
Membrane-Based Pulsed Sampling Method for Extended Dynamic Range of Ion Mobility Spectrometry


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
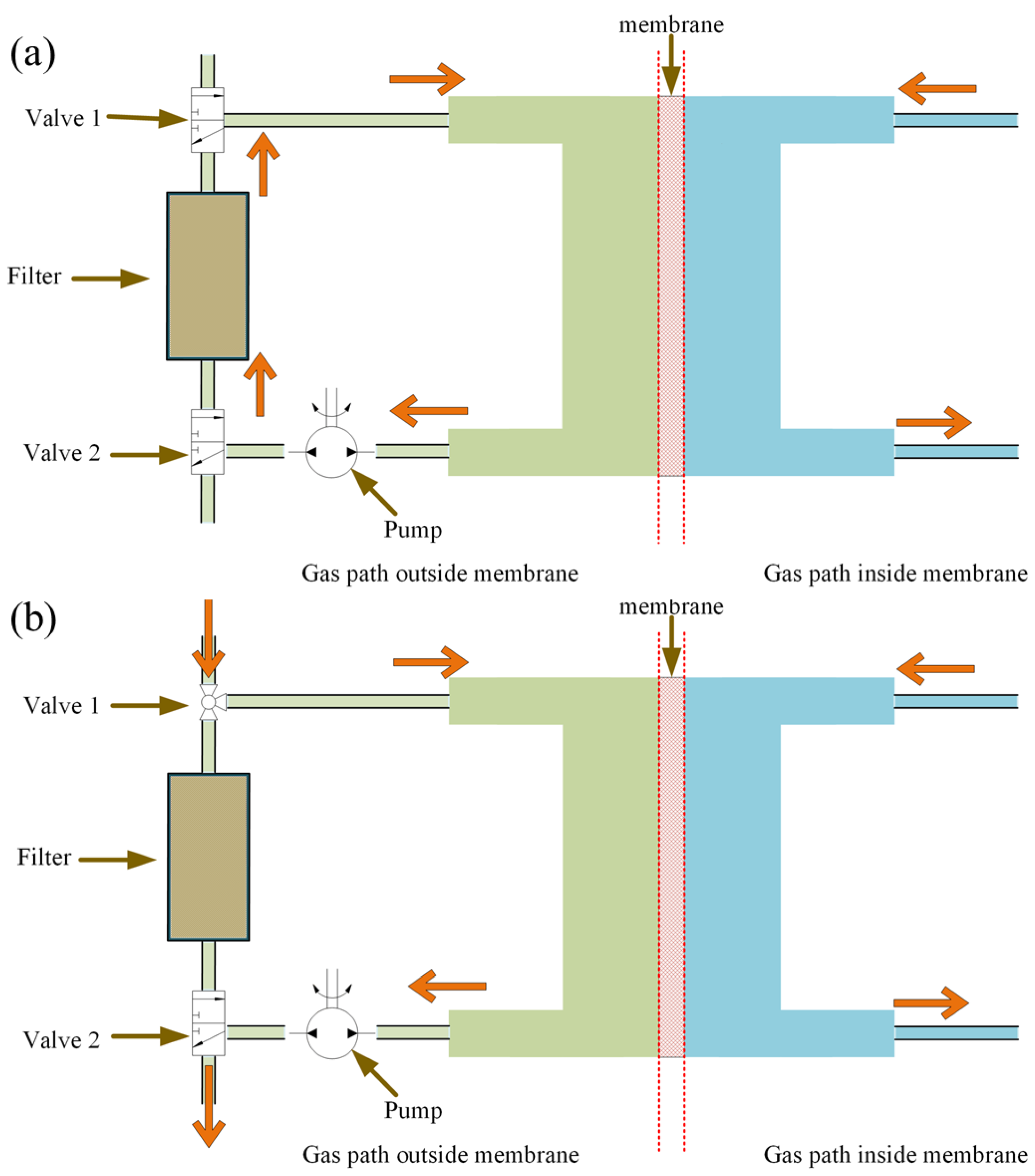
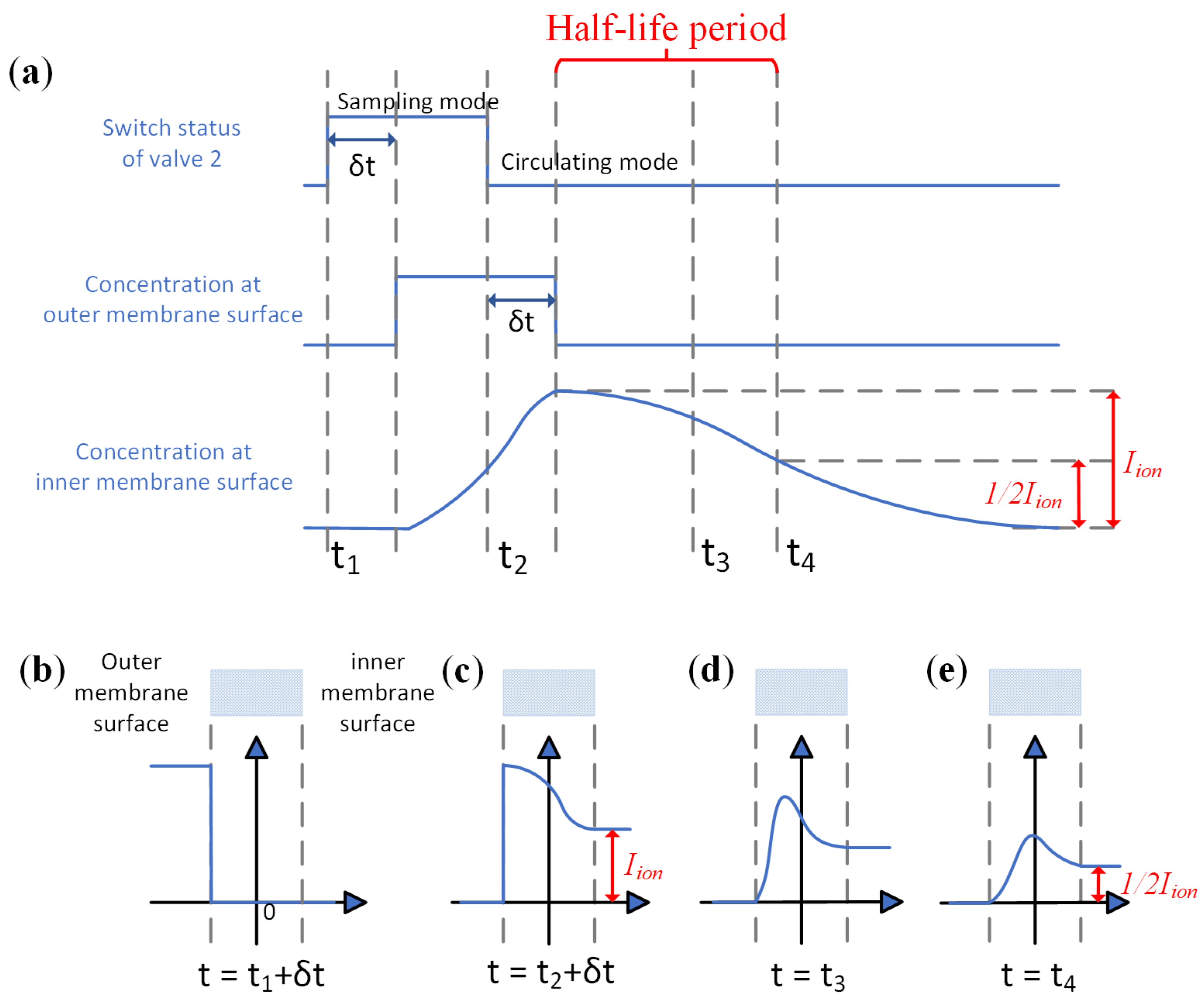
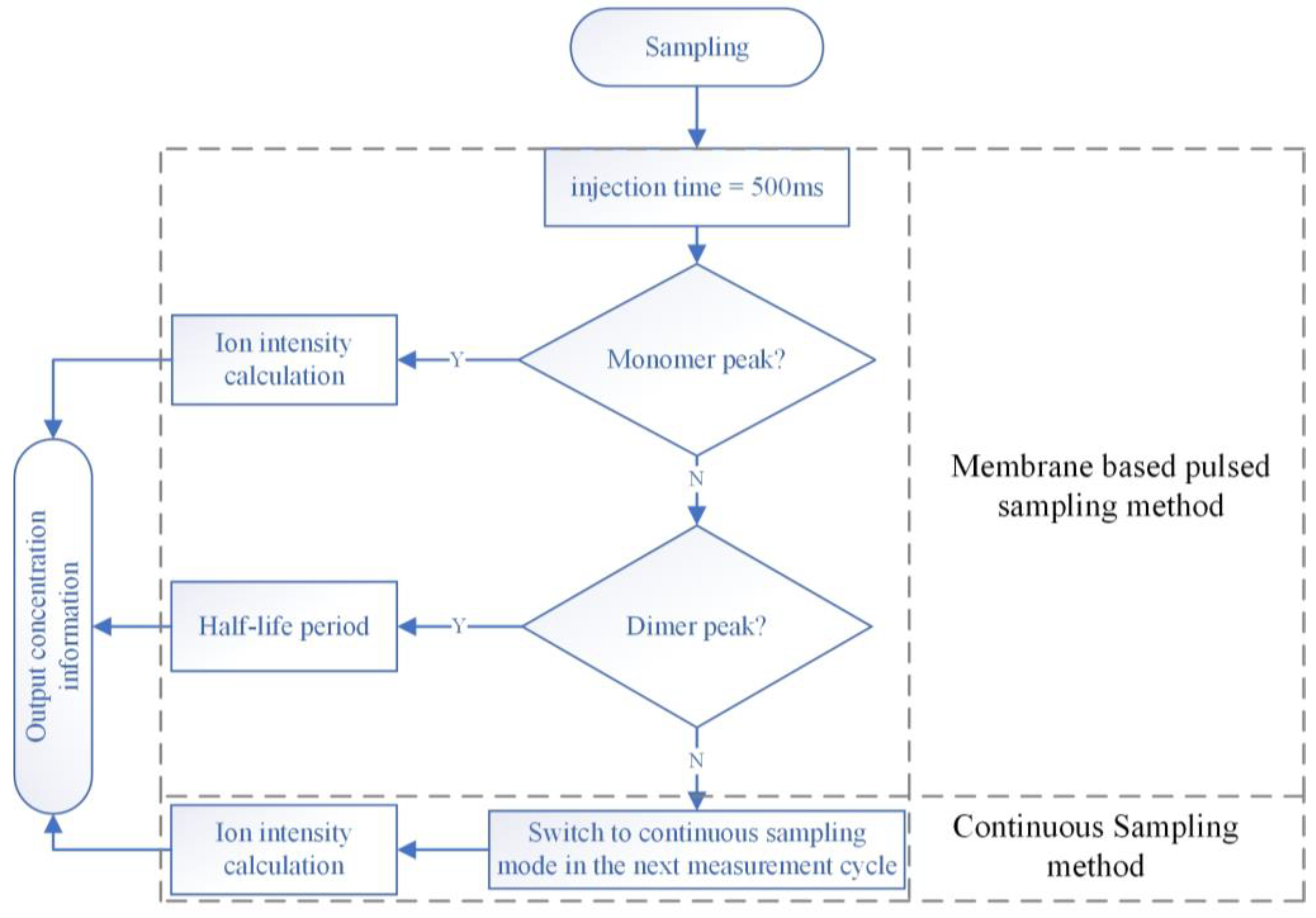
2. The Principle of the Membrane-Based Pulsed Sampling Method
3. Experiment
3.1. Reagents and Instruments
3.1.1. Reagents
3.1.2. Instruments
3.2. Sample Preparation
4. Results and Discussion
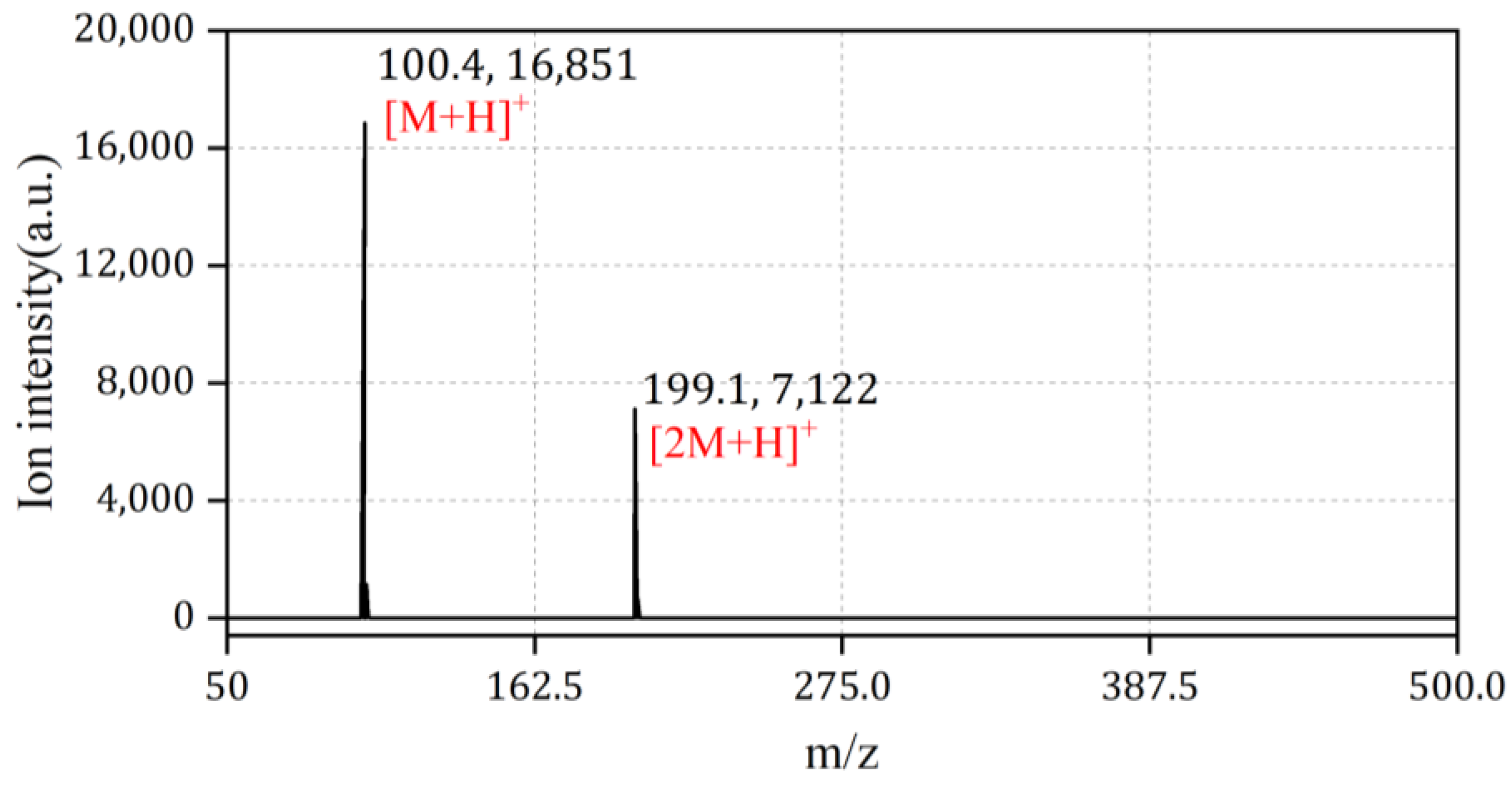
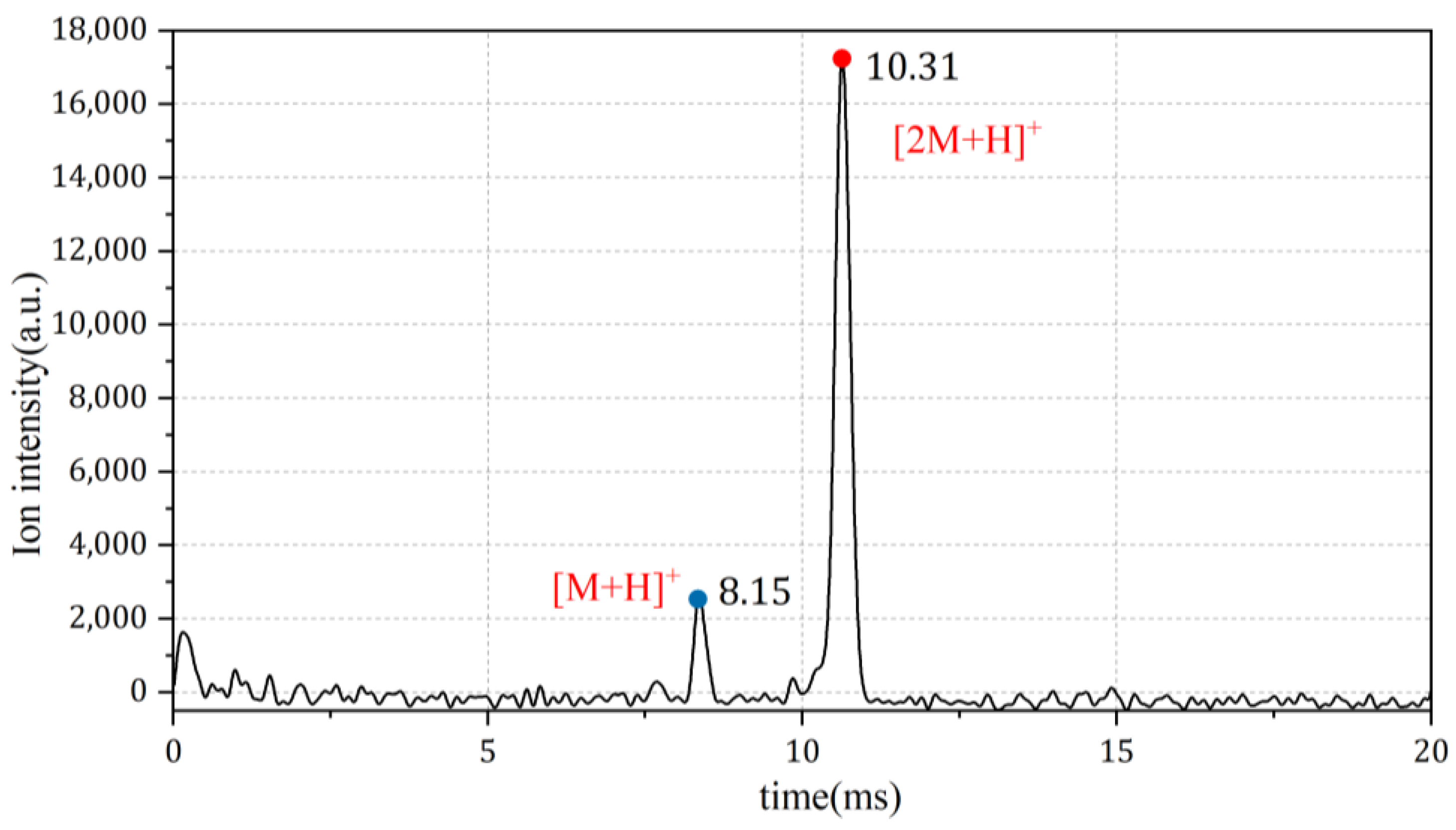
4.1. Discussion of Ion Peak Positions
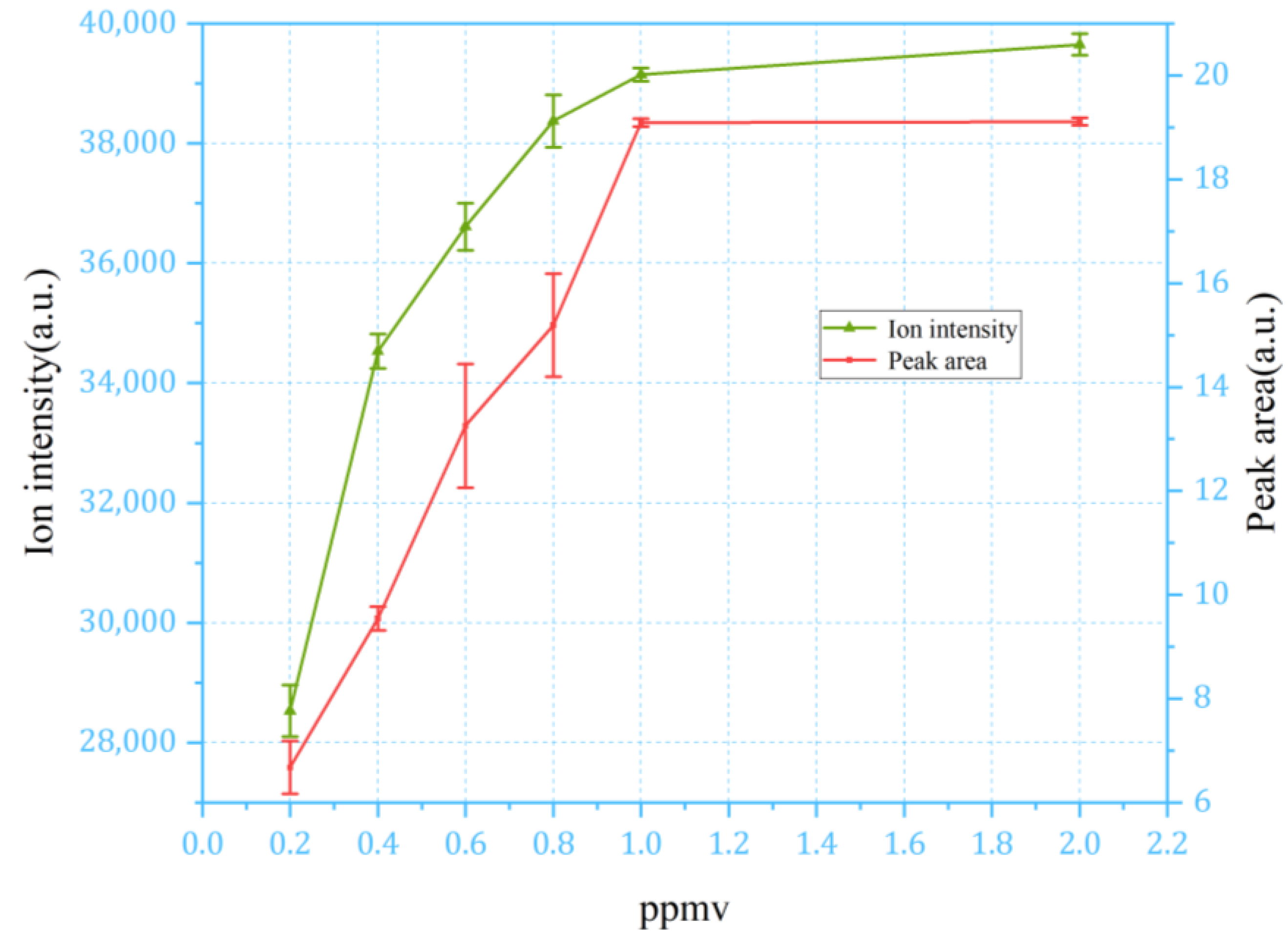
4.2. Discussion of Continuous Sampling Results
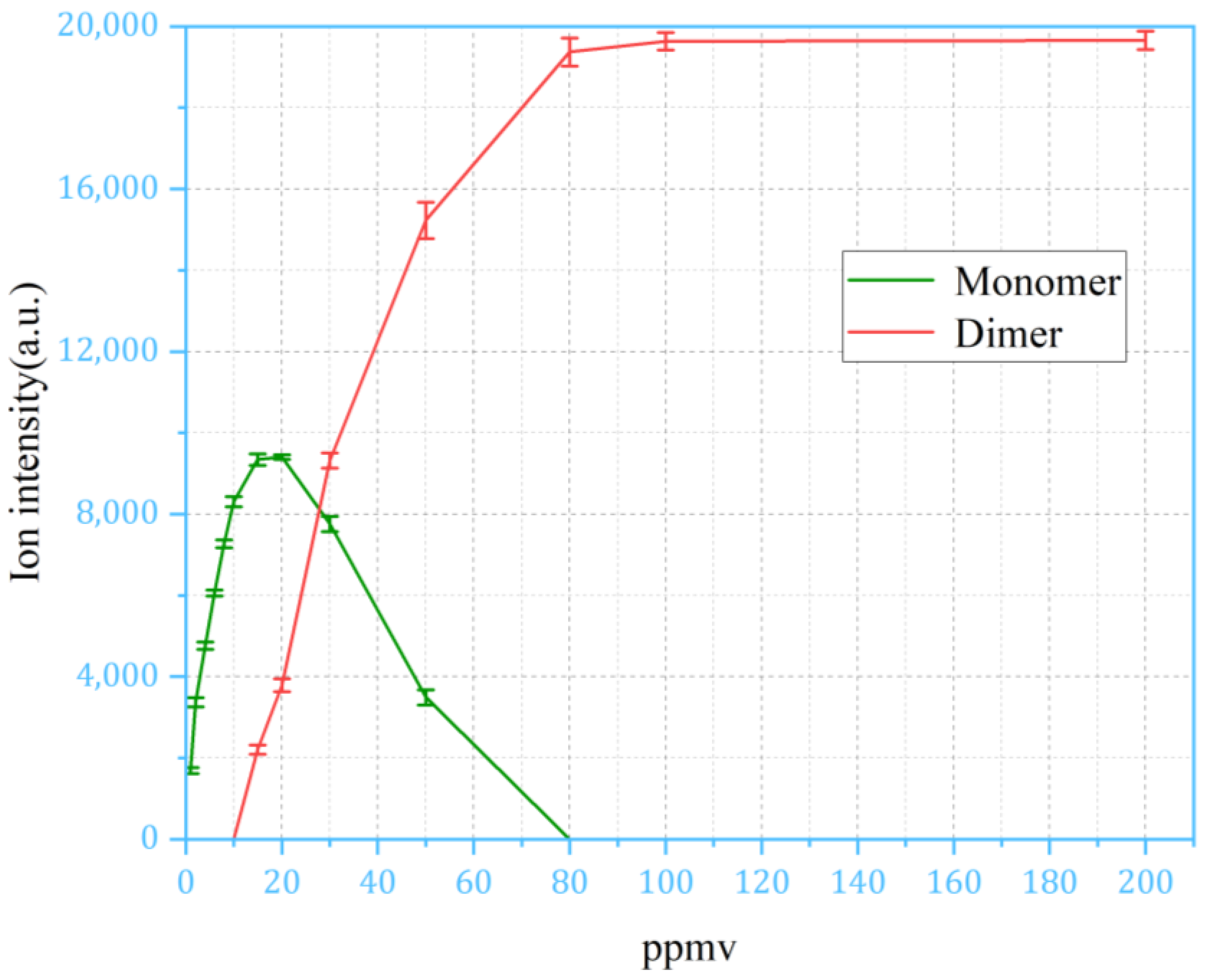
4.3. Discussion of Pulsed Sampling Results
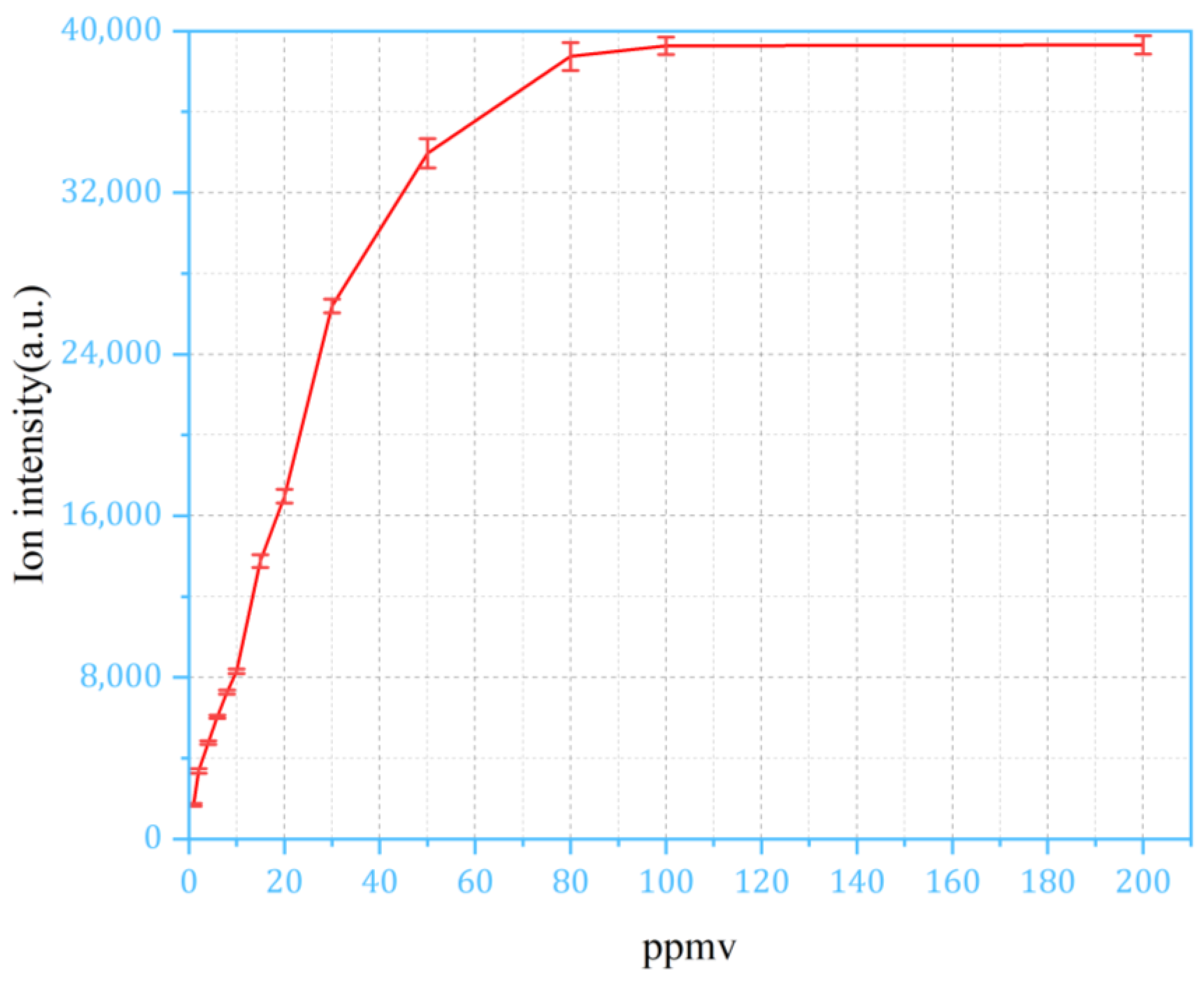
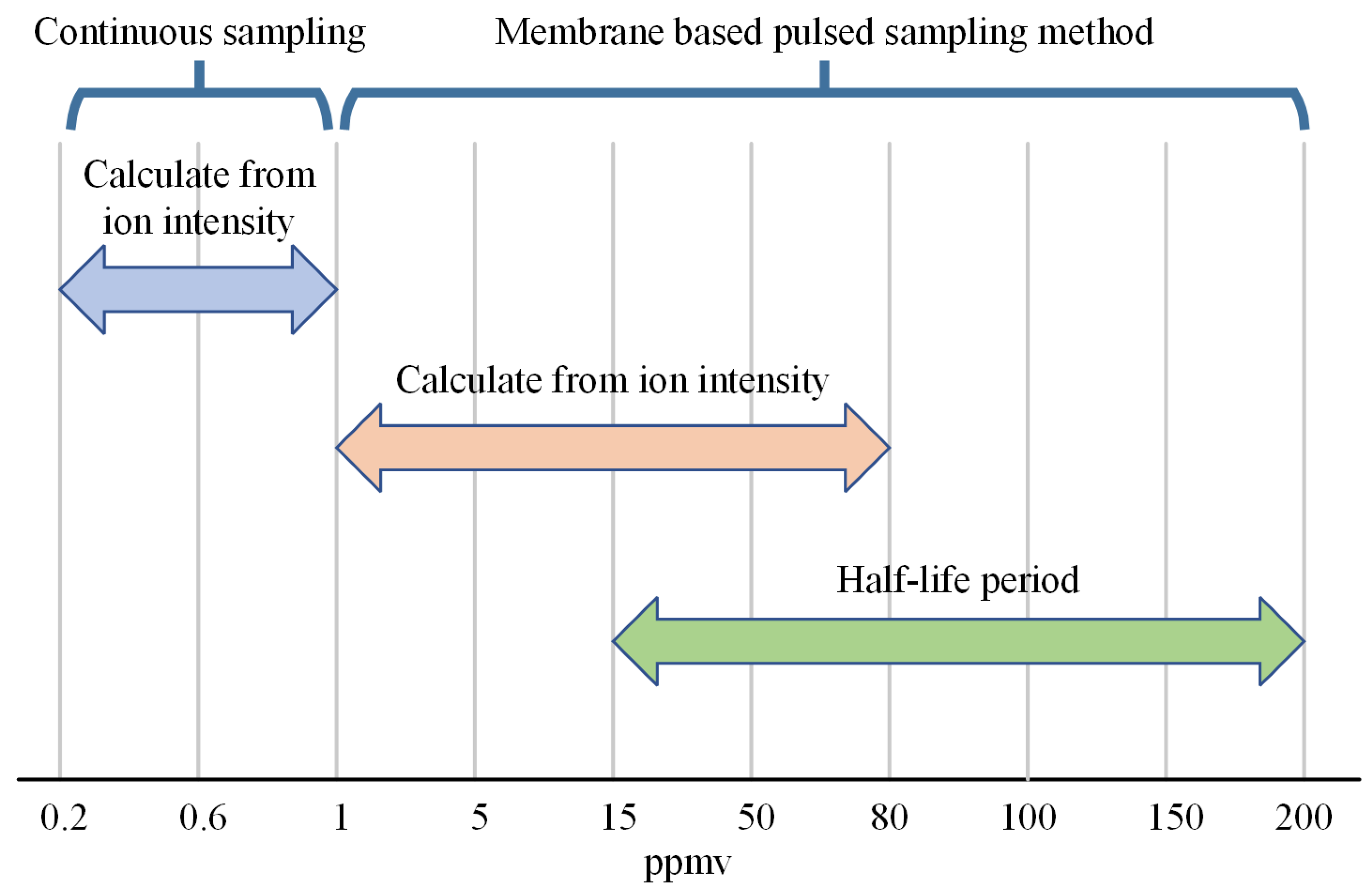
4.4. Integration of Sampling Method for Extending Dynamic Range
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Epping, R.; Koch, M. On-Site Detection of Volatile Organic Compounds (VOCs). Molecules 2023, 28, 1598. [Google Scholar] [CrossRef]
- Reimann, S.; Lewis, A.C. Anthropogenic VOCs. In Volatile Organic Compounds in the Atmosphere; Wiley: Hoboken, NJ, USA, 2007; pp. 33–81. [Google Scholar]
- Mizukoshi, A.; Kumagai, K.; Yamamoto, N.; Noguchi, M.; Yoshiuchi, K.; Kumano, H.; Yanagisawa, Y. A novel methodology to evaluate health impacts caused by VOC exposures using real-time VOC and Holter monitors. Int. J. Env. Res. Public. Health 2010, 7, 4127–4138. [Google Scholar] [CrossRef] [PubMed]
- Rumchev, K.; Brown, H.; Spickett, J. Volatile organic compounds: Do they present a risk to our health? Rev. Environ. Health 2007, 22, 39–55. [Google Scholar] [CrossRef]
- Koppmann, R. Volatile organic compounds in the atmosphere: An overview. In Volatile Organic Compounds in the Atmosphere; Wiley: Hoboken, NJ, USA, 2007; pp. 3–5. [Google Scholar] [CrossRef]
- Verma, G.; Gokarna, A.; Kadiri, H.; Nomenyo, K.; Lerondel, G.; Gupta, A. Multiplexed Gas Sensor: Fabrication Strategies, Recent Progress, and Challenges. ACS Sens. 2023, 8, 3320–3337. [Google Scholar] [CrossRef] [PubMed]
- Blanco-Rodriguez, A.; Camara, V.F.; Campo, F.; Becheran, L.; Duran, A.; Vieira, V.D.; de Melo, H.; Garcia-Ramirez, A.R. Development of an electronic nose to characterize odours emitted from different stages in a wastewater treatment plant. Water Res. 2018, 134, 92–100. [Google Scholar] [CrossRef] [PubMed]
- Park, S.J.; Park, C.S.; Yoon, H. Chemo-Electrical Gas Sensors Based on Conducting Polymer Hybrids. Polymers 2017, 9, 155. [Google Scholar] [CrossRef] [PubMed]
- Nath, N.; Kumar, A.; Chakroborty, S.; Soren, S.; Barik, A.; Pal, K.; de Souza, F.G. Carbon Nanostructure Embedded Novel Sensor Implementation for Detection of Aromatic Volatile Organic Compounds: An Organized Review. ACS Omega 2023, 8, 4436–4452. [Google Scholar] [CrossRef] [PubMed]
- Poole, C.F. Alkylsilyl derivatives for gas chromatography. J. Chromatogr. A 2013, 1296, 2–14. [Google Scholar] [CrossRef]
- Santos, F.J.; Galceran, M.T. The application of gas chromatography to environmental analysis. TrAC Trends Anal. Chem. 2002, 21, 672–685. [Google Scholar] [CrossRef]
- Lim, Y.M.; Swamy, V.; Ramakrishnan, N.; Chan, E.S.; Kesuma, H.P. Volatile organic compounds (VOCs) in wastewater: Recent advances in detection and quantification. Microchem. J. 2023, 195, 109537. [Google Scholar] [CrossRef]
- Li, T.; Zhu, X.; Hai, X.; Bi, S.; Zhang, X. Recent Progress in Sensor Arrays: From Construction Principles of Sensing Elements to Applications. ACS Sens. 2023, 8, 994–1016. [Google Scholar] [CrossRef] [PubMed]
- Ziyatdinov, A.; Marco, S.; Chaudry, A.; Persaud, K.; Caminal, P.; Perera, A. Drift compensation of gas sensor array data by common principal component analysis. Sens. Actuators B Chem. 2010, 146, 460–465. [Google Scholar] [CrossRef]
- Wilson, A.D. Review of Electronic-nose Technologies and Algorithms to Detect Hazardous Chemicals in the Environment. Procedia Technol. 2012, 1, 453–463. [Google Scholar] [CrossRef]
- Lee, J.; Sayler, S.K.; Zhou, M.; Zhu, H.; Richardson, R.J.; Neitzel, R.L.; Kurabayashi, K.; Fan, X. On-site monitoring of occupational exposure to volatile organic compounds by a portable comprehensive 2-dimensional gas chromatography device. Anal. Methods 2018, 10, 237–244. [Google Scholar] [CrossRef]
- Costanzo, M.T.; Boock, J.J.; Kemperman, R.H.J.; Wei, M.S.; Beekman, C.R.; Yost, R.A. Portable FAIMS: Applications and Future Perspectives. Int. J. Mass. Spectrom. 2017, 422, 188–196. [Google Scholar] [CrossRef] [PubMed]
- Williamson, D.L.; Nagy, G. Evaluating the Utility of Temporal Compression in High-Resolution Traveling Wave-Based Cyclic Ion Mobility Separations. ACS Meas. Sci. Au 2022, 2, 361–369. [Google Scholar] [CrossRef]
- Conant, C.R.; Attah, I.K.; Garimella, S.V.B.; Nagy, G.; Bilbao, A.; Smith, R.D.; Ibrahim, Y.M. Evaluation of Waveform Profiles for Traveling Wave Ion Mobility Separations in Structures for Lossless Ion Manipulations. J. Am. Soc. Mass. Spectrom. 2020, 32, 225–236. [Google Scholar] [CrossRef]
- Ahrens, A.; Hitzemann, M.; Zimmermann, S. Miniaturized high-performance drift tube ion mobility spectrometer. Int. J. Ion. Mobil. Spectrom. 2019, 22, 77–83. [Google Scholar] [CrossRef]
- To, K.C.; Ben-Jaber, S.; Parkin, I.P. Recent Developments in the Field of Explosive Trace Detection. ACS Nano 2020, 14, 10804–10833. [Google Scholar] [CrossRef]
- Li, M.; Wang, S.; Xu, C.; Ruan, H.; Wang, W.; Chen, C.; Li, H. Parallel Coupling of Ion Mobility Spectrometry and Ion Trap Mass Spectrometry for the Real-Time Alarm Triggering and Identification of Hazardous Chemical Leakages. Anal. Chem. 2021, 93, 11852–11858. [Google Scholar] [CrossRef]
- Fisher, D.; Lukow, S.R.; Berezutskiy, G.; Gil, I.; Levy, T.; Zeiri, Y. Machine Learning Improves Trace Explosive Selectivity: Application to Nitrate-Based Explosives. J. Phys. Chem. A 2020, 124, 9656–9664. [Google Scholar] [CrossRef] [PubMed]
- Bell, R.J.; Short, R.T.; van Amerom, F.H.W.; Byrne, R.H. Calibration of an In Situ Membrane Inlet Mass Spectrometer for Measurements of Dissolved Gases and Volatile Organics in Seawater. Environ. Sci. Technol. 2007, 41, 8123–8128. [Google Scholar] [CrossRef] [PubMed]
- Eiceman, G.A.; Karpas, Z.; Hill, H.H., Jr. Ion Mobility Spectrometry, 3rd ed.; Routledge: London, UK, 2016. [Google Scholar]
- Sliz, R.; Valikangas, J.; Silva Santos, H.; Vilmi, P.; Rieppo, L.; Hu, T.; Lassi, U.; Fabritius, T. Suitable Cathode NMP Replacement for Efficient Sustainable Printed Li-Ion Batteries. ACS Appl. Energy Mater. 2022, 5, 4047–4058. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, X.; Lu, W.; Lan, D.; Zhang, B.; Gu, H.; Shen, M.; Li, L.; Li, P. Membrane-Based Pulsed Sampling Method for Extended Dynamic Range of Ion Mobility Spectrometry. Sensors 2024, 24, 3106. https://doi.org/10.3390/s24103106
Chen X, Lu W, Lan D, Zhang B, Gu H, Shen M, Li L, Li P. Membrane-Based Pulsed Sampling Method for Extended Dynamic Range of Ion Mobility Spectrometry. Sensors. 2024; 24(10):3106. https://doi.org/10.3390/s24103106
Chicago/Turabian StyleChen, Xinzhi, Wencheng Lu, Di Lan, Bo Zhang, Hao Gu, Mutong Shen, Lingfeng Li, and Peng Li. 2024. "Membrane-Based Pulsed Sampling Method for Extended Dynamic Range of Ion Mobility Spectrometry" Sensors 24, no. 10: 3106. https://doi.org/10.3390/s24103106
APA StyleChen, X., Lu, W., Lan, D., Zhang, B., Gu, H., Shen, M., Li, L., & Li, P. (2024). Membrane-Based Pulsed Sampling Method for Extended Dynamic Range of Ion Mobility Spectrometry. Sensors, 24(10), 3106. https://doi.org/10.3390/s24103106