Enzyme Electrode with Low Interference Level. Electroanalysis 2001, 13, 370–374. [Google Scholar]
- Sasso, S.; Pierce, R.; Walla, R.; Yacynych, A. Electropolymerized 1,2-diaminobenzene as a means to prevent interferences and fouling and to stabilize immobilized enzymes in electrochemical biosensors. Anal. Chem. 1990, 62, 1111–1117. [Google Scholar]
- Reynolds, E. R.; Yacynych, A. M. Direct sensing platinum ultramicrobiosensors for glucose. Biosens. Bioelectron. 1994, 9, 283–293. [Google Scholar]
- Geise, R. J.; Adams, J. M.; Barone, N. J.; Yacynych, A. M. Electropolymerized films to prevent interferences and electrode fouling in biosensors. Biosens. Bioelectron. 1991, 6, 151–160. [Google Scholar]
- Kirwan, S. M.; Rocchitta, G.; McMahon, C. P.; Craig, J. D.; Killoran, S. J.; O'Brien, K. B.; Serra, P. A.; Lowry, J. P.; O'Neill, R. D. Modifications of Poly(o-phenylenediamine) Permselective Layer on Pt-Ir for Biosensor Application in Neurochemical Monitoring. Sensors 2007, 7, 420–437. [Google Scholar]
- Vaidya, R.; Wilkins, E. Effect of interference on amperometric glucose biosensors with cellulose acetate membranes. Electroanalysis 1994, 6, 677–682. [Google Scholar]
- Gunasingham, H.; Teo, P. Y. T.; Lai, Y.-H.; Tan, S.-G. Chemically modified cellulose acetate membrane for biosensor applications. Biosensors 1989, 4, 349–359. [Google Scholar]
- Palecek, E.; Billová, S.; Havran, L.; Kizek, R.; Miculková, A.; Jelen, F. DNA hybridization at microbeads with cathodic stripping voltammetric detection. Talanta 2002, 56, 919–930. [Google Scholar]
- Adam, V.; Petrlova, J.; Potesil, D.; Zehnalek, J.; Sures, B.; Trnkova, L.; Jelen, F.; Kizek, R. Study of Metallothionein Modified Electrode Surface Behavior in the Presence of Heavy Metal Ions-Biosensor. Electroanalysis 2005, 17, 1649–1657. [Google Scholar]
- Palmiter, R. D. Regulation of metallothionein genes by heavy metals appears to be mediated by a zinc-sensitive inhibitor that interacts with a constitutively active transcription factor, MTF-1. Proc. Natl. Acad. Sci. USA 1994, 91, 1219–1223. [Google Scholar]
- Petrlova, J.; Potesil, D.; Mikelova, R.; Blastik, O.; Adam, V.; Trnkova, L.; Jelen, F.; Prusa, R.; Kukacka, J.; Kizek, R. Attomole voltammetric determination of metallothionein. Electrochim. Acta 2006, 51, 5112–5119. [Google Scholar]
- Renault, N. J.; Martelet, C.; Chevolot, Y.; Cloarec, J. P. Biosensors and Bio-Bar Code Assays Based on Biofunctionalized Magnetic Microbeads. Sensors 2007, 7, 589–614. [Google Scholar]
- Wang, J.; Kawde, A. B. Amplified label-free electrical detection of DNA hybridazation. Analyst 2002, 127, 383–386. [Google Scholar]
- Gorton, G.; Csoregi, E.; Dominguez, E.; Emneus, J.; Jonsson Pettersson, G.; Marko-Varga, G.; Persson, B. Selective detection in flow analysis based on combination of immobilized enzymes and chemically modified electrodes. Anal. Chim. Acta 1991, 250, 203–248. [Google Scholar]
- Cass, A. E.; Davis, G.; Francis, G.; Hill, H.; Aston, W.; Higgins, I. J.; Plotkin, E.; Scott, L.; Turner, A. P. Ferrocene-mediated enzyme electrode for amperometric determination of glucose. Anal. Chem. 1984, 56, 667–671. [Google Scholar]
- Ivanova, E. V.; Sergeeva, V. S.; Oni, J.; Kurzawa, C.; Ryabov, A. D.; Schuhmann, W. Evaluation of redox mediators for amperometric biosensors: Ru-complex modified carbon-paste/enzyme electrodes. Bioelectrochemistry 2003, 60, 65–71. [Google Scholar]
- Ryabov, A. D.; Sukharev, V. S.; Alexandrova, L.; Le Lagadec, R.; Pfeffer, M. New Synthesis and New Bio-Application of Cyclometalated Ruthenium(II) Complexes for Fast Mediated Electron Transfer with Peroxidase and Glucose Oxidase. Inorg. Chem. 2001, 40, 6529–6532. [Google Scholar]
- Cardosi, M. F.; Turner, A. P. F. The realisation of electron transfer from biological molecules to electrodes. In Biosensors: Fundamentals and Applications; Turner, A., Karube, I., Wilson, G., Eds.; Oxford University Press: New York, 1987; pp. 257–75. [Google Scholar]
- Alves, W. A.; Fiorito, P. A.; Córdoba de Torresi, S. I.; Torresi, R. M. Design of molecular wires based on supramolecular structures for application in glucose biosensors. Biosens. Bioelectron. 2006, 22, 298–305. [Google Scholar]
- Pandey, P. C.; Upadhyay, S.; Upadhyay, B. Peroxide biosensors and mediated electrochemical regeneration of redox enzymes. Anal. Biochem. 1997, 252, 136–142. [Google Scholar]
- Cass, A. E.; Davis, G.; Francis, G. D.; Hill, H. A.; Aston, W. J.; Higgins, I. J.; Plotkin, E. V.; Scott, L. D.; Turner, A. P. Ferrocene mediated enzyme electrode for amperometric determination of glucose. Anal. Chem. 1984, 56, 667–671. [Google Scholar]
- García-Ruiz, E.; Vidal, J. C.; Aramendía, M. T.; Castillo, J. R. Design of an Interference-Free Cholesterol Amperometric Biosensor Based on the Electrosynthesis of Polymeric Films of Diaminonaphthalene Isomers. Electroanalysis 2004, 16, 497–504. [Google Scholar]
- Rondeau, A.; Larsson, N.; Boujtita, M.; Gorton, L.; El Murr, N. The synergetic effect of redox mediators and peroxidase in a bienzymatic biosensor for glucose assays in FIA. Analusis 1999, 27, 649–656. [Google Scholar]
- Ohara, T. J.; Rajagopalan, R.; Heller, A. “Wired” enzyme electrodes for amperometric determination of glucose or lactate in the presence of interfering substances. Anal. Chem. 1994, 66, 2451–2457. [Google Scholar]
- Carano, M.; Cosnier, S.; Kordatos, K.; Marcaccio, M.; Margotti, M.; Paolucci, F.; Prato, M.; Roffia, S. A glutathione amperometric biosensor based on an amphiphilic fullerene redox mediator immobilised within an amphiphilic polypyrrole film. J. Mater. Chem. 2002, 12, 1996–2000. [Google Scholar]
- Ghica, M. E.; Brett, C. M. A. A glucose biosensor using methyl viologen redox mediator on carbon film electrodes. Anal. Chim. Acta 2005, 532, 145–151. [Google Scholar]
- De Lacey, A. L.; Bes, M. T.; Moreno, C. G.; Moreno, C. G.; Fernandez, V. M. Amperometric enzyme electrode for NADP+ based on a ferrodoxin-NADP+ reductase and viologen-modified glassy carbon electrode. J. Electroanal. Chem. 1995, 390, 69–76. [Google Scholar]
- Qian, J.; Li, X.; Huang, H. Sol-gel-derived amperometric glucose biosensor based on covalent attachment of toluidine blue O to carrier. Polym. Advan. Technol. 2003, 14, 207–211. [Google Scholar]
- Fei, J.; Wu, Y.; Ji, X.; Wang, J.; Hu, S.; Gao, Z. An Amperometric Biosensor for Glucose Based on Electrodeposited Redox Polymer/Glucose Oxidase Film on a Gold Electrode. Anal. Sci. 2003, 19, 1259–1263. [Google Scholar]
- Calvo, E. J.; Battaglini, F.; Danilowicz, C.; Wolosiuk, A.; Otero, M. Layer-by-layer electrostatic deposition of biomolecules on surfaces for molecular recognition, redox mediation and signal generation. Faraday Discuss. Chem. Soc. 2000, 116, 47–65. [Google Scholar]
- Danilowicz, C.; Corton, E.; Battaglini, F. Osmium complexes bearing functional groups: Building blocks for integrated chemical systems. J. Electroanal. Chem. 1998, 445, 89–94. [Google Scholar]
- Battaglini, F.; Bartlett, P. N.; Wang, J. H. Covalent Attachment of Osmium Complexes to Glucose Oxidase and the Application of the Resulting Modified Enzyme in an Enzyme Switch Responsive to Glucose. Anal. Chem. 2000, 72, 502–509. [Google Scholar]
- Pishko, M. V.; Revzin, A.; Simonian, A. L. Mass Transfer in Amperometric Biosensors Based on Nanocomposite Thin Films of Redox Polymers and Oxidoreductases. Sensors 2002, 2, 79–90. [Google Scholar]
- Hodak, J.; Etchenique, R.; Calvo, E. J. Layer-by-layer Self Assembley of Glucose Oxidase with a Poly(allylamine)ferrocene Redox Mediator. Langmuir 1997, 13, 2708–2716. [Google Scholar]
- Calvo, E. J.; Etchenique, R.; Pietrasanta, L.; Wolosiuk, A.; Danilowicz, C. Layer-by-layer Self Assembly of Glucose Oxidase and Os(bpy)2ClPyCH2NH-Poly(allylamine). Bioelectrode. Anal. Chem. 2001, 73, 1161–1168. [Google Scholar]
- Seo, K. D.; Lee, K. P.; Gopalan, A. I.; Chung, S. J.; Lim, Y. T.; Choi, S. H. Horseradish Peroxidase (HRP) Immobilized Poly(aniline-co-m-aminophenol) Film Electrodes–fabrication and Evaluation as Hydrogen Peroxide Sensor. Sensors 2007, 7, 719–729. [Google Scholar]
- Wang, J. Electrochemical Glucose Biosensors. Chem. Rev. 2007. 10.1021/cr068123a. [Google Scholar]
- Khayyami, M.; Pérez Pita, M. T; Peña Garcia, N.; Johansson, G.; Danielsson, B.; Larsson, P.-O. Development of an amperometric biosensor based on acetylcholine esterase covalently bound to a new support material. Talanta 1998, 45, 557–563. [Google Scholar]
- Pasco, N.; Baronian, K.; Jeffries, C.; Webber, J.; Hay, J. MICREDOX-development of a ferricyanide-mediated rapid biochemical oxygen demand method using an immobilised Proteus vulgaris biocomponent. Biosens. Bioelectron. 2004, 20, 524–532. [Google Scholar]
- Gros, P.; Comtat, M. A bioelectrochemical polypyrrole-containing Fe(CN)63− interface for the design of a NAD-dependent reagentless biosensor. Biosens. Bioelectron. 2004, 20, 204–210. [Google Scholar]
- Chaubey, A.; Malhotra, B. D. Mediated biosensors. Biosens. Bioelectron. 2002, 17, 441–456. [Google Scholar]
- Takenaka, S.; Uto, Y.; Kondo, H.; Ihara, T.; Takegi, M. Electrochemically active DNA Probes: Detection of Target DNA Sequences at Femtomole Level by High-Performance Liquid Chromatography with Electrochemical Detection. Anal. Biochem. 1994, 218, 436–443. [Google Scholar]
- Fojta, M.; Havran, L.; Kizek, R.; Billová, S. Voltammetric microanalysis of DNA adducts with osmium tetroxide,2,2′-bipyridine using pyrolytic graphite electrode. Talanta 2002, 56, 867–874. [Google Scholar]
- Scheller, F. W.; Wollenberger, U.; Warsinke, A.; Lisdat, F. Research and development in biosensors. Curr. Opin. Biotech. 2001, 12, 35–40. [Google Scholar]
- Johnston, D. H.; Glasgow, K. C.; Thorp, H. H. Electrochemical Measurement of the Solvent Accessibility of Nucleobases Using Electron Transfer between DNA and Metal Complexes. J. Am. Chem. Soc. 1995, 117, 8933–8938. [Google Scholar]
- Koehne, J. E.; Chen, H.; Cassell, A. M.; Ye, Q.; Han, J.; Meyyappan, M.; Li, J. Miniaturized Multiplex Label-Free Electronic Chip for Rapid Nucleic Acid Analysis Based on Carbon Nanotube Nanoelectrode Arrays. Clin. Chem. 2004, 50, 1886–1893. [Google Scholar]
- Tijsen, P. Practice and theory of enzyme immunoassays; Elsevier Science Publishers: Amsterdam, 1985; Volume 15. [Google Scholar]
- Guilbault, G. G. Handbook of immobilized enzymes; Marcel-Dekker: New York, 1984. [Google Scholar]
- Le, C.-X.; Wu, J.; Wang, H.; Shen, G.-L.; Yu, R.-Q. A new electrochemical immunoassay strategy for detection of transferrin based on electrostatic interaction of natural polymers. Talanta 2004, 63, 469–474. [Google Scholar]
- Hsiue, G.-H.; Lu, P.-L.; Chen, J.-C. Multienzyme-immobilized modified polypropylene membrane for an amperometric creatinine biosensor. J. Appl. Polym. Sci. 2004, 92, 3126–3134. [Google Scholar]
- Bruckenstein, S.; Brzezinska, K.; Hillman, A. R. EQCM Studies of Polypyrrole Films. 1. Exposure to Aqueous Sodium Tosylate Solutions under Thermodynamically Permselective Conditions. Electrochim. Acta 2000, 45, 3801–3811. [Google Scholar]
- Bruckenstein, S.; Brzezinska, K.; Hillman, A. R. EQCM Studies of Polypyrrole Films. 2. Exposure to Aqueous Sodium Tosylate Solutions under Thermodynamically Non-Permselective Conditions. Chem. Phys. 2000, 2, 1221–1229. [Google Scholar]
- Varela, H.; Malta, M.; Torresi, R. M. Microgravimetric study of the influence of the solvent on the redox properties of polypyrrol modified electrodes. J. Power Sources 2001, 92, 50–55. [Google Scholar]
- Lagier, C. M.; Efimov, I.; Hillman, A. R. Film resonance on acoustic wave devices: the roles of frequency and contacting fluid. Anal. Chem. 2005, 77, 335–343. [Google Scholar]
- Li, G.; Wang, Y.; Xu, H. A Hydrogen Peroxide Sensor Prepared by Electropolymerization of Pyrrole Based on Screen-Printed Carbon Paste Electrodes. Sensors 2007, 7, 239–250. [Google Scholar]
- Rajesh; Bisht, V.; Takashima, W.; Kaneto, K. An amperometric urea biosensor based on covalent immobilization of urease onto an electrochemically prepared copolymer poly (N-3-aminopropyl pyrrole-co-pyrrole) film. Biomaterials 2005, 26, 3683–3690. [Google Scholar]
- Fiorito, P. A.; Córdoba de Torresi, S. I. Hybrid nickel hexacyanoferrate/polypyrrole composite as mediator for hydrogen peroxide detection and its application in oxidase-based biosensors. J. Electroanal. Chem. 2005, 581, 31–37. [Google Scholar]
- Fiorito, P. A.; Brett, C. M. A.; Córdoba de Torresi, S. I. Polypyrrole/copper hexacyanoferrate hybrid as redox mediator for glucose biosensors. Talanta 2006, 69, 403–408. [Google Scholar]
- Tahir, Z. M.; Alocilja, E. C.; Grooms, D. L. Indium Tin Oxide-Polyaniline Biosensor: Fabrication and Characterization. Sensors 2007, 7, 1123–1140. [Google Scholar]
- Calvo, E. J.; Danilowicz, C.; Lagier, C. M.; Manrique, J.; Otero, M. Characterization of self-assembled redox polymer and antibody molecules on thiolated gold electrodes. Biosens. Bioelectron. 2004, 19, 1219–1228. [Google Scholar]
- Wang, G.; Xu, J. J.; Chen, H. Y.; Lu, Z. H. Amperometric hydrogen peroxide biosensor with sol-gel/chitosan network-like film as immobilization matrix. Biosens. Bioelectron. 2003, 18, 335–343. [Google Scholar]
- Yu, H.; Yan, F.; Dai, Z.; Ju, H. A disposable amperometric immunosensor for α-1-fetoprotein based on enzyme-labeled antibody/chitosan-membrane-modified screen-printed carbon electrode. Anal. Biochem. 2004, 331, 98–105. [Google Scholar]
- Teles, F. R.; Prazeres, D. M.; Lima-Filho, J. L. Electrochemical Detection of a Dengue-related Oligonucleotide Sequence Using Ferrocenium as a Hybridization Indicator. Sensors 2007, 7, 2510–2518. [Google Scholar]
- Yang, M.; Yang, Y.; Yang, Y.; Shen, G.; Yu, R. Bienzymatic amperometric biosensor for choline based on mediator thionine in situ electropolymerized within a carbon paste electrode. Anal. Biochem. 2004, 334, 127–134. [Google Scholar]
- Emr, S. A.; Yacynych, A. M. Use of polymer films in amperometric biosensors. Electroanalysis 1995, 7, 913–923. [Google Scholar]
- Palmisano, F.; Zambonin, P. G.; Centonze, D. Amperometric biosensors based on electrosynthesised polymeric films. Fresenius. J Anal Chem. 2000, 366, 586–601. [Google Scholar]
- Vidal, J. C.; Garcia-Ruiz, E.; Castillo, J. R. Recent Advances in Electropolymerized Conducting Polymers in Amperometric Biosensors. Microchim. Acta 2003, 143, 93–111. [Google Scholar]
- Ghindilis, A. L.; Makower, A.; Bauer, C. G.; Bier, F. F.; Scheller, F. W. Determination of p-aminophenol and catecholamines at picomolar concentrations based on recycling enzyme amplification. Anal. Chim. Acta 1995, 304, 25–31. [Google Scholar]
- Bier, F. F.; Ehrentreich-Forster, E.; Scheller, F. W.; Makower, A.; Eremenko, A.; Wollenberger, U.; Bauer, C. G.; Pfeiffer, D.; Michael, N. Ultrasensitive biosensors. Sensor. Actuat. B-Chem 1996, 33, 5–12. [Google Scholar]
- Asouzu, M. U.; Nonidez, W. K.; Ho, M. H. Flow Injection Analysis of L-Lactate with Enzyme Amplification and Amperometric Detection. Anal. Chem. 1990, 62, 708–712. [Google Scholar]
- Chaubey, A.; Pande, K. K.; Pandey, M. K.; Singh, V. S.; Malhotra, B. D. Co-immobilization of lactate oxidase and lactate dehydrogenase on conducting polyaniline films. Anal. Chim. Acta 2000, 407, 97–103. [Google Scholar]
- Conrath, N.; Gründig, B.; Hüwel, St.; Camman, K. A novel enzyme sensor for the determination of inorganic phosphate. Anal. Chim. Acta 1995, 309, 47–52. [Google Scholar]
- DiMeglio, L. A.; White, K. E.; Econs, M. J. Disorders of phosphate metabolism. Endocrinol. Metab. Clin. North. Am. 2000, 29, 591–609. [Google Scholar]
- Caruso, F.; Niikura, K.; Furlong, D. N.; Okahata, Y. Assembly of alternating polyelectrolyte and protein multilayer films for immunosensing. Langmuir 1997, 13, 3427–3433. [Google Scholar]
- Caruso, F.; Furlong, D N.; Ariga, K.; Ichinose, I.; Kunitake, T. Characterization of polyelectrolyte-protein multilayer films by atomic force microscopy, scanning electron microscopy and Fourier transform infrared reflection-absorption spectroscopy. Langmuir 1998, 14, 4559–4565. [Google Scholar]
- Danilowicz, C.; Manrique, J. M. A new self-assembled modified electrode for competitive immunoassay. Electrochem. Commun. 1999, 1, 22–25. [Google Scholar]
- Chen, H.; Lee, M.; Choi, S.; Kim, J. H.; Choi, H. J.; Kim, S. H.; Lee, J.; Koh, K. Comparative study of protein immobilization properties on calixarene monolayers. Sensors 2007, 7, 1091–1107. [Google Scholar]
- Chen, H.; Kim, Y. S.; Lee, J.; Yoon, S. J.; Lim, D. S.; Choi, H. J.; Koh, K. Enhancement of BSA Binding on Au Surfaces by calix[4]bisazacrown Monolayer. Sensors 2007, 7, 2263–2272. [Google Scholar]
- Caruso, F.; Rodda, E.; Furlong, D. N. Orientational aspects of antibody immobilization and immunological activity on quartz crystal microbalance electrodes. J. Colloid Interf. Sci. 1996, 178, 104–115. [Google Scholar]
- Chatrathi, M. P.; Wang, J.; Collins, G. E. Sandwich electrochemical immunoassay for the detection of Staphylococcal enterotoxin B based on immobilized thiolated antibodies. Biosens. Bioelecton. 2007, 22, 2932–2938. [Google Scholar]
- Wang, J. Nanomaterial-based electrochemical biosensors. Analyst 2005, 130, 421–426. [Google Scholar]
- Patolsky, F.; Zheng, G.; Liebner, C. M. Nanowire-based biosensors. Anal. Chem. 2006, 78, 4260–4269. [Google Scholar]
- Pumera, M.; Sánchez, S.; Ichinose, I.; Tang, J. Electrochemical nanobiosensors. Sensor. Actuat. B-Chem. 2007, 123, 1195–1205. [Google Scholar]
- Wang, J. From DNA biosensors to gene chips. Nucleic Acids Res. 2000, 28, 3011–3016. [Google Scholar]
- Niemeyer, C. M. Nanoparticles, Proteins, and Nucleic Acids: Biotechnology Meets Materials Science. Angew. Chem. Int. Ed. 2001, 40, 4129–4158. [Google Scholar]
- Wanekaya, A. K.; Chen, W.; Myung, N. V.; Mulchandani, Ashok. Nanowire-Based Electrochemical Biosensors. Electroanalysis 2006, 18, 533–550. [Google Scholar]
- Dequaire, M.; Degrand, C.; Limoges, B. An electrochemical metalloimmunoassay based on colloidal gold label. Anal. Chem. 2000, 72, 5521–5528. [Google Scholar]
- Tang, D.; Yuan, R.; Chai, Y.; Fu, Y.; Dai, J.; Liu, Y.; Zhong, X. New amperometric and potentiometric immunosensors based on gold nanoparticles/tris (2,2′-bipyridyl)cobalt(III) multilayer films for hepatitis B surface antigen determinations. Biosens. Bioelectron. 2005, 21, 539–548. [Google Scholar]
- Jena, B. K.; Raj, C. R. Amperometric L-Lactate Biosensor Based on Gold Nanoparticles. Electroanalysis 2007, 19, 816–822. [Google Scholar]
- Delvaux, M.; Demoustier-Champagne, S. Immobilisation of glucose oxidase within metallic nanotube arrays for application to enzyme biosensors. Biosens. Bioelectron. 2003, 18, 943–951. [Google Scholar]
- Delvaux, M.; Walcarius, A.; Demoustier- Champagne, S. Electrocatalytic H2O2 amperometric detection using gold nanotube electrode ensembles. Anal. Chim. Acta 2004, 525, 221–230. [Google Scholar]
- Li, J.; Ng, H. T.; Cassell, A.; Fan, W.; Chen, H.; Ye, Q.; Koehne, J.; Han, J.; Meyyappan, M. Carbon nanotube nanoelectrode array for ultrasensitive DNA detection. Nano Lett. 2003, 3, 597–602. [Google Scholar]
- Wang, J.; Músame, M.; Lin, Y. Solubilization of Carbon Nanotubes by Nafion toward the Preparation of Amperometric Biosensors. J. Am. Chem. Soc. 2003, 125, 2408–2409. [Google Scholar]
- Rubianes, M. D.; Rivas, G. A. Carbon nanotubes paste electrode. Electrochem. Commun. 2003, 5, 689–694. [Google Scholar]
- Jain, K. K. Nanotechnolgy in clinical laboratory diagnosis. Clin. Chim. Acta 2005, 358, 37–54. [Google Scholar]
- Duan, C.; Meyerhoff, M. E. Separation-free sandwich enzyme immunoassays using microporous gold electrodes and self-assembled monolayer/immobilized capture antibodies. Anal. Chem. 1994, 66, 1369–1377. [Google Scholar]
- Meyerhoff, M. E.; Duan, C.; Meusel, M. Novel nonseparation sandwich-type electrochemical enzyme immunoassay system for detecting marker proteins in undiluted blood. Clin. Chem. 1995, 41, 1378–84. [Google Scholar]
- Zhang, F.; Yang, S. H.; Kang, T. Y.; Cha, G. S.; Nam, H.; Meyerhoff, M. E. A rapid competitive binding nonseparation electrochemical enzyme immunoassay (NEEIA) test strip for microcystin-LR (MCLR) determination. Biosens. Bioelectron. 2007, 22, 1419–1425. [Google Scholar]
- Zhang, F.; Cho, S. S.; Yang, S. H.; Seo, S. S.; Cha, G. Sig; Nam, H. Gold Nanoparticle-Based Mediatorless Biosensor Prepared on Microporous Electrode. Electroanalysis 2006, 18, 217–22. [Google Scholar]
- Patolsky, F.; Wizemann, Y.; Willner, J. Long-range electrical contacting of redox enzymes by SWCNT connectors. Angew. Chem. Int. Ed. 2004, 43, 2113–2117. [Google Scholar]
- Willner, B.; Katz, E.; Willner, I. Electrical contacting of redox proteins by nanotechnological means. Curr. Opin. Biotech. 2006, 17, 589–596. [Google Scholar]
- Kerman, K.; Nagatani, N.; Chikae, M.; Yuhi, T.; Takamura, Y.; Tamiya, E. Label-free electrochemical immunoassay for the detection of human chorionic gonadotropin hormone. Anal. Chem. 2006, 78, 5612–5616. [Google Scholar]
- Vestergaard, M.; Kerman, K.; Tamiya, E. An overview of Label-free Electrochemical Protein Sensors. Sensors 2007, 7, 3442–3458. [Google Scholar]
- Katakis, I.; Mir, M. Towards a fast-responding, label-free electrochemical DNA biosensor. Ana. Bioanal. Chem. 2005, 381, 1033–1035. [Google Scholar]
- Wang, J.; Kawde, A. Pencil-based renewable biosensor for label-free electrochemical detection of DNA hybridization. Anal. Chim. Acta 2001, 431, 219–224. [Google Scholar]
- Kerman, K.; Morita, Y.; Takamura, Y.; Tamiya, E. Label-free electrochemical detection of DNA hybridization on gold electrode. Electrochem. Commun. 2003, 5, 887–891. [Google Scholar]
- Takata, M.; Kerman, K.; Nagataki, N.; Konaka, H.; Namiki, M.; Tamiya, E. Label-free bioelectronic immunoassy for the detection of human telomerase reverse transcriptase in urine. J. Electroanal. Chem. 2006, 596, 109–116. [Google Scholar]
- Janasek, D.; Franzke, J.; Manz, A. Scaling and the design of miniaturized chemical-analysis systems. Nature 2006, 442, 374–380. [Google Scholar]
- Schabmueller, C. G. J.; Loppow, D.; Piechotta, G.; Schutze, B.; Albers, J.; Hintsche, R. Micromachined sensor for lactate monitoring in saliva. Biosens. Bioelectron. 2006, 21, 1770–1776. [Google Scholar]
- de Mello, A. J.; Beard, N. Dealing with “real” samples: sample pre-treatmment in mycrofluidic systems. Lab Chip 2003, 3, 11N–19N. [Google Scholar]
- Wang, J.; Chatrathi, M. P.; Ibanez, A. Glucose biochip: dual analyte response in connection to two pre-column enzymatic reactions. Analyst 2001, 126, 1203–1206. [Google Scholar]
- Wang, J.; Chatrathi, M. P.; Tian, B. Microseparation chips for performing multienzymatic dehydrogenase/oxidase assays: simultaneous electrochemical measurement of ethanol and glucose. Anal. Chem. 2001, 73, 1296–1300. [Google Scholar]
- Wang, Y. C.; Stevens, A. L.; Han, J. Million-fold Preconcentration of Proteins and Peptides by Nanofluidic Filter. Anal. Chem. 2005, 77, 4293–4299. [Google Scholar]
- Wang, J.; Ibanez, A.; Chatrathi, M. P.; Escarpa, A. Electrochemical enzyme immunoassays on microchip platforms. Anal. Chem. 2001, 73, 5323–5327. [Google Scholar]
- Goral, V. N.; Zaytseva, N. V.; Baeumner, A. J. Electrochemical microfluidic biosensor for the detection of nucleic acid sequences. Lab Chip 2006, 6, 414–421. [Google Scholar]
- Soper, S. A.; Brown, K.; Ellington, A.; Frazier, B.; Garcia-Manero, G.; Gau, V.; Gutman, S. I.; Hayes, D. F.; Korte, B.; Landers, J. L.; Larson, D.; Ligler, F.; Majumdar, A.; Mascini, M.; Nolte, D.; Rosenzweig, Z.; Wang, J.; Wilson, D. Point-of-care biosensor systems for cancer diagnostics/prognostics. Biosens. Bioelectron. 2006, 21, 1932–42. [Google Scholar]
- Rasooly, A.; Jacobson, J. Development of biosensors for cancer clinical testing. Biosens. Bioelectron. 2006, 21, 1851–1858. [Google Scholar]
- Petrlova, J.; Potesil, D.; Zehnalek, J.; Sures, B.; Adam, V.; Trnkova, L.; Kizek, R. Cisplatin electrochemical biosensor. Electrochim. Acta 2006, 51, 5169–5173. [Google Scholar]
- Daniels, J. S.; Pourmand, N. Label-Free Impedance Biosensors: Opportunities and Challenges. Electroanalysis 2007, 19, 1239–1257. [Google Scholar]
- Barrettino, D. Design considerations and recent advances in CMOS-based microsystems for point-of-care clinical diagnostics. ISCAS 2006. Proc. IEEE Int. Symp. 2006, 4359–4362. [Google Scholar]
- Tüdos, A. J.; Besselink, G. A. J.; Schasfoort, R. B. M. Trends in miniaturized total analysis systems for point-of-care testing in clinical chemistry. Lab Chip 2001, 1, 83–95. [Google Scholar]
- Yager, P.; Edwards, T.; Fu, E.; Helton, K.; Nelson, K.; Tam, M. R.; Weigl, B H. Microfluidic diagnostic technologies for global public health. Nature 2006, 442, 412–418. [Google Scholar]
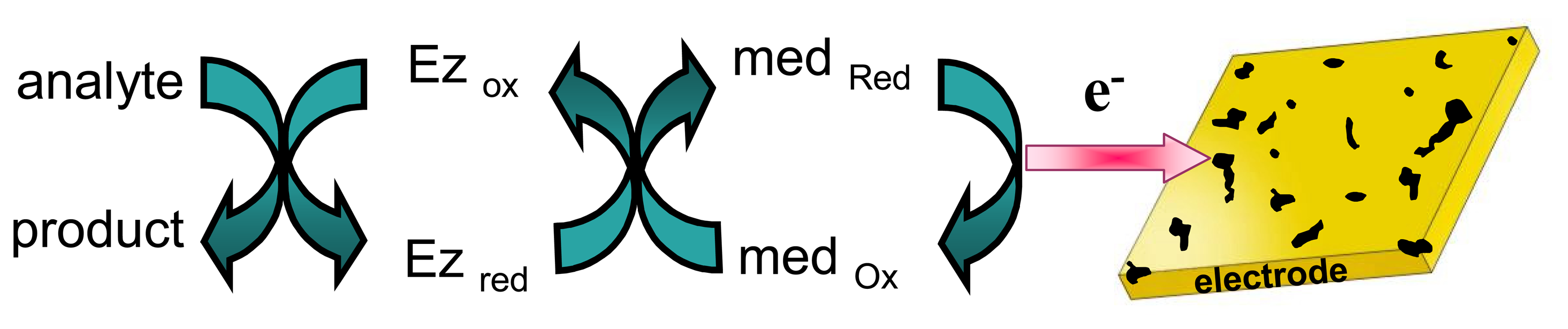
 ) is recycled as long the electroactive species (
) is recycled as long the electroactive species (
 ) is consumed.
) is recycled as long the electroactive species (
) is consumed.
) is consumed.
) is recycled as long the electroactive species (
) is consumed.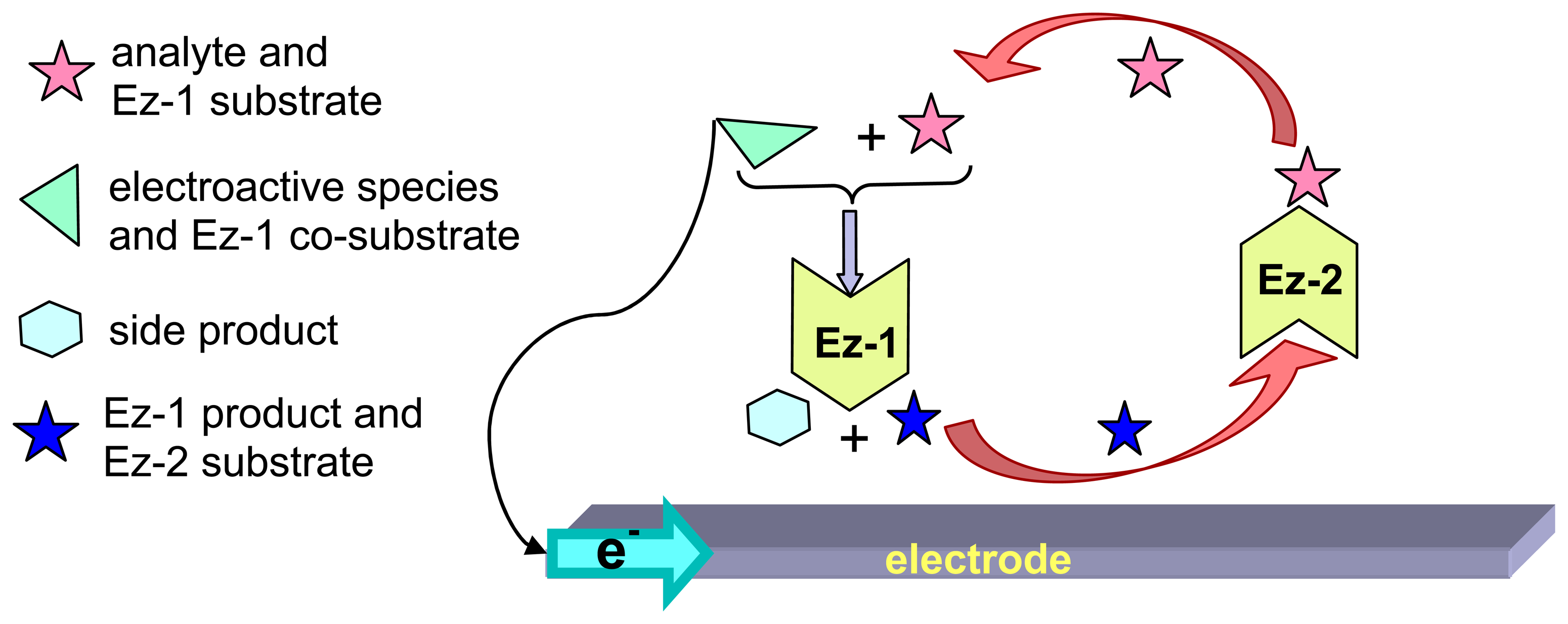

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
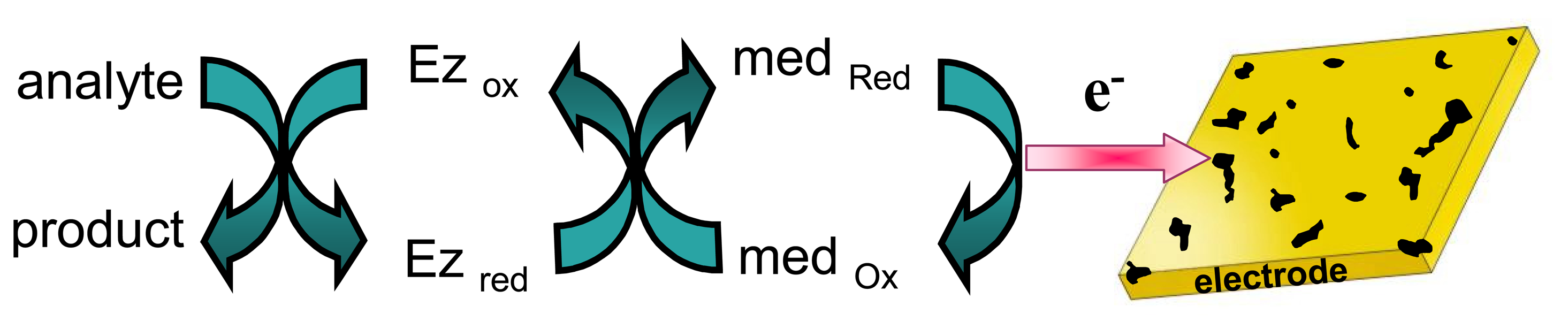{kind=link}
{kind=link}
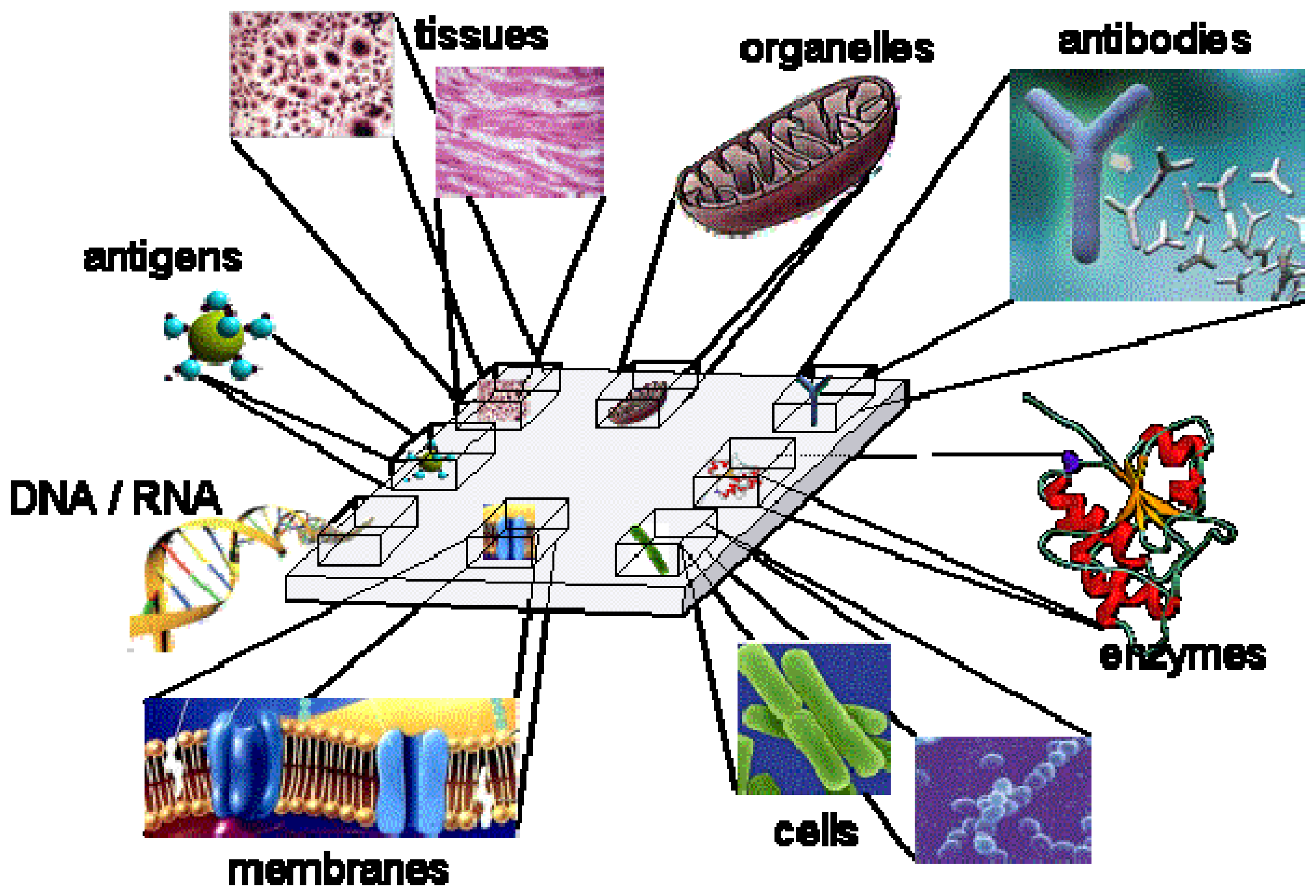
| Biorecognition layer (complexity order increases to the bottom) | Main requests for functional integrity (the needs usually add up to the bottom) | |
|---|---|---|
| Single molecule | Aptamer / ion carrier (ionophore) | Appropriate withholding |
| Antibody / DNA | pH and saline stability | |
| Enzyme / high molecular weigh protein | Absence of certain poisoning products (e.g. heavy metals) | |
| Membrane receptor | Mechanic protection | |
| Organelle (chloroplast, mitochondrion, etc) | Appropriate osmotic pressure, oxygen | |
| Cell | Nutrients | |
| Tissue | ||
| Organ | Undamaged tissue | |
© 2008 by MDPI Reproduction is permitted for noncommercial purposes.
Share and Cite
Belluzo, M.S.; Ribone, M.E.; Lagier, C.M. Assembling Amperometric Biosensors for Clinical Diagnostics. Sensors 2008, 8, 1366-1399. https://doi.org/10.3390/s8031366
Belluzo MS, Ribone ME, Lagier CM. Assembling Amperometric Biosensors for Clinical Diagnostics. Sensors. 2008; 8(3):1366-1399. https://doi.org/10.3390/s8031366
Chicago/Turabian StyleBelluzo, María Soledad, María Elida Ribone, and Claudia Marina Lagier. 2008. "Assembling Amperometric Biosensors for Clinical Diagnostics" Sensors 8, no. 3: 1366-1399. https://doi.org/10.3390/s8031366
APA StyleBelluzo, M. S., Ribone, M. E., & Lagier, C. M. (2008). Assembling Amperometric Biosensors for Clinical Diagnostics. Sensors, 8(3), 1366-1399. https://doi.org/10.3390/s8031366