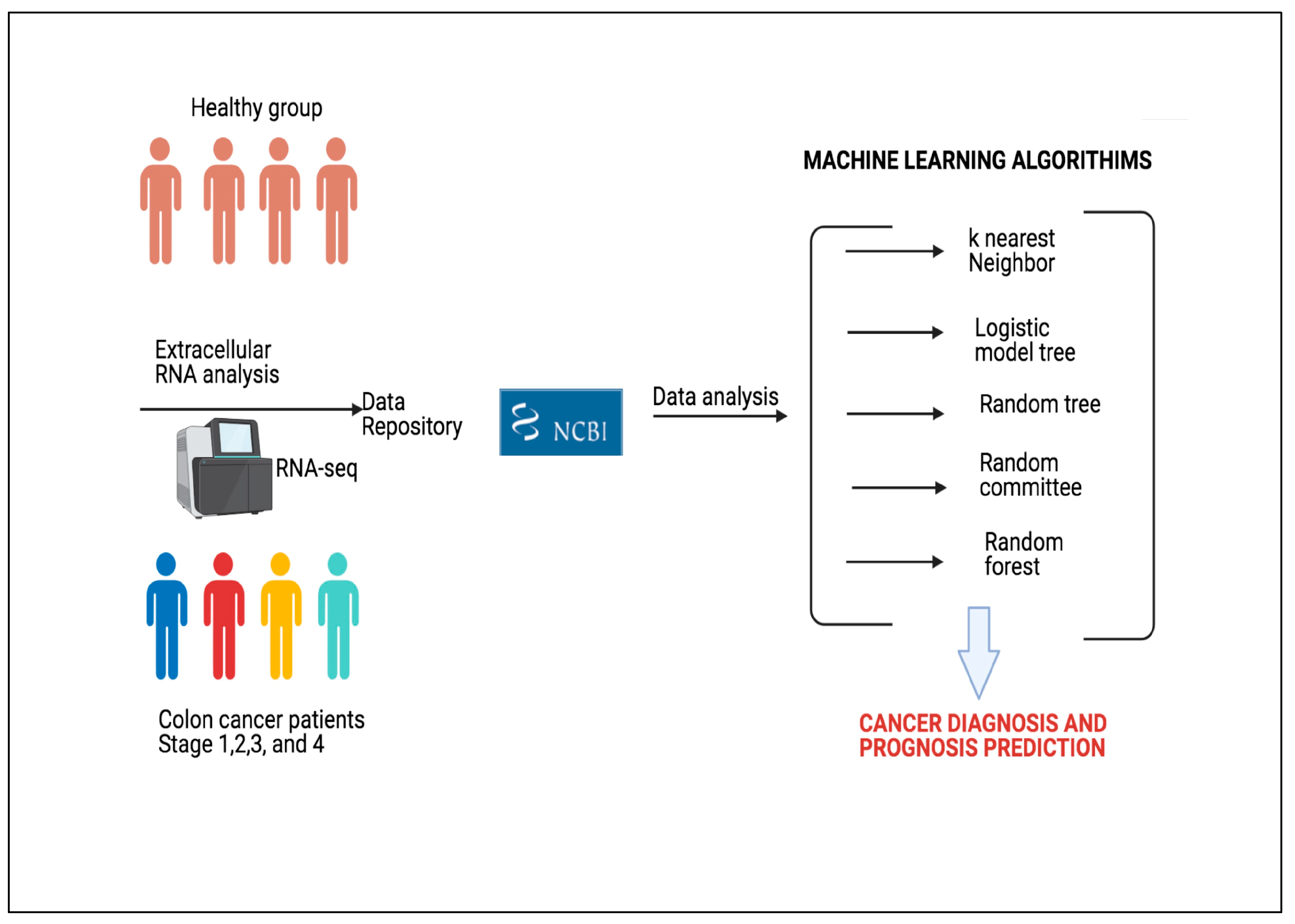
3.2. Data
A total of 150 subjects were separated as shown in
Figure 2. As can be seen in
Figure 2a, 100 of 150 subjects were cancer patients at a certain level. As seen in
Figure 2b, 100 patient individuals were equally divided in 4 stages of the disease.
The ML methods generally require large datasets. The data we used in this study were from 150 subjects and may be insufficient for this type of work, and an augmentation of the data was required to achieve a satisfactory performance of ML classifiers [
33]. For this purpose, the data were augmented to include 300 samples. Later, considering the large number of features compared with the sample number of the data, a certain number of features were selected, considering that some features may be more important than others. In addition, it is important to note that selecting features reduces the complexity of the data and shortens the training time [
34].
For data augmentation, the Synthetic Minority Over-sampling Technique (SMOTE) algorithm was utilized in the study [
35,
36]. SMOTE was originally developed for imbalanced datasets to oversample the minority class. However, it can also be used to oversample the whole dataset. SMOTE oversamples the minority class by generating synthetic data by working on feature space. This method oversamples by taking every minority class example into account and presenting synthetic examples and joining nearest neighbors to that class. The nearest neighbor count depends on the size of the oversampling process. The first step of generating synthetic examples is calculating the difference between the feature vector of current example and its nearest neighbor. The second step includes multiplying the calculated difference by a randomly generated number between 0 and 1. In the third step, the calculated vector is added to the feature vector of the current example.
In our study, first, for a randomly selected healthy sample, 50 augmented healthy samples were generated, while 50 randomly selected cancerous samples were used. Therefore, a total number of 100 healthy samples were obtained. Secondly, for a randomly selected cancerous sample, 100 augmented cancerous samples were generated, while all healthy samples were used. As a result, a total number of 200 cancerous samples were achieved. In total, the size of the dataset was increased to 300 samples while keeping the imbalanced ratio.
On the data, normalization was applied to reduce the effect of outliers and guarantee that all attributes have the same scale for both canonical ML and DL algorithms. In our study, min–max normalization was used for data normalization process. Min–max normalization can be seen in the following Equation (1):
In the equation above, and represent the new normalized and the current values of the attribute, respectively, whereas and represent the current minimum and the current maximum values, respectively, in the related attribute column of all samples; and represent the new minimum and the new maximum values, respectively, in the new normalized range.
In this study, standard [0–1] min–max normalization is applied for canonical ML algorithms, while [0–255] min–max normalization is preferred for DL algorithms. The reason for this choice is that CNN architecture accepts images as input (in the experiments 1-D CNN is fed by gray scale images). In addition, LSTM and BiLSTM algorithms were fed by input values having a range between 0 and 255.
In the dataset that is used in the study, there existed 493 attributes for each sample. To observe the effect of the number of attributes that will be given as inputs to ML and DL algorithms on the performance, a feature selection algorithm was applied. Feature selection can help to reduce dimensionality and, therefore, reduce computational load of ML frameworks. In addition, by selecting relevant features, accuracy of predictions can be increased [
37]. Simply, the algorithm calculated the information gain (
IG) for each attribute.
IG can be defined as expectation of entropy reduction while splitting the samples according to an attribute. In other words, IG determines how much information an attribute supplies about a class. Therefore, the higher value of
IG of an attribute, the more informative it is.
IG can be calculated as in the following Equations (2) and (3):
In the equations above, represents the target or class, represents the attribute vector, and represents each value of the attribute vector . While calculating entropy, represents the number of the cases of the target or briefly the number of classes. Finally, represents the probability of a value occurring in the target data.
For the experiments, n attributes with the highest IG values were selected to feed the algorithm for training process. In our study 10, 20, 30, 40, and 50 attributes having the highest IG scores were selected, and all experiments were conducted by using these attributes. In addition, the experiments were repeated and compared on the basis of performance by including all attributes.
In all experiments, to calculate the performance of the models, based on the evaluation metrics, the 10-fold cross validation technique was employed. According to this technique, the dataset was split into 10 equal parts while maintaining the class ratio. In the next step, the first part was excluded, while the remaining part was used to train the ML or DL algorithm. After the training phase, the obtained model was validated with the excluded part. These processes were repeated until all parts were used to validate the models (
Figure 3). To evaluate the final accuracy of a model after 10-fold cross validation, the accuracy results of all folds were taken into consideration. The final accuracy was calculated by averaging the accuracy results of the 10 folds.
3.3. Machine Learning Analysis
The popularity of ML has tremendously increased over the last decade. This increase has enabled ML to be applied to increasingly more areas. One of the most important applications of ML is the prediction of diseases. By analyzing the data obtained from individuals, the probability of disease can be predicted with high accuracy.
The literature presents different approaches of ML in medical domain and disease prediction. In this study, we used 5 canonical ML approaches to predict whether an individual has colon cancer. The selected methods are kNN, LMT, RT, RC, and RF. All canonical ML algorithms were employed with default parameters (
Table 1). In addition, DL algorithms were utilized to predict the stage of the cancer and whether an individual has colon cancer. 1-D CNN, LSTM, and BiLSTM DL algorithms were applied in the study and optimized.
kNN is the one of the most used approaches of ML. This supervised learning method was presented in 1967 by Cover and Hart [
38]. This approach classifies a sample by looking at its previously classified neighbor samples and is independent of the hidden joint distribution on other samples and their classification. The literature has different applications of kNN on cancer diagnosis, particularly in breast cancer [
39,
40,
41,
42].
LMT is a supervised classification algorithm, which is the combination of two learning approaches with complementary superiority and weakness: DT and Logistic Regression [
43]. The LogitBoost algorithm is used to generate a logistic regression model at each node of these classification trees with logistic reduction functions on their leaves. In this way, it is ensured that the child nodes contain information about the main nodes and that probability estimates are formed for each class. The resulting model is simplified by dividing it according to C4.5 criteria. LMT algorithm is used for disease classification [
44,
45] and predicting cancer and cancer proteins [
46,
47].
RT is a DT-based supervised classifier that randomly selects the k number of attributes at each node [
48]. The algorithm has no pruning to decrease the error and is very effective on classification and regression tasks. The classifier depends mainly on the single model tree and Random Forest [
49]. Previous studies demonstrate that RT classifier is easy to implement, effective, and does not overfit [
50,
51].
RC is an ensemble classifier which uses base classifiers with the same data but a different number of seed values to make a predictions separately [
52]. The algorithm forges final prediction by averaging the results of these individual base classifiers [
53]. In the literature, RC is used in disease prediction tasks [
54].
RF is another widely used classifier that utilizes a group of unpruned DTs and is accurate on large volumes of data in classification and regression tasks [
55]. This group of DTs is built from a training data set and determines the output. Each DT in this group is a separate classifier and has its own predictions from a sample. This algorithm combines all the results from DTs to decide the final prediction [
56]. The RF classifier is used to predict different cancer types, such as esophageal [
57], breast [
58,
59], prostate [
60], colorectal [
56], lung [
61], and cervical [
62].
LSTM networks are an upgraded version of recurrent neural networks (RNN) [
63]. In recent years, they output better classification results when compared with other DL networks on various research areas, such as time series and genome data [
64,
65]. In order to comprehend LSTM structure, RNN structure needs to be defined. RNNs are neural networks that also have memory and are able to recall all the information that is sequentially captured in the previous element. In other words, RNNs are an efficient way to use data from relatively long series since they perform similar tasks for each element in the series, with output dependent on all previous computations. A network with a feed-forward architecture and an extra cyclic loop is considered as RNN. By using this cyclic loop, RNN carries information throughout the network one time step to the next one. A form of short-term memory, cyclic loops are used to store and retrieve historical data throughout time steps.
An RNN that learns temporal patterns estimates the current time-step by using the prior state and the present state. However, RNN architectures come with a disadvantage—vanishing gradients. The issue of vanishing gradients arises when recurrent neural networks are required to learn long-term relationships in time steps. For this requirement, the gradient vector increases or decreases exponentially as it propagates through multiple layers of the RNN to learn long-term dependencies over time steps. LSTM aims to solve this issue. In order to tackle vanishing gradient problem, LSTM uses memory blocks instead of traditional RNN units [
65]. Its main advantage over RNNs is that it incorporates a cell state to store long-term states. An LSTM network can remember and connect information from the past to current information [
64]. An updated version of LSTM called BiLSTM has emerged in recent years [
66]. This architecture enables LSTM to analyze input data both forward and backwards. It actually adds two layers of memory cells to analyze data on both ways. The binding process of hidden states of backward and forward layers creates the representation of input data [
67].
1-D CNN is a modified version of CNN DL model [
68]. In this version, one dimensional convolutional layers and sub-samplings are used to build feature space [
69]. The one-dimensional convolution patch is handled by a number of convolution and pooling layers in the model, which extract features from one-dimensional input using a local receptive field and shared weights. These shared weights adjust the number of training parameters to be less than traditional CNN architectures. Through the use of several convolution filters, feature maps in the convolution and sub-sampling layers derive discriminant feature representations from many input vector segments. The 1-D CNN classifier is constructed with sample class information in the training process, and the gradient descent algorithm is utilized for adjusting network parameters [
70].
The general structure of CNN consists of convolution, pooling, and a fully connected layer [
69]. In the convolution layer, several convolution filters are employed to extract representative information from the raw data. Neurons are connected locally, thus reducing calculation load. In pooling layer, a process called sub-sampling is used to obtain more detailed feature maps at a lower resolution. The fully connected layer generally comes before the output layer to forward features to final classification phase [
71].
The experimental setup for canonical ML algorithms can be seen in
Table 1,
Table 2,
Table 3 and
Table 4, which indicate the hyper-parameters of the proposed 1-D CNN architecture.
Table 5 and
Table 6 show hyper-parameters to build LSTM and BiLSTM architectures from scratch. Therefore, the results can be reobtained for each model by utilizing the optimized hyper-parameters.
All canonical ML and DL algorithms have some advantages and disadvantages. Their performances are closely related to the utilized dataset. The advantages and disadvantages of the algorithms utilized in this study are explained briefly. In our study, kNN is chosen since it is easy to implement and it makes no assumptions about the data. However, it has a disadvantage in dealing with imbalanced data. LMT algorithm is expected to provide accurate results since it combines decision tree and logistic regression algorithms. In contrast, due to its high computational cost, it is not a preferred algorithm. The advantage of RC is that it takes into account the results of different classifiers. Likewise, this situation can lead to a disadvantage. If the majority of the classifiers make an incorrect prediction, the algorithm’s prediction will also be incorrect. RF’s advantage is that it is composed of uncorrelated decision trees. In other words, the trees that form the forest are not similar. Therefore, the algorithm has a high generalization capacity and handles imbalanced data. Nevertheless, if a dataset does not have some informative attributes, prediction performance of RF will suffer. As with RF, the performance of the RT algorithm directly depends on whether there are some informative attributes in the dataset. Consequently, if a dataset is an imbalanced one and some of the attributes have importance, it will be more likely expected that RF yields better accuracy than other ML algorithms.
CNN is chosen because it exhibits high performance when classifying images. Since an image is a matrix, we can build a model using CNN architecture if we express each sample as a 1-D matrix. LSTM and BiLSTM are efficient in processing sequential data. In addition, if we have 1-D matrices as inputs, we can feed these algorithms. All DL algorithms utilized in the study suffer from the training time to build a model.
To compare the algorithms, some evaluation metrics are needed. One metric is not sufficient to reveal the superiority of an algorithm. To support the accuracy of the algorithms, statistical tests are applied on the results. In this study, the Kappa statistic and McNemar’s test were utilized to validate the results.
While experimenting with DL algorithms, values of some hyper-parameters needed to be optimized. Therefore, for each DL algorithm, GA, a meta-heuristic approach, was utilized for optimization.
GA is a meta-heuristic search algorithm that mimics the evolutionary process, having the principle of the survival of the fittest. Especially in cancer diagnosis, GA has a wide range of use [
27,
72]. In this study, GA was utilized to optimize hyper-parameters of DL algorithms. Each possible solution was represented by a chromosome in GA. A chromosome is composed of genes that represent the hyper-parameters to be optimized of a DL architecture. All chromosomes form a population where the optimal chromosome, which satisfies the fitness function, is attempted to be found. Firstly, a population is initialized randomly. Secondly, fitness value of all chromosomes is evaluated in the population. Thirdly, the parent chromosomes that will form the next generation are chosen. Crossover and mutation operations are applied on chosen chromosomes. The third step is repeated until a stopping criterion is met. Some of the chromosomes pass on the next generation directly; these chromosomes are called elites. In our study, the number of generations was selected as 100, and it was used as the stopping criterion. The percentage of elites was selected as 5% of the population. The crossover operation that determines the fraction of the next generation was applied on 80% of the population. The rest of the population was mutated while surviving to the next generation. Since GA does not guarantee the global minimum, a large population size of 200 was selected to reduce the probability of obtaining a local minimum while increasing the run time of the algorithm. To produce children chromosomes, scattered crossover was utilized for crossover operation (
Figure 4). In scattered crossover, after selecting the parent chromosomes, a randomly created binary vector determines the genes of the child chromosome (Equation (4)).
In Equation (4), represents the ith gene in the child chromosome () and parent chromosomes ( and ), while represents the ith value in the random binary vector.
For 1-D CNN DL, hyper-parameters, such as filter size and number of filters, were optimized for each convolutional layer by applying GA. General structure of the 1-D CNN architecture is shown in
Figure 5.
The number of convolutional layers to be added in the 1-D CNN architecture was determined by the size of the input (attributes). Therefore, for each number of attributes (10, 20, 30, 40, 50, and all attributes that form the feature vector) different numbers of convolutional and max pooling layers existed in the related 1-D CNN architecture (
Table 2). For each convolutional layer, the stride parameter was selected as 1 and zero padding was applied, when necessary, to make the output as the same size as the input. After each convolutional layer, there existed a max pooling layer in the architecture. Max pooling layers halve the size of the input to perform down sampling. To ensure that, the stride parameter and pool size parameter were selected as 2 and 3, respectively, and zero padding was applied, when necessary. Consequently, different numbers of convolutional and max pooling layers were added according to the size of the input in the architecture until the output size was 1.
Optimized values by applying GA for 1-D CNN architecture can be seen in
Table 3 and
Table 4 for both cancer prediction and cancer stage classification.
In order to make a consistent comparison, the number of layers obtained for 1-D CNN was also used for LSTM and BiLSTM models. For LSTM and BiLSTM DL algorithms, the number of hidden units in each LSTM and BiLSTM layers was optimized by applying GA (
Table 5 and
Table 6). The general structure of the LSTM and BiLSTM architectures is shown in
Figure 6.
For each of the DL algorithms, adaptive moment estimation (Adam) optimizer was utilized, and early stopping was applied to prevent overfitting. In addition, data shuffling was enabled before each training epoch.


 ,
, 


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}










