
Enhanced Tumor Diagnostics via Cyber-Physical Workflow: Integrating Morphology, Morphometry, and Genomic MultimodalData Analysis and Visualization in Digital Pathology
 , , and
, , and
Abstract
1. Introduction
1.1. 10X Genomics
1.2. Bruker Corporation
1.3. Oncompass
1.4. TurbineAI
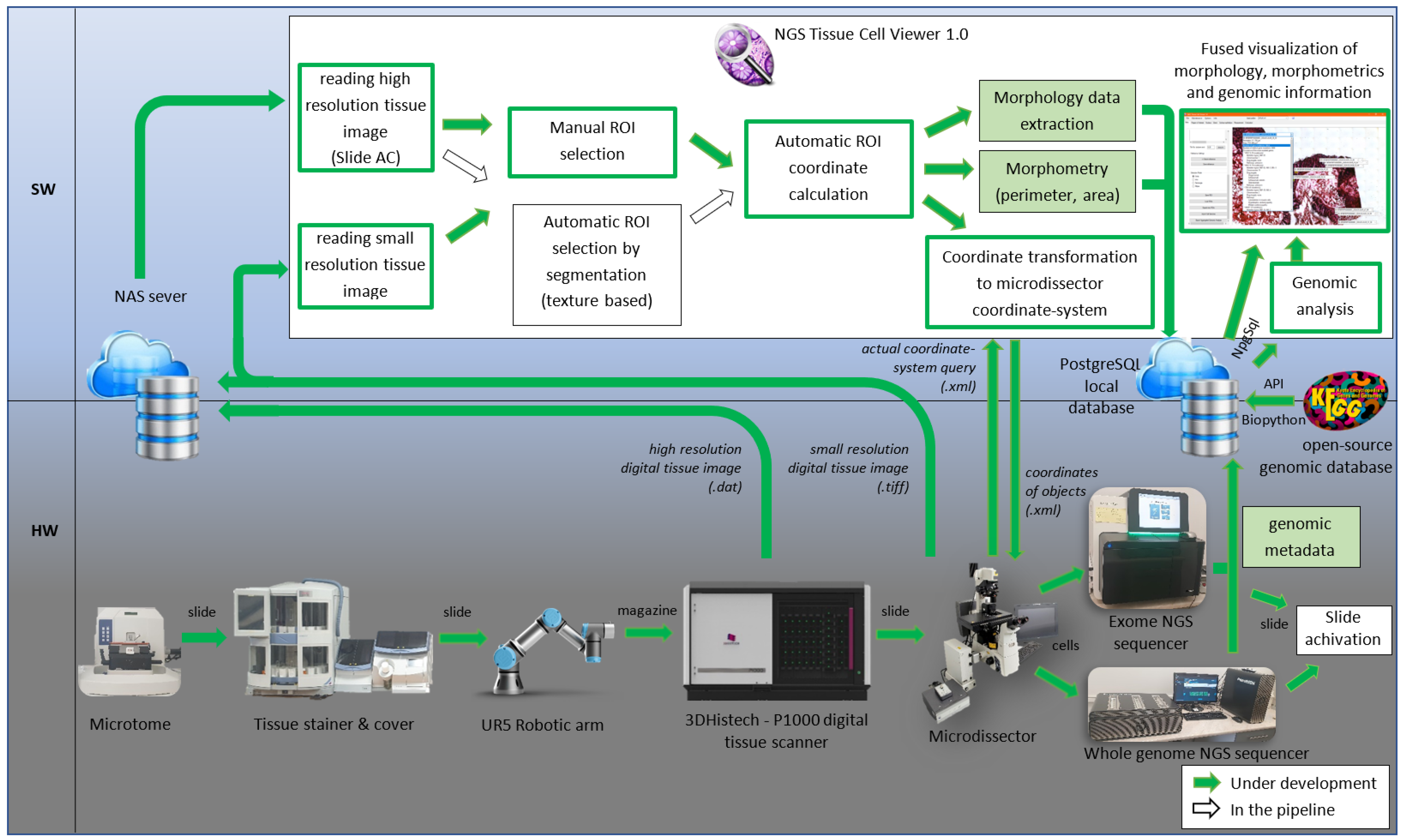
2. Materials and Methods
2.1. Tissue Preparation on Membrane Slides
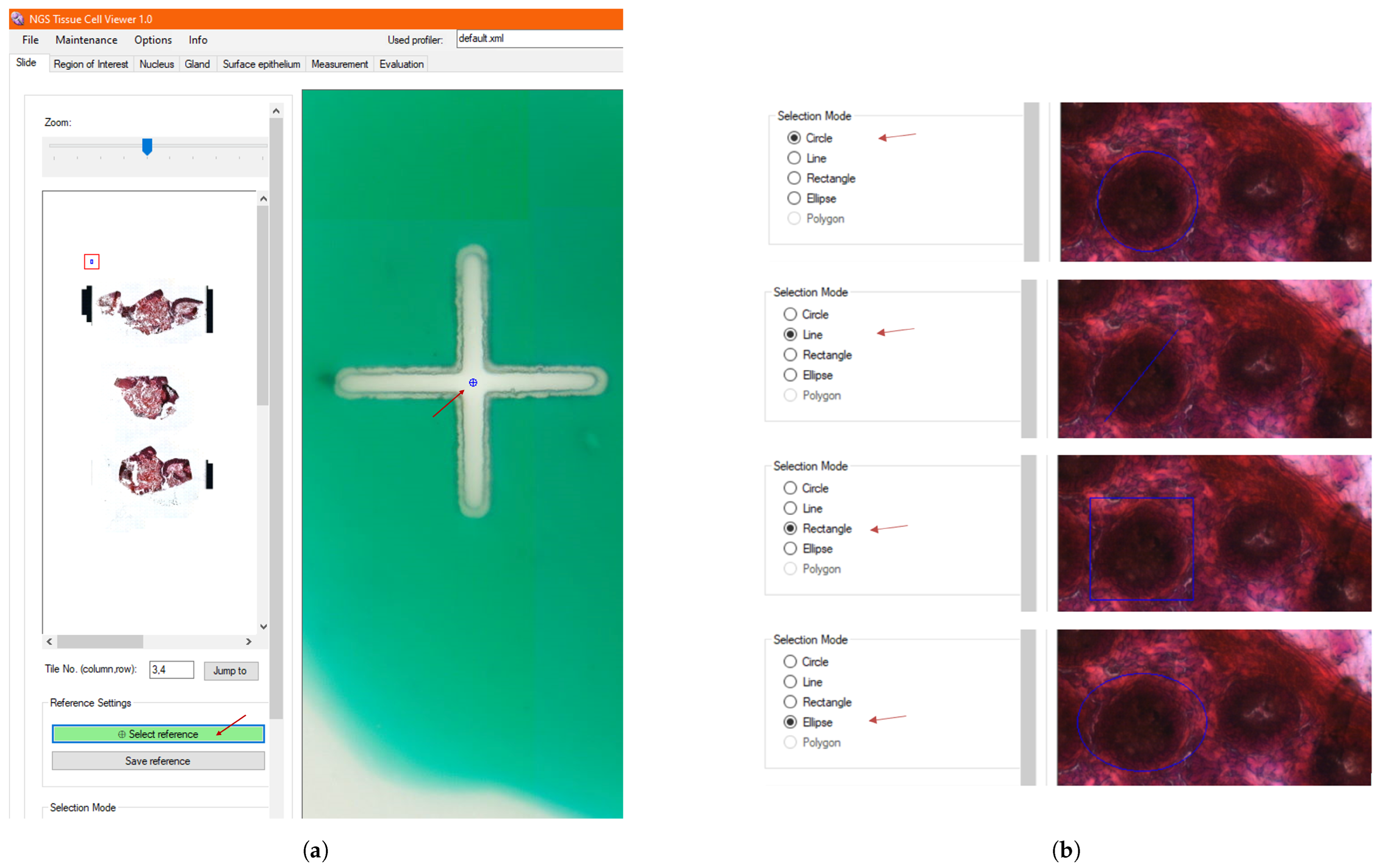
2.2. Slide Detection and Manipulation
2.3. Scanning Slides
2.4. Image Processing of Scanned Morphology
2.5. Morphometry Calculation
2.6. Laser Microdissection
2.7. DNA Sequencing
- DRAGENSnpHardQUAL: For SNP variants, if the QUAL score is below 10.41, the variant is filtered out.
- DRAGENIndelHardQUAL: For INDEL variants, if the QUAL score is below 7.83, the variant is filtered out.
- LowDepth: Variants with a depth (DP) less than or equal to 1 (DP ≤ 1) are filtered out.
- PloidyConflict: Variants whose genotypic calls are inconsistent with the chromosome ploidy are filtered out.
- base_quality: Variants are filtered if the median base quality of alternative reads at the locus falls below a threshold.
- lod_fstar: For mitochondrial contigs, if the LOD (log-likelihood) score does not exceed the threshold of 6.3, the variant is filtered out.
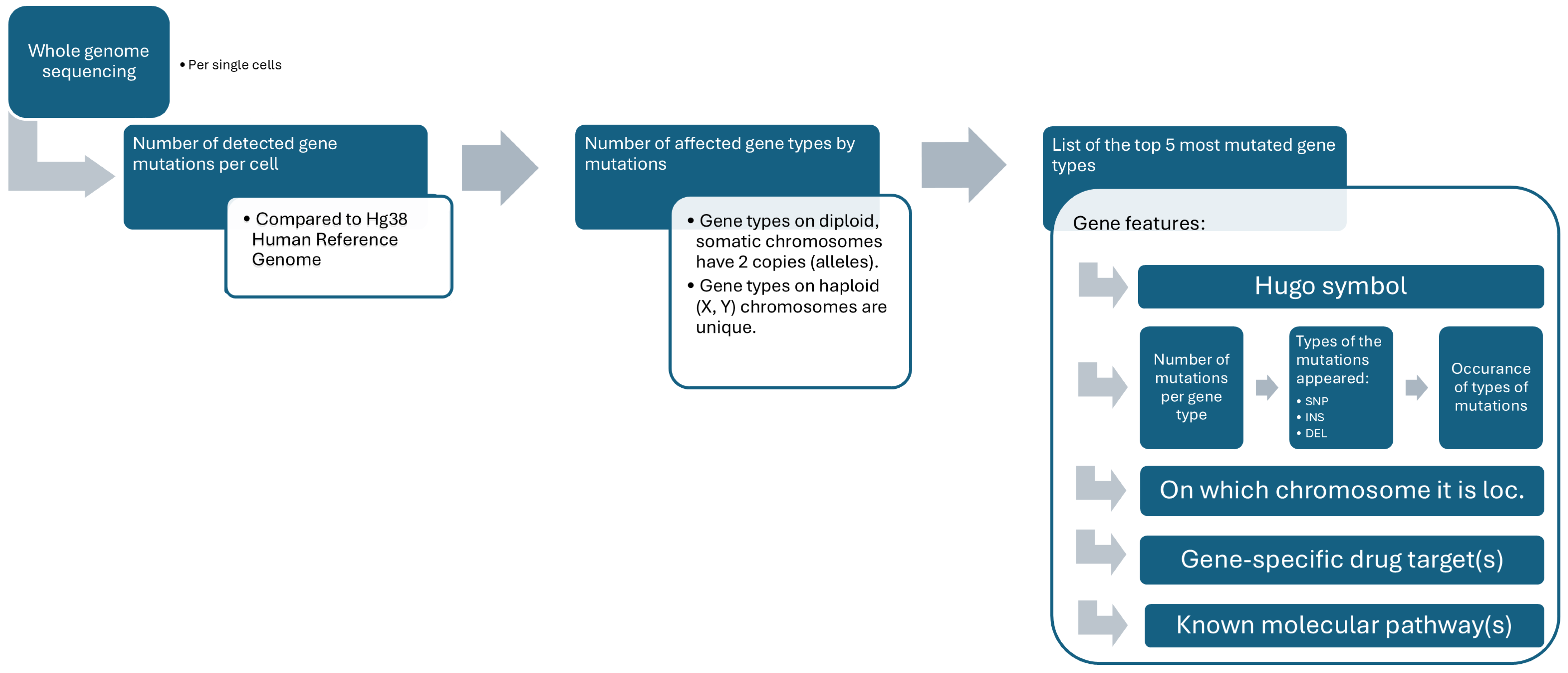
2.8. Genomic Analysis
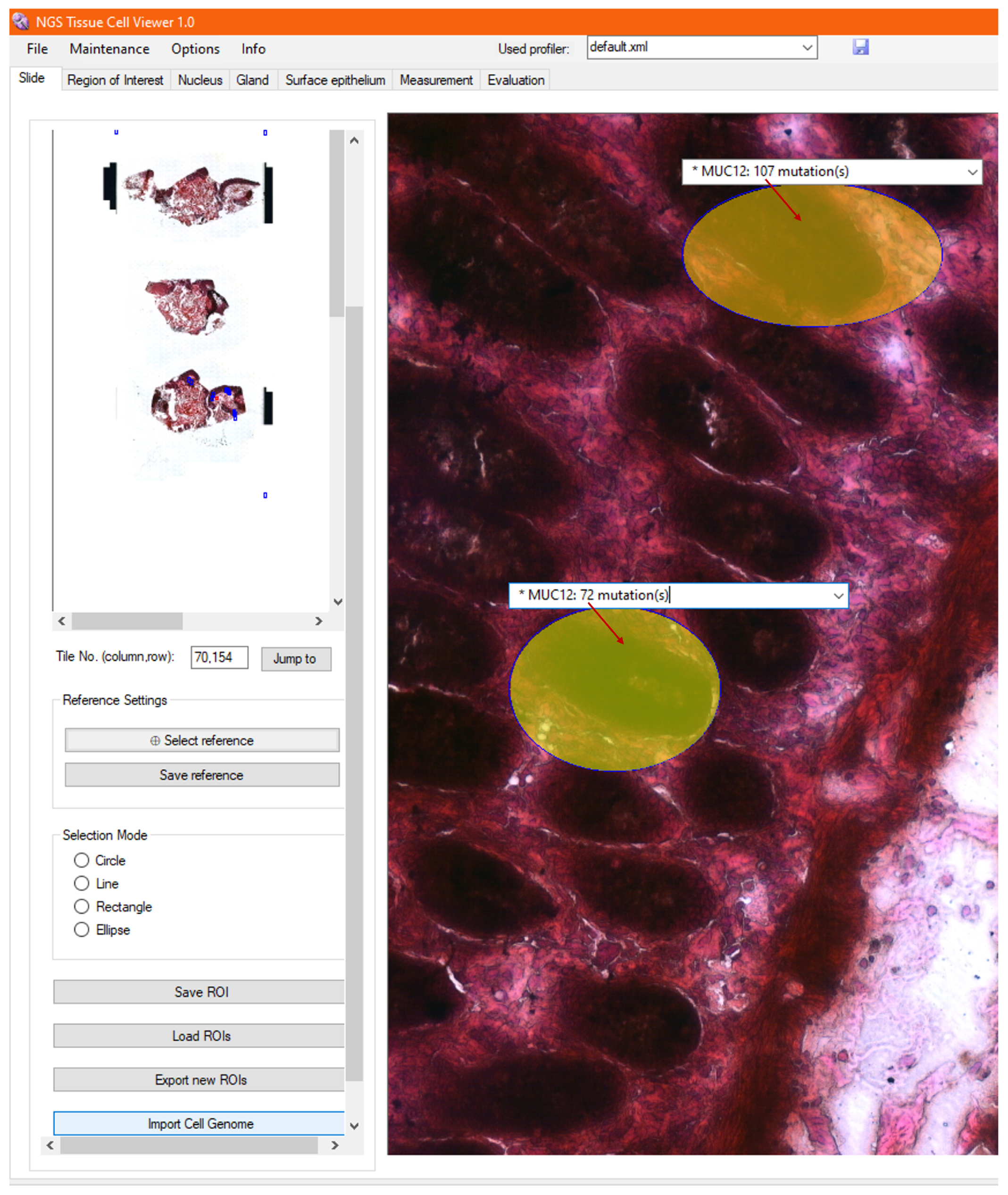
2.9. Visualization of Fused Data
2.9.1. Annotation
2.9.2. Heatmap
3. Results
3.1. System Feasibility
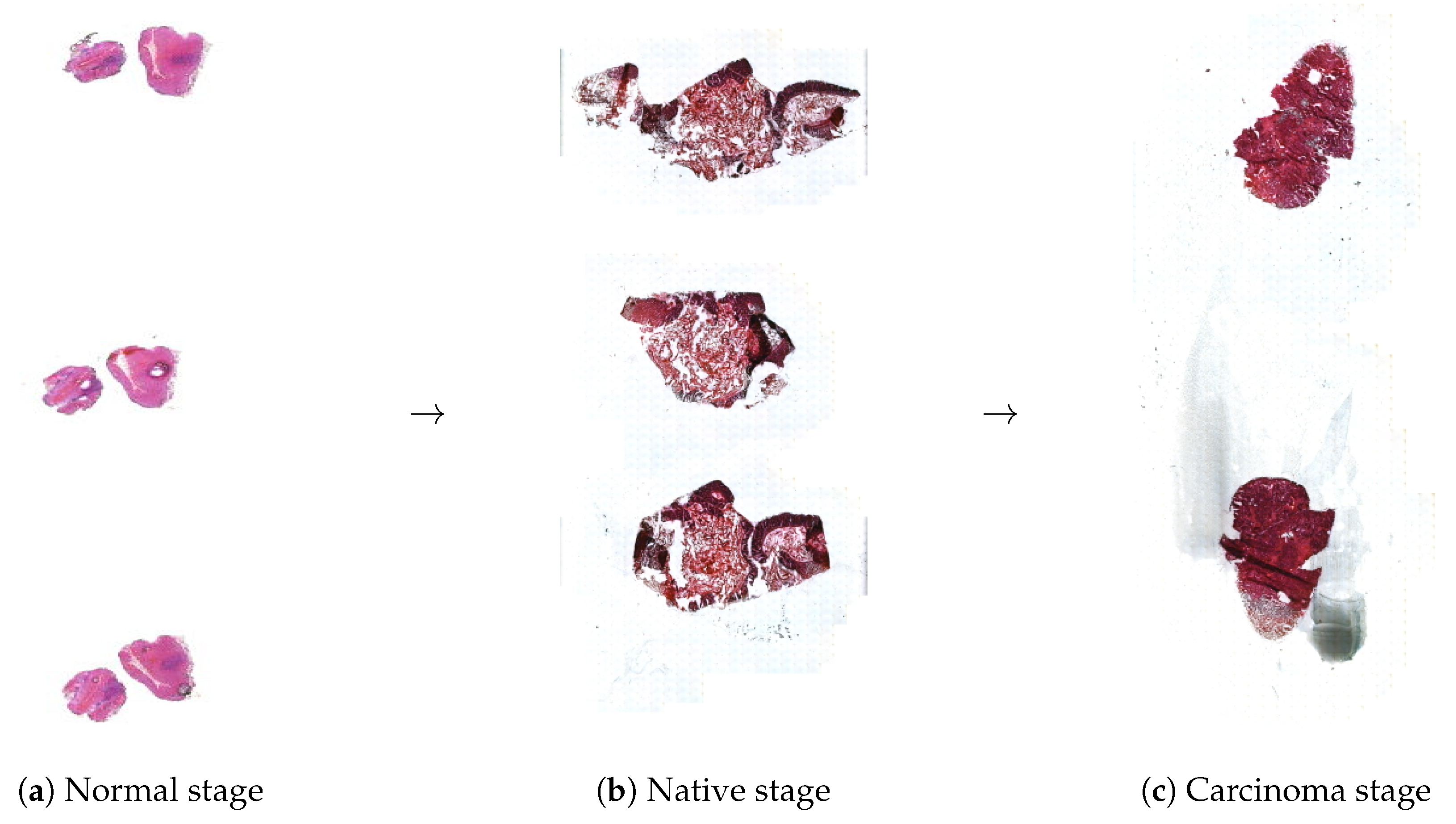
3.2. Scanned Membrane Slides
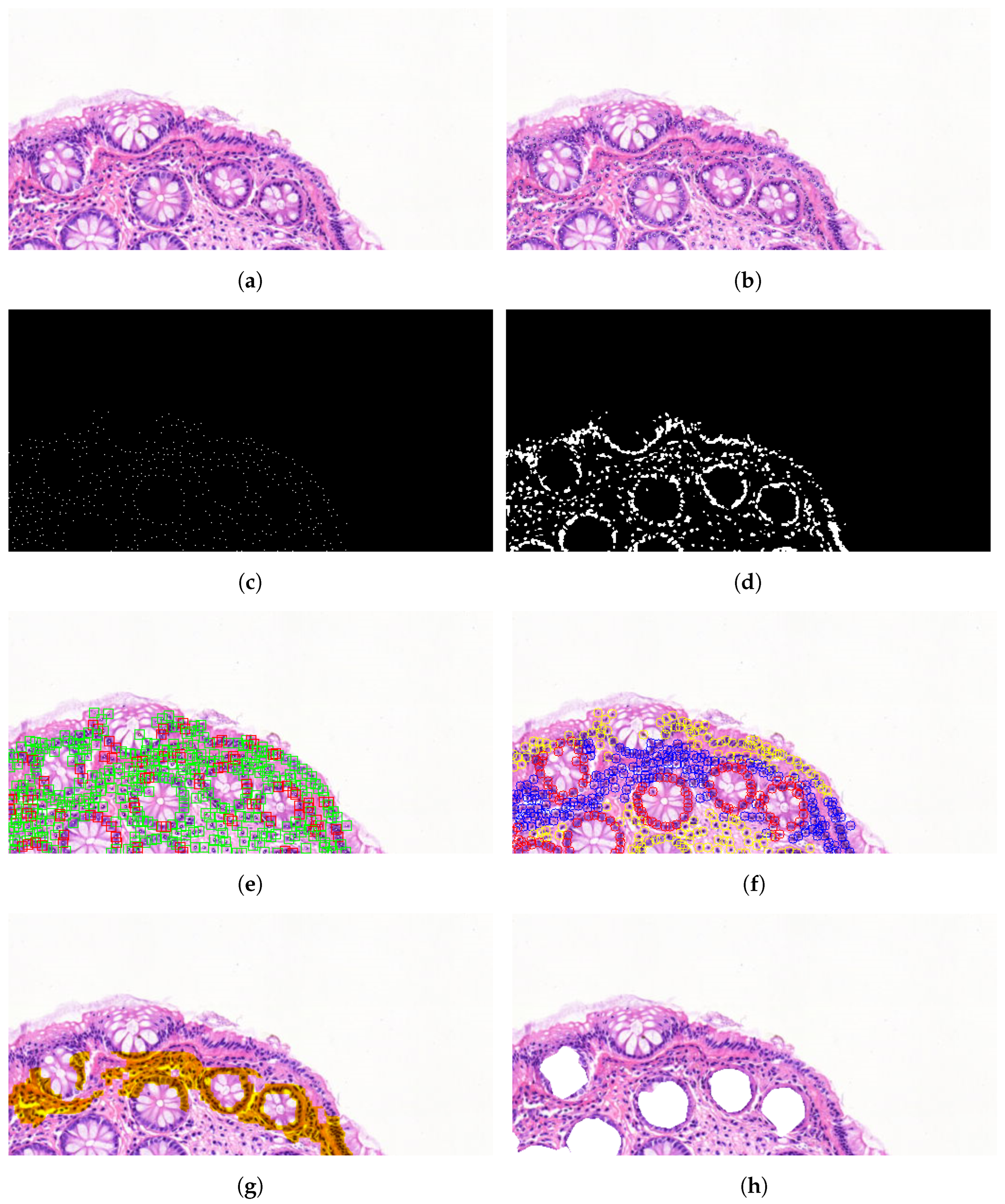
3.3. Algorithmic Selection
3.4. Manual Selection
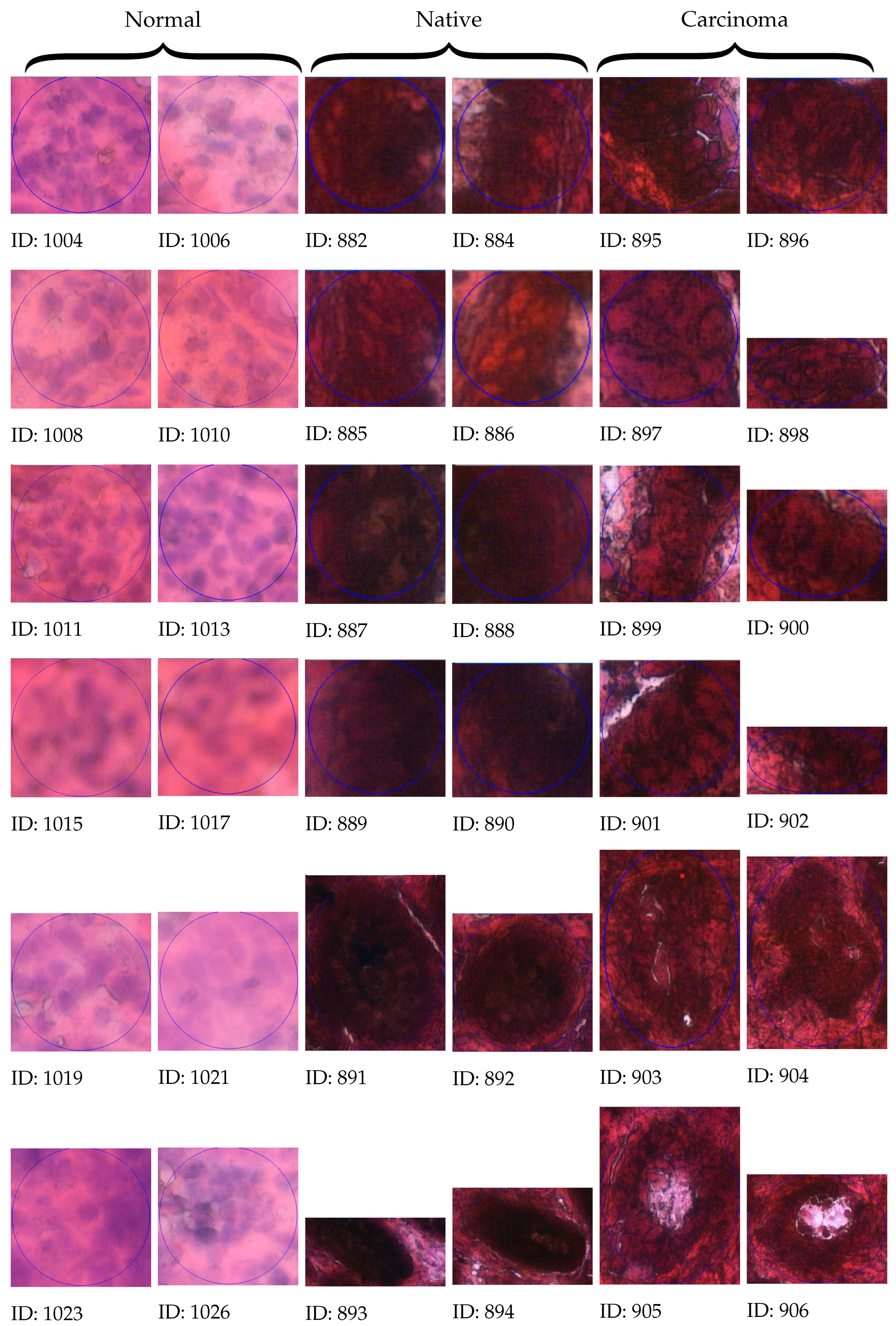
3.5. Calculated Morphometry
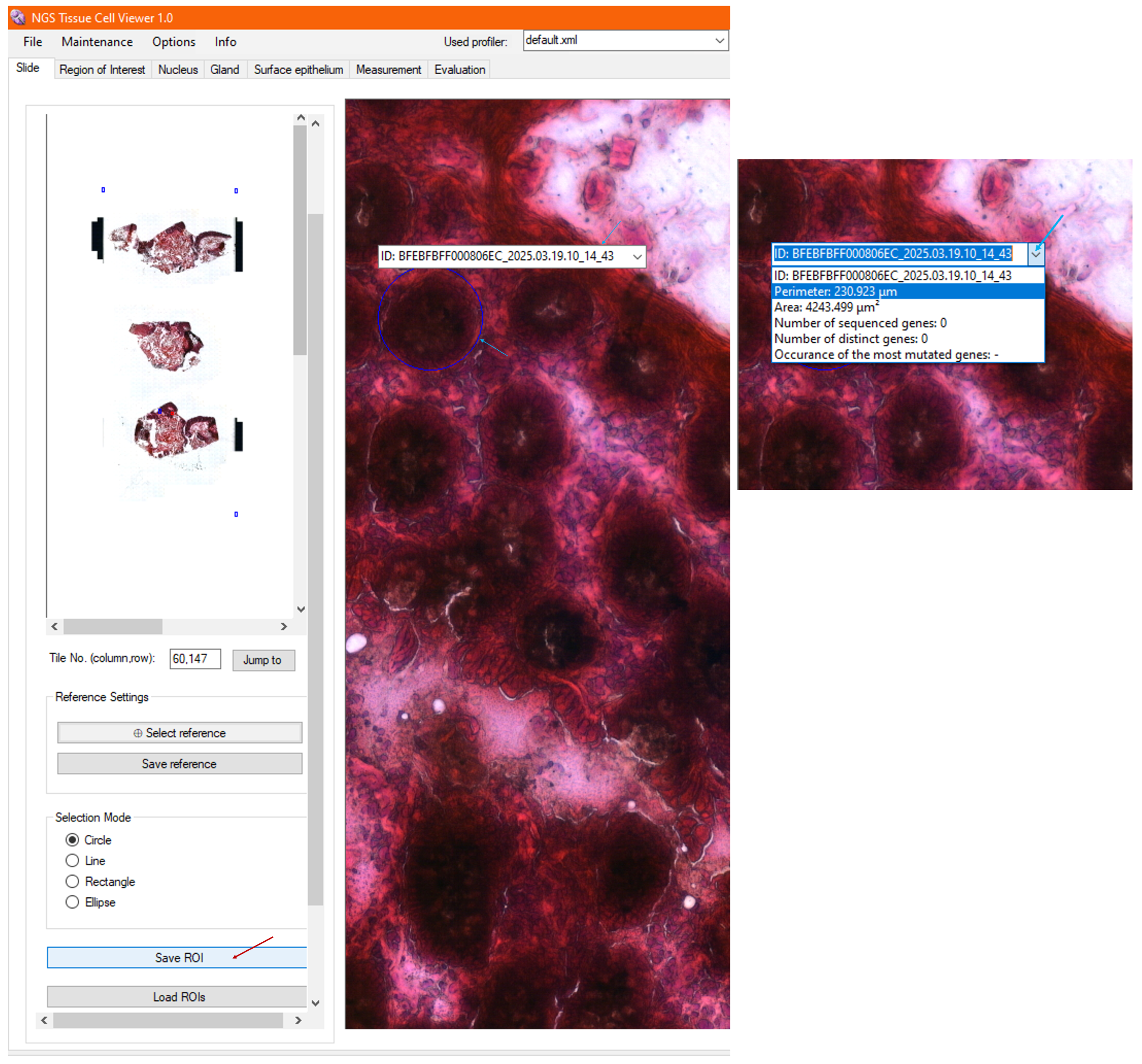
3.6. Exporting Single-Cell Morphology and Morphometry to LMD
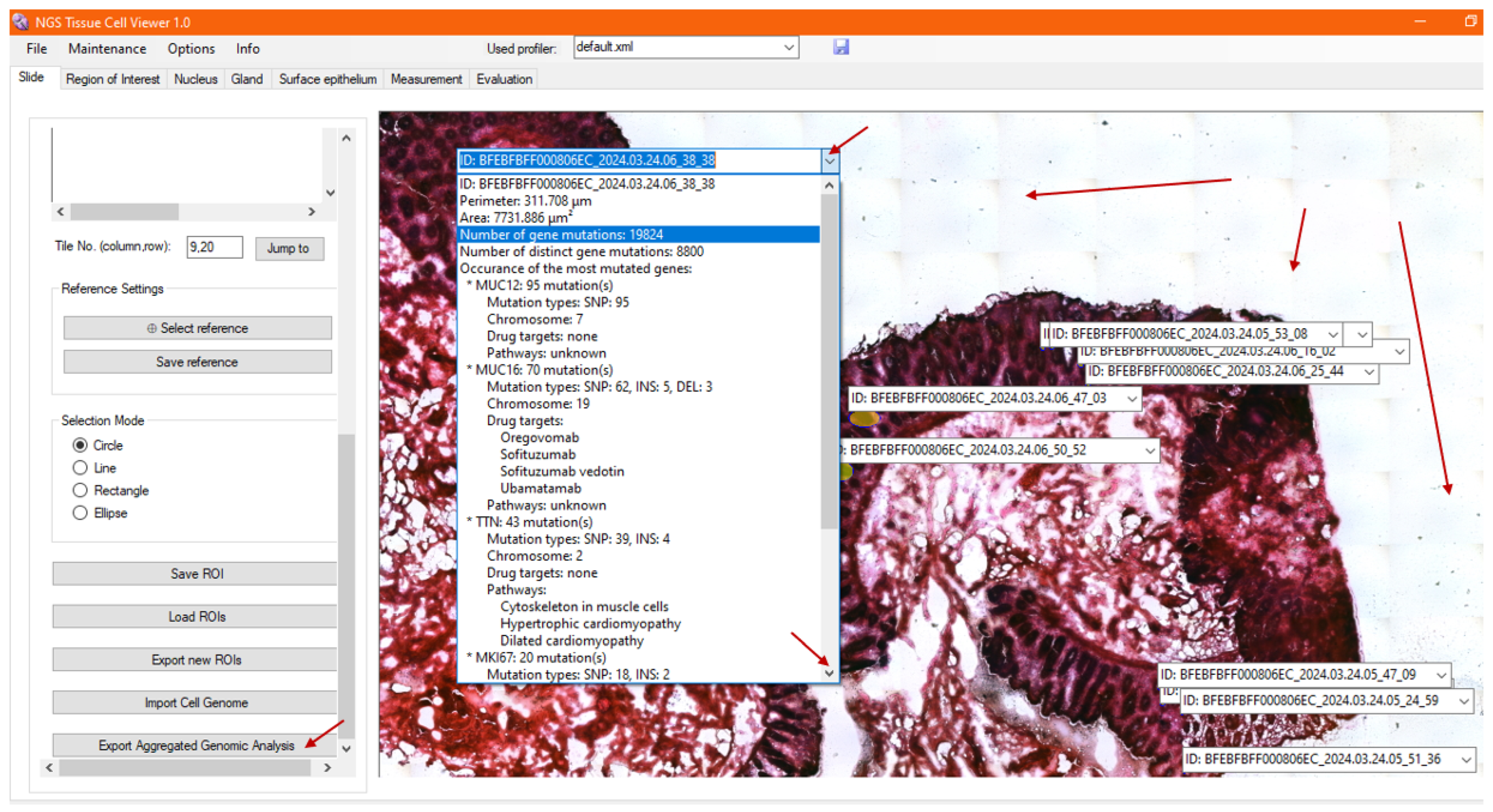
3.7. Importing Genomic Metadata
3.8. Loading Archived ROIs
3.9. Visualization of Genomic Analysis, Together with Morphology and Morphometry
3.10. Visualization of Tumor Foci
3.11. NGS Viewer System Testing
4. Discussion
4.1. Workflow Integration in Laboratories
4.2. Clinical Relevance of the Proposed Framework
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| A | Adenine |
| API | Application programming interface |
| C | Cytosine |
| CRC | Carcinoma (cancerous tissue) |
| DB | Database |
| DEL | Deletion of nucleotide sequence |
| DNA | Deoxyribonucleic acid |
| DSC | Dice similarity coefficient |
| FFPE | Formalin-fixed and paraffin-embedded |
| G | Guanine |
| GUI | Graphical user interface |
| HG38 | Human reference genome 38 |
| IHC | Immunohistochemical |
| INS | Insertion of nucleotide sequence |
| LMD | Laser microdissection machine |
| MMI | Molecular Machines & Industries Ltd. |
| NAT | Native tissue (the nearest intact tissue sample to cancer) |
| NGS | Next-generation sequencing |
| NORM | Normal tissue (healthy) |
| ROI | Region of interest |
| SNP | Single-nucleotide polymorphism |
| T | Thymine |
| TME | Tumor microenvironment |
| TTN | Titin protein |
| ULT | Ultra-low temperature |
| U | Uracil |
References
- Fischer, E. Nuclear Morphology and the Biology of Cancer Cells. Acta Cytol. 2020, 64, 511–516. [Google Scholar] [CrossRef] [PubMed]
- Folkman, J. Tumor Angiogenesis: Therapeutic Implications. N. Engl. J. Med. 1971, 285, 1181–1186. [Google Scholar] [CrossRef]
- Kinzler, K.; Vogelstein, B. Lessons from Hereditary Colorectal Cancer. Cell 1996, 87, 159–170. [Google Scholar] [CrossRef] [PubMed]
- Garber, M.E.; Troyanskaya, O.G.; Schluens, K.; Petersen, S.; Thaesler, Z.; Pacyna-Gengelbach, M.; van de Rijn, M.; Rosen, G.D.; Perou, C.M.; Whyte, R.I.; et al. Diversity of gene expression in adenocarcinoma of the lung. Proc. Natl. Acad. Sci. USA 2001, 98, 13784–13789. [Google Scholar] [CrossRef] [PubMed]
- Perou, C.; Sørlie, T.; Eisen, M.; van de Rijn, M.; Jeffrey, S.; Rees, C.; Pollack, J.; Ross, D.; Johnsen, H.; Aksle, N.L.; et al. Molecular portraits of human breast tumours. Nature 2000, 406, 747–752. [Google Scholar] [CrossRef] [PubMed]
- Yachida, S.; Jones, S.; Bozic, I.; Antal, T.; Leary, R.; Fu, B.; Kamiyama, M.; Hruban, R.; Eshleman, J.; Nowak, M.; et al. Distant Metastasis Occurs Late during the Genetic Evolution of Pancreatic Cancer. Nature 2010, 467, 1114–1117. [Google Scholar] [CrossRef] [PubMed]
- Jones, S.; Zhang, X.; Parsons, D.; Lin, J.; Leary, R.; Angenendt, P.; Mankoo, P.; Carter, H.; Kamiyama, H.; Jimeno, A.; et al. Core Signaling Pathways in Human Pancreatic Cancers Revealed by Global Genomic Analyses. Science 2008, 321, 1081–1086. [Google Scholar] [CrossRef] [PubMed]
- Burrell, R.A.; McGranahan, N.; Bartek, J.; Swanton, C. The Causes and Consequences of Genetic Heterogeneity in Cancer Evolution. Nature 2013, 501, 338–345. [Google Scholar] [CrossRef] [PubMed]
- Gerlinger, M.; Horswell, S.; Larkin, J.; Rowan, A.; Salm, M.; Varela, I.; Fisher, R.; McGranahan, N.; Matthews, N.; Santos, C.; et al. Genomic Architecture and Evolution of Clear Cell Renal Cell Carcinomas Defined by Multiregion Sequencing. Nat. Genet. 2014, 46, 225–233. [Google Scholar] [CrossRef] [PubMed]
- Knoche, S.M.; Larson, A.C.; Sliker, B.H.; Poelaert, B.J.; Solheim, J.C. The role of tumor heterogeneity in immune-tumor interactions. Cancer Metastasis Rev. 2021, 40, 377–389. [Google Scholar] [CrossRef] [PubMed]
- Germanà, E.; Ludovica, P.; Pizzimenti, C.; Ballato, M.; Pierconti, F.; Tuccari, G.; Ieni, A.; Giuffrè, G.; Fadda, G.; Fiorentino, V.; et al. Programmed Cell Death Ligand 1 (PD-L1) Immunohistochemical Expression in Advanced Urothelial Bladder Carcinoma: An Updated Review with Clinical and Pathological Implications. Int. J. Mol. Sci. 2024, 25, 6750. [Google Scholar] [CrossRef] [PubMed]
- Beca, F.; Polyak, K. Intratumor Heterogeneity in Breast Cancer. In Novel Biomarkers in the Continuum of Breast Cancer; Springer International Publishing: Cham, Switzerland, 2016; pp. 169–189. [Google Scholar] [CrossRef]
- 10X Genomics. Compare Products. Available online: https://www.10xgenomics.com/products (accessed on 11 May 2024).
- 10X Genomics. In Situ Gene Experssion. Available online: https://www.10xgenomics.com/support/in-situ-gene-expression (accessed on 11 May 2024).
- 10X Genomics. Find Product User Guide for In Situ Gene Expression. Available online: https://www.10xgenomics.com/support/user-guides/in-situ-gene-expression?menu%5bproductNames%5d=In%20Situ%20Gene%20Expression (accessed on 11 May 2024).
- NanoString, A Bruker Company. What is Spatial Biology? 2022. Available online: https://nanostring.com/blog/how-do-i-choose-the-right-spatial-biology-technology/ (accessed on 11 May 2024).
- NanoString, A Bruker Company. What is Spatial Transcriptomics? Available online: https://nanostring.com/research-focus/spatial-transcriptomics/ (accessed on 11 May 2024).
- Oncompass Gmbh. Precision Oncology Program. Available online: https://oncompassmedicine.com/about-the-process# (accessed on 11 May 2024).
- Papp, O.; Jordán, V.; Hetey, S.; Balázs, R.; Kaszás, V.; Bartha, Á.; Ordasi, N.N.; Kamp, S.; Farkas, B.; Mettetal, J.; et al. Network-driven cancer cell avatars for combination discovery and biomarker identification for DNA damage response inhibitors. NPJ Syst. Biol. Appl. 2024, 10, 68. [Google Scholar] [CrossRef]
- Mareček-Kolibiský, M.; Janík, S.; Mĺkva, M.; Szabó, P.; Czifra, G. Human-Machine Co-Working for Socially Sustainable Manufacturing in Industry 4.0. Acta Polytech. Hung. 2024, 21, 33–53. [Google Scholar] [CrossRef]
- Kozlovszky, M.; Hegedűs, K.; Szenasi, S.; Kiszler, G.; Wichmann, B.; Bándi, I.; Eigner, G.; Sas, P.; Kovács, L.; Garaguly, Z.; et al. Parameter assisted HE colored tissue image classification. In Proceedings of the 2013 IEEE 17th International Conference on Intelligent Engineering Systems (INES), San Jose, Costa Rica, 19–21 June 2013. [Google Scholar] [CrossRef]
- Thermo Scientific. Thermo Scientific Gemini AS Operator Guide Issue 10. Available online: https://www.medwrench.com/documents/view/15154/thermo-scientific-gemini-as-operator-guide-issue-10 (accessed on 28 April 2025).
- Cardiff, R.D.; Miller, C.H.; Munn, R.J. Manual Hematoxylin and Eosin Staining of Mouse Tissue Sections. Cold Spring Harb. Protoc. 2014, 6, 654–658. [Google Scholar] [CrossRef] [PubMed]
- Epredia. ClearVue Coverslipper. Available online: https://www.epredia.com/products/histology-instruments/staining-and-coverslipping/clearvue (accessed on 28 April 2025).
- Universal Robots A/S. UR5e. Available online: https://www.universal-robots.com/products/ur5e/ (accessed on 28 April 2025).
- Kucarov, M.D.; Molnár, B.; Kozlovszky, M. Robot Instead of Laboratory Technicians - Slide Holder Detection and 3D Position Determination by Robotic Arm. In Proceedings of the 2022 IEEE 26th International Conference on Intelligent Engineering Systems (INES), Georgioupolis Chania, Greece, 12–15 August 2022; pp. 97–102. [Google Scholar] [CrossRef]
- Kucarov, M.D.; Takács, M.; Molnár, B.; Kozlovszky, M. Transparent Slide Detection and Gripper Design for Slide Transport by Robotic Arm. In Proceedings of the 2022 IEEE 22nd International Symposium on Computational Intelligence and Informatics and 8th IEEE International Conference on Recent Achievements in Mechatronics, Automation, Computer Science and Robotics (CINTI-MACRo), Budapest, Hungary, 21–22 November 2022; pp. 31–36. [Google Scholar] [CrossRef]
- Kucarov, M.D.; Molnár, B.; Kozlovszky, M. Calibration of Robotic Arm for Workstation Installation in Changing Environment—Comparison of Manual, Mechanic, and Automatic Calibration. In Proceedings of the 2023 IEEE 17th International Symposium on Applied Computational Intelligence and Informatics (SACI), Timisoara, Romania, 23–26 May 2023; pp. 593–598. [Google Scholar] [CrossRef]
- 3DHISTECH. Pannoramic 1000 User’s Guide. 2018. Available online: https://assets.thermofisher.com/TFS-Assets/APD/Product-Guides/US-Only-P1000-BF-Users-Guide-EN-2018-04.pdf (accessed on 11 May 2024).
- Kucarov, M.D.; Molnár, B.; Kozlovszky, M. Localization and Conversion of Single Cell Positions from Static High-Resolution Digital Images to Lasermicrodissector Coordinate System through Utilization of References and 2D Transformation Techniques. Acta Polytech. Hung. 2024, 20, 257–279. [Google Scholar] [CrossRef]
- Kucarov, M.D.; Molnár, B.; Kozlovszky, M. Single Cell Position Determination and Transformation From Static High-resolution Digital Image To Laser-microdissector Coordinate System Using Image Processing Techniques. In Proceedings of the 2023 IEEE 17th International Symposium on Applied Computational Intelligence and Informatics (SACI), Timisoara, Romania, 23–26 May 2023; pp. 195–201. [Google Scholar] [CrossRef]
- Molecular Machines; Industries GmbH. mmi CellCut User Manual. 2020. Available online: https://scopem.ethz.ch/center/instruments-alphabetical/nikon-mm1-md.html (accessed on 11 May 2024).
- Illumina, Incorporation. MiniSeq Sequencing System Applications and Methods. Available online: https://www.illumina.com/systems/sequencing-platforms/miniseq/applications.html (accessed on 11 May 2024).
- Oxford Nanopore Technologies plc. PromethION 24/48. Available online: https://nanoporetech.com/products/sequence/promethion-24-48 (accessed on 11 May 2024).
- Illumina, Incorporation. Illumina DRAGEN Bio-IT Platform v3.6 User Guide. Available online: https://support.illumina.com/content/dam/illumina-support/documents/documentation/software_documentation/dragen-bio-it/dragen-bio-it-platform-v3.6-user-guide-1000000128306-00.pdf (accessed on 5 March 2025).
- Illumina, Incorporation. Germline Variant Small Hard Filtering. Available online: https://jp.support.illumina.com/content/dam/illumina-support/help/Illumina_DRAGEN_Bio_IT_Platform_v3_7_1000000141465/Content/SW/Informatics/Dragen/GPipelineVarCalFilt_fDG.htm (accessed on 5 March 2025).
- Kucarov, M.D.; Molnár, B.; Kozlovszky, M. Patho-Genomics Fusioned Database Schema and Optimisation for Automatic Pathology Workflow. In Proceedings of the 2023 IEEE 21st World Symposium on Applied Machine Intelligence and Informatics (SAMI), Herl’any, Slovakia, 19–21 January 2023; pp. 317–323. [Google Scholar] [CrossRef]
- Kanehisa Laboratories. KEGG: Kyoto Encyclopedia of Genes and Genomes. Available online: https://www.genome.jp/kegg/ (accessed on 25 June 2024).
- Kucarov, M.D.; Molnár, B.; Kozlovszky, M. Integration of NGS Genomic Metadata Analysis with Open-source Genomic Databases for Single Cell Tissue Samples. In Proceedings of the 2025 IEEE 12th International Conference on Computational Cybernetics and Cyber-Medical Systems (ICCC), Beau Vallon, Seychelles, 9–11 April 2025. [Google Scholar]
- DocCheck Community GmbH. Chromosom. 2024. Available online: https://flexikon.doccheck.com/de/Chromosom (accessed on 8 August 2024).
- National Center for Biotechnology Information (NCBI). Chromosome Map. 2016. Available online: https://www.ncbi.nlm.nih.gov/books/NBK22266/ (accessed on 8 August 2024).
- The Npgsql Development Team. Npgsql Getting Started. Available online: https://www.npgsql.org/doc/index.html (accessed on 25 June 2024).


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Vendor | Product | Intended Purpose | Key Features |
|---|---|---|---|
| 10X Genomics (Pleasanton, CA, USA) | Chromium Single Cell | Gene expression profiling | Multiomic options in fresh, frozen, and formalin-fixed and paraffin-embedded (FFPE) tissue |
| 10X Genomics | Xenium In Situ platform | Targeting 100s to 1000s of RNAs in individual cells | In fresh, frozen, or FFPE tissue; subcellular profiling; and gene expression |
| 10X Genomics | Visium Spacial | Performing unbiased spatial discovery | Probe-based: HD spatial gene expression |
| Poly-A-based: spatial gene expression | |||
| 10X Genomics | Loupe Browser | Visualization of gene expression | For single-cell transcriptomic data, aligning gene expression spots to histology images, marking, annotating populations, and clustering |
| Bruker Corporation (Billerica, MA, USA) (NanoString Technologies (Seattle, WA, USA)) | nCounter | Tissue object detection, readout creation | Analysis system, panels, and assays |
| Bruker Corporation (NanoString Technologies) | GeoMx | Highly multiplexed profiling | Digital spatial profiler, panels, and assays |
| Bruker Corporation (NanoString Technologies) | CosMx | Quantification of up to 1000 RNAs or up to 100 proteins | Smart cyclic in situ hybridization chemistry, single-cell and subcellular resolution, spatial molecular imager, panels, and assays |
| Bruker Corporation (NanoString Technologies) | AtoMx | Analysis and visualization of spatial multiomics data | Cloud-based spatial informatics platform |
| Oncompass (Schindellegi, Schwyz, Switzerland) | Precision oncology service | Personalized targeted treatment planning | AI-based determination of both the molecular targets and their associated targeted compounds |
| Turbine AI (Budapest, Hungary) | Cell model | Drug–cell response simulation | AI-driven predictive modeling for drug discovery and optimization of clinical trial designs |
| Nucleus Segmentation | Gland Segmentation | Surface Epithelium Segmentation |
|---|---|---|
| K-means algorithm | Density-based Hysteresis Snail (DBHS) algorithm | Surface Nucleus Chain-based Algorithm (SNCA) |
| Gauss filter = true | Use own cell map = true | Minimum sample size = 6000 |
| Wallis filter = false | Maximum distance = 18 | Maximum sample size = 50,000,000 |
| Cell centralization = true | Maximum connection = 14 | Full image threshold open mask size = 5 |
| Neighborhood = true | Maximum hysteresis distance = 25 | Full image threshold open iteration = 5 |
| Mask X size = 5.76 | Hysteresis threshold = 80 | Maximum hole size = 15,000 |
| Mask Y size = 5.76 | Maximum pixel count = 10,000 | |
| Zoom level = 2 (Postcalculation) | Get mask = true | |
| Dilate mask size = 5 | Minimum gland area = 1900 | |
| Dilate iteration = 1 | Maximum gland area = 16,000 | |
| Minimum nucleus pixel count = 250 | Check compactness = true | |
| Close iteration 3 | Compactness factor = 1.71 | |
| Full slide zoom level = 6 | Brightness average delta = 15 | |
| Global threshold = 240 | Use rectangular density mask = true | |
| Surface dilate mask size = 21 | Minimum density = 15 | |
| Surface dilate iteration = 1 | Density mask size = 20 | |
| Density map minimum gland area = 5 | ||
| Density map maximum gland area = 10,000 | ||
| Close mask size = 3 | ||
| Close iteration = 0 |
| Tumor Class: NORM/NAT/CRC | ROI | ROI | All Cells in Given Tumor Class | |||||
|---|---|---|---|---|---|---|---|---|
|
Chrom.
Name |
Approximate
Gene Number of Chromosome |
Total
Gene Mutations |
Affected
Distinct Genes |
Proportion of
Mutated Genes [%] |
Approximate
Mean of Mutations/Gene |
Measured Data
of Cells |
Median of
Mutated Gene Proportions |
Median of
Mutations per Affected Gene |
| 1 2 | 3000 2 | … | median () | median () | ||||
| 2 2 | 2500 2 | … | median () | median () | ||||
| 3 2 | 1900 2 | … | median () | median () | ||||
| further chromosomes… | … | … | … | |||||
| 21 2 | 400 2 | … | median () | median () | ||||
| X 1 | 1400 1 | … | median () | median () | ||||
| Y 1 | 200 1 | … | median () | median () | ||||
| PC Component | Details |
|---|---|
| Operating system | Microsoft Windows 10 (64-bit recommended) |
| Execution environment | Microsoft .Net Framework, ≥version 4.8.04084 |
| Disk space | 300 MB free space for program and 10 GB for local slide storage (1 digital slide size is 0.5–8 GB, depending on resolution) |
| RAM | ≥2 GB, 1.6 GHz (4 GB recommended) |
| CPU | ≥Intel(R) Core(TM) i5 or equivalent AMD processor |
| Graphics card memory | 512 MB (1 GB recommended) |
| Screen resolution | 19″, 1280 × 1024 resolution, 141 DPI |
| Tissue Sample | Number of Detected Cells | Manual Number of Cell Annotations | Number of Filtered, Missed Cells (False Negative) | DSC |
|---|---|---|---|---|
| Sample A | 160 | 207 | 47 | 0.872 |
| Sample B | 261 | 269 | 8 | 0.985 |
| Sample C | 176 | 197 | 21 | 0.944 |
| Sample D | 367 | 444 | 77 | 0.905 |
| Sample E | 545 | 706 | 161 | 0.871 |
| Sample F | 716 | 872 | 156 | 0.902 |
| Sample G | 285 | 410 | 125 | 0.820 |
| Sample H | 431 | 484 | 53 | 0.890 |
| Sample I | 256 | 288 | 32 | 0.889 |
| Sample J | 89 | 117 | 28 | 0.864 |
| Total | 3286 | 3994 | 708 | 0.891 |
| Workflow Phase | Specialized Equipment, Chemistry | Approx. Mean Price [USD] (in 2025) | Important Settings | Output | Technical Expertise | Time Requirement |
|---|---|---|---|---|---|---|
| Biopsy | Clinical setup, scalpel | N/A | Standard biopsy protocol, resection margins including native region | Raw tissue sample | Surgeon, nurse, staff | 1–2 h |
| Tissue preservation | Ultra-low-temperature (ULT) freezer, cryogenic tube/formalin, paraffin | ∼15,000/∼850 | Snap-frozen tissue stored at −80 °C, while paraffin block stored at −4 °C in a freezer. | Snap-frozen/FFPE tissue block | Lab technician | 3 days |
| Tissue sectioning | Automatic microtome | ∼30,000 | Preventing tissue thawing during handling. Section thickness proportionality to examining cell type (Snap-frozen: 10–20 µm, FFPE: 4–6 µm) and uniformity of sections. | Tissue sections | Histologist supervision | 0.5 h |
| Section mounting | Warm water bath for surface stretching of tissue sections, slide-drying hot plate | ∼1000 + 1600 | Membrane slides (MMI) used instead of classic glass slides for LMD compatibility. To ensure that no air bubbles are trapped, a brush is used to flatten. | Sections fixed on slides | Lab technician | 1.5 h |
| Slide transferring | UR5 robotic arm (Universal Robots USA, Novi, MI, USA) with custom magazine and slide manipulator | ∼50,000 | Accurate, damage-free membrane slide placement into downstream modules. | Loaded slides | Robotics engineer supervision | 10 min |
| Slide staining | Gemini automated stainer (Thermo Fisher Scientific, Waltham, MA, USA), H&E stainer kit/IHC panel kit | ∼60,000 + ∼150–5000 | Staining and IHC protocols | Contrast-enhanced, stained sections | Immunologist supervision | 3 h (protocol-dependent) |
| Slide scanning | Pannoramic P1000 scanner (3DHistech, Budapest, Hungary) | ∼350,000 | Use of obtained custom settings profile for membrane slides. | High-resolution digital slide images (.MRXS) | Lab technician supervision | 5–30 min (resolution-dependent) |
| Image analysis: segmentation and classification | NGS Viewer | N/A | Fine-tuning of algorithm parameters for examined tissue type. | Selected ROIs (e.g., cells), their morphology, calculated morphometry saved in DB and exportable ROI parameters for excision (.xml) | Bioinformatician supervision | 10–20 min |
| Cell extraction (step required only for single-cell-level analysis) | CellCut LMD (MMI) | ∼150,000–200,000 | Laser velocity, focus, and power adjustment based on tissue type and section thickness. 2x cutting repeat. | Isolated cells in Eppendorf capsules | Lab technician | 0.5 h |
| DNA sequencing: electrical signal reading and base calling | NextSeq 500 (Illumina, Inc., San Diego, CA, USA)+kit for exome of single cells/PromethION (Nanopore, Oxford, UK)+kit for whole genome of bulk + server | ∼275,000+3000/run/ ∼450,000+1000/run + 10000 | ≥40 TB data storage. Quality checking for sequencing depth and uniformity of coverage. | Raw signal reads (.POD5) and nucleotide sequences (.FASTQ) | Lab technician | 5–10 days |
| Variant calling: contig alignment and variant calling | BaseSpace (Illumina)/Epi2Me (Nanopore) | Usage-based (0.035/GB)/free | ≥20 TB data storage. Flowcell + chemistry compatible model use. Comparison with the latest human reference genome. | Exportable genomic metadata of ClinVar DB annotated gene mutations (.vcf) | Bioinformatician supervision | 0.5 day |
| Diagnosis reporting | NGS Viewer | N/A | Fused genomic metadata analysis and visualization by reopening saved ROI morphology and morphometry data and importing .vcf variants. Correlation examination via the listed molecular pathways of the mutated genes. | Support of diagnostic conclusion establishment | Pathologists | 30–40 min |
| Therapy planning | NGS Viewer | N/A | Selection of listed gene-specific drugs that target the most critically mutated genes. | Personalized treatment plan support | Oncologist board and attending physician | 20–30 min |
| Total | ≥1.15M | Staff of ≥10 people | ∼10–15 days |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kucarov, M.D.; Szakállas, N.; Molnár, B.; Kozlovszky, M. Enhanced Tumor Diagnostics via Cyber-Physical Workflow: Integrating Morphology, Morphometry, and Genomic MultimodalData Analysis and Visualization in Digital Pathology. Sensors 2025, 25, 4465. https://doi.org/10.3390/s25144465
Kucarov MD, Szakállas N, Molnár B, Kozlovszky M. Enhanced Tumor Diagnostics via Cyber-Physical Workflow: Integrating Morphology, Morphometry, and Genomic MultimodalData Analysis and Visualization in Digital Pathology. Sensors. 2025; 25(14):4465. https://doi.org/10.3390/s25144465
Chicago/Turabian StyleKucarov, Marianna Dimitrova, Niklolett Szakállas, Béla Molnár, and Miklos Kozlovszky. 2025. "Enhanced Tumor Diagnostics via Cyber-Physical Workflow: Integrating Morphology, Morphometry, and Genomic MultimodalData Analysis and Visualization in Digital Pathology" Sensors 25, no. 14: 4465. https://doi.org/10.3390/s25144465
APA StyleKucarov, M. D., Szakállas, N., Molnár, B., & Kozlovszky, M. (2025). Enhanced Tumor Diagnostics via Cyber-Physical Workflow: Integrating Morphology, Morphometry, and Genomic MultimodalData Analysis and Visualization in Digital Pathology. Sensors, 25(14), 4465. https://doi.org/10.3390/s25144465