
In Situ Real-Time Quantitative Determination in Electrochemical Nuclear Magnetic Resonance Spectroscopy


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Experimental
2.1. Reagents and Instruments
2.2. Electrochemical Quantitative Nuclear Magnetic Resonance (EC-qNMR) Cell with Palisade Gold Film (PGF)
3. Results and Discussion
3.1. Intrinsic Effect on the NMR Spectra by the Samples with Different Conductivity
3.2. Choice of the External Standard and Its Concentration in EC-qNMR
3.3. Method of Calibration in EC-qNMR
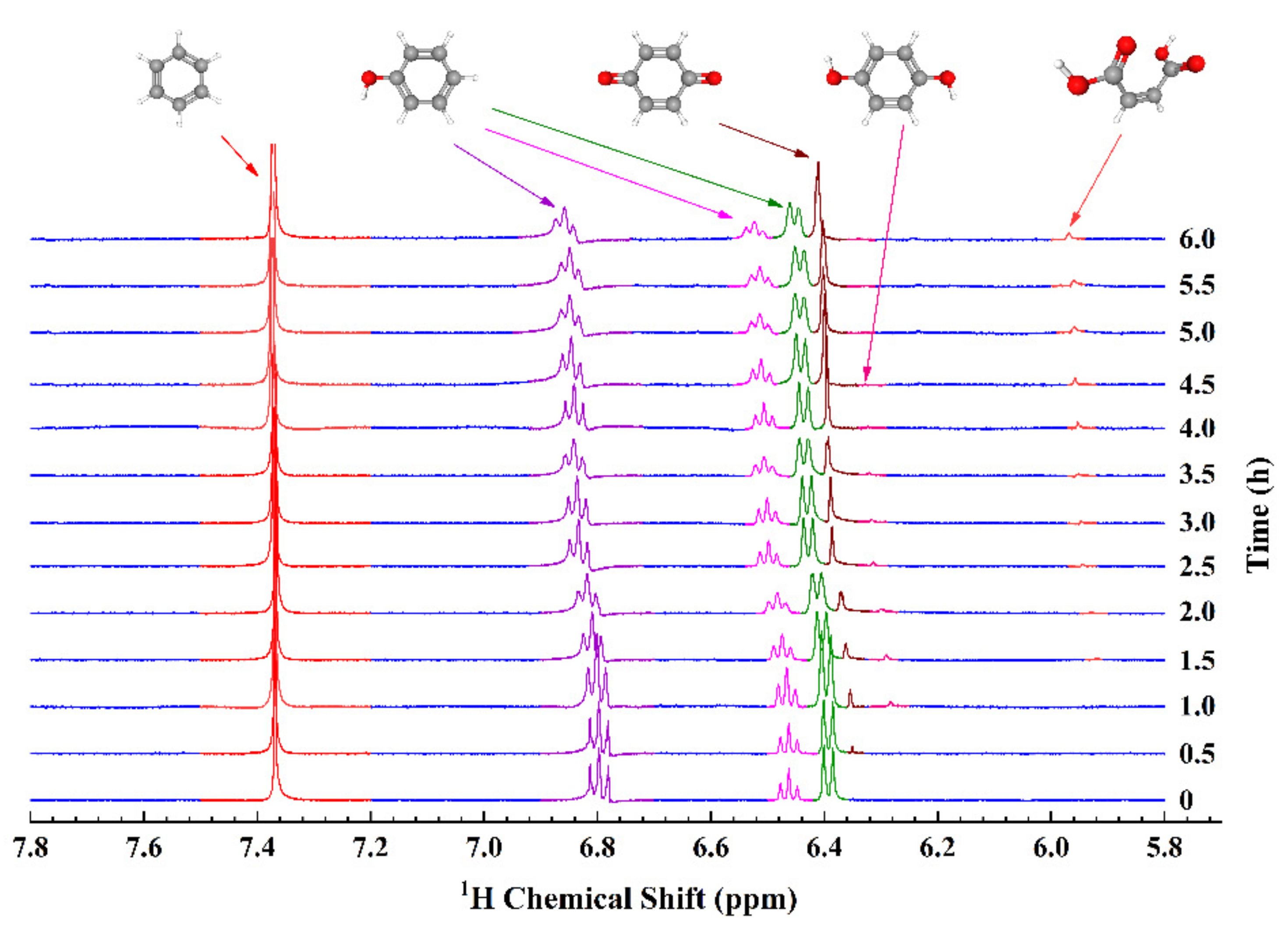
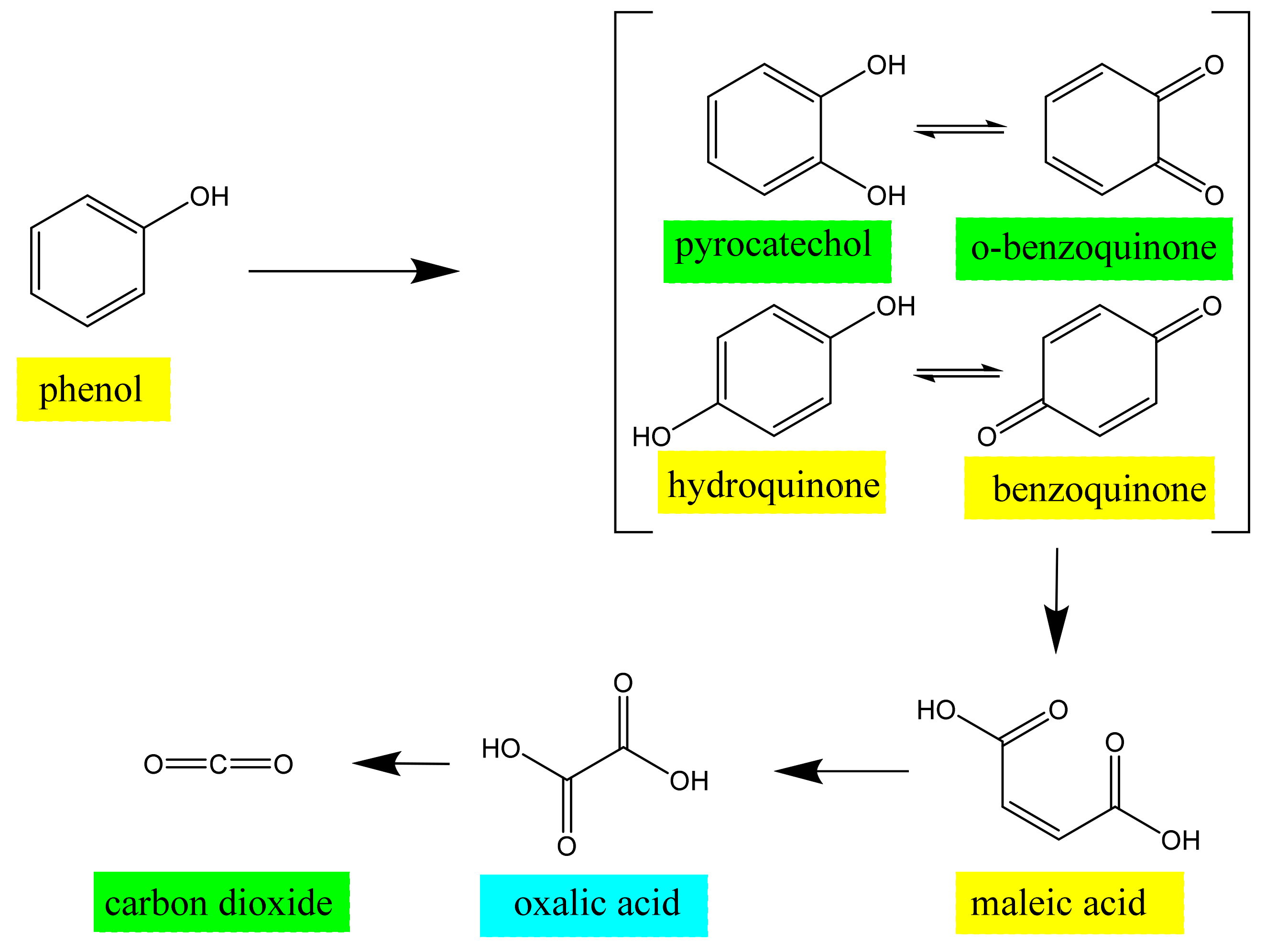
3.4. EC-qNMR Performance: Advanced Electrochemical Oxidation Process for Phenol Degradation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wu, J.-J.; Lorenzo, P.; Zhong, S.-Y.; Ali, M.; Butts, C.P.; Myers, E.L.; Aggarwal, V. Synergy of synthesis, computation and NMR reveals correct baulamycin structures. Nature 2017, 547, 436–440. [Google Scholar] [CrossRef]
- Khelifa, M.; Mounier, D.; Yaakoubi, N. Design of high performance scroll microcoils for nuclear magnetic resonance spectroscopy of nanoliter and subnanoliter samples. Sensors 2021, 21, 170. [Google Scholar] [CrossRef]
- Ban, D.; Smith, C.A.; de Groot, B.L.; Griesinger, C.; Lee, D. Recent advances in measuring the kinetics of biomolecules by NMR relaxation dispersion spectroscopy. Arch. Biochem. Biophys. 2017, 628, 81–91. [Google Scholar] [CrossRef]
- Giraudeau, P. Challenges and perspectives in quantitative NMR. Magn. Reson. Chem. 2017, 55, 61–69. [Google Scholar] [CrossRef]
- Huang, Y.-D.; Su, B.-N.; Ye, Q.-M.; Palaniswamy, V.A.; Bolgar, M.S.; Raglione, T.V. Improving the efficiency of quantitative 1H NMR: An innovative external standard-internal reference approach. J. Pharm. Biomed. Anal. 2014, 88, 1–6. [Google Scholar] [CrossRef]
- Watanabe, R.; Sugai, C.; Yamazaki, T.; Matsushima, R.; Uchida, H.; Matsumiya, M.; Takatsu, A.; Suzuki, T. Quantitative nuclear magnetic resonance spectroscopy based on PULCON methodology: Application to quantification of invaluable marine toxin, okadaic acid. Toxins 2016, 8, 294. [Google Scholar] [CrossRef] [Green Version]
- Maniara, G.; Rajamoorthi, K.; Rajan, S.; Stockton, G.W. Method performance and validation for quantitative analysis by 1H and 31P NMR spectroscopy. Applications to analytical standards and agricultural chemicals. Anal. Chem. 1998, 70, 4921–4928. [Google Scholar] [CrossRef]
- Pietrzak, M.; Jopa, S.; Mames, A.; Urbanczyk, M.; Wozny, M.; Ratajczyk, T. Recent progress in liquid state electrochemistry coupled with NMR spectroscopy. Chemelectrochem 2021, 8, 4181–4198. [Google Scholar] [CrossRef]
- Huang, L.; Sun, J.-Y.; Cao, S.-H.; Zhan, M.; Ni, Z.-R.; Sun, H.-J.; Chen, Z.; Zhou, Z.-Y.; Sorte, E.G.; Tong, Y.J.; et al. Combined EC-NMR and in situ FTIR spectroscopic studies of glycerol electrooxidation on Pt/C, PtRu/C, and PtRh/C. ACS Catal. 2016, 6, 7686–7695. [Google Scholar] [CrossRef]
- Zhang, X.-C.; Zwanziger, J.W. Design and applications of an in situ electrochemical NMR cell. J. Magn. Reson. 2011, 208, 136–147. [Google Scholar] [CrossRef]
- Boisseau, R.; Bussy, U.; Giraudeau, P.; Boujtita, M. In situ ultrafast 2D NMR spectroelectrochemistry for real-time monitoring of redox reactions. Anal. Chem. 2015, 87, 372–375. [Google Scholar] [CrossRef]
- Cao, S.-H.; Liu, S.; Sun, H.-J.; Huang, L.; Ni, Z.-R.; Jiang, W.-L.; Zhan, M.; Zhou, Z.-Y.; Sun, S.-G.; Chen, Z. Versatile, robust, and facile approach for in situ monitoring electrocatalytic processes through liquid electrochemical NMR spectroscopy. Anal. Chem. 2019, 91, 1686–1691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.-L.; You, X.-Q.; Xiao, C.; Zhang, X.-P.; Cai, S.-H.; Jiang, W.-L.; Guo, S.-S.; Cao, S.-H.; Chen, Z. Small-sized Pt nanoparticles supported on hybrid structures of MoS2 nanoflowers/graphene nanosheets: Highly active composite catalyst toward efficient ethanol oxidation reaction studied by in situ electrochemical NMR spectroscopy. Appl. Catal. B-Environ. 2019, 259, 118060. [Google Scholar] [CrossRef]
- Richter, J.B.; Essbach, C.; Senkovska, I.; Kaskel, S.; Brunner, E. Quantitative in situ 13C NMR studies of the electro-catalytic oxidation of ethanol. Chem. Commun. 2019, 55, 6042–6045. [Google Scholar] [CrossRef] [Green Version]
- Yahnke, M.S.; Rush, B.M.; Reimer, J.A.; Cairns, E.J. Quantitative solid-state NMR spectra of CO adsorbed from aqueous solution onto a commercial electrode. J. Am. Chem. Soc. 1996, 118, 12250–12251. [Google Scholar] [CrossRef]
- Bussy, U.; Giraudeau, P.; Tea, I.; Boujtita, M. Understanding the degradation of electrochemically-generated reactive drug metabolites by quantitative NMR. Talanta 2013, 116, 554–558. [Google Scholar] [CrossRef]
- Richards, J.A.; Evans, D.H. Flow cell for electrolysis within the probe of a nuclear magnetic resonance spectrometer. Anal. Chem. 1975, 47, 964–966. [Google Scholar] [CrossRef]
- Davoodi, H.; Nordin, N.; Bordonali, L.; Korvink, J.G.; MacKinnon, N.; Badilita, V. An NMR-compatible microfluidic platform enablingin situelectrochemistry. Lab Chip 2020, 20, 3202–3212. [Google Scholar] [CrossRef] [PubMed]
- Ni, Z.-R.; Cui, X.-H.; Sun, S.-G.; Chen, Z. Coupling liquid phase electrochemistry with nuclear magnetic resonance spectroscopy and its applications. Spectrosc. Spectr. Anal. 2011, 31, 1–6. [Google Scholar] [CrossRef]
- Webster, R.D. In situ electrochemical-NMR spectroscopy. Reduction of aromatic halides. Anal. Chem. 2004, 76, 1603–1610. [Google Scholar] [CrossRef]
- Da Silva, P.F.; Gomes, B.F.; Silva Lobo, C.M.; Keng Queiroz Júnior, L.H.; Danieli, E.; Carmo, M.; Blüemich, B.; Colnago, L.A. Electrochemical NMR spectroscopy: Electrode construction and magnetic sample stirring. Microchem. J. 2019, 146, 658–663. [Google Scholar] [CrossRef]
- Zhou, L.-N.; Leskes, M.; Ilott, A.J.; Trease, N.M.; Grey, C.P. Paramagnetic electrodes and bulk magnetic susceptibility effects in the in situ NMR studies of batteries: Application to Li1.08Mn1.92O4 spinels. J. Magn. Reson. 2013, 234, 44–57. [Google Scholar] [CrossRef]
- Ni, Z.-R.; Cui, X.-H.; Cao, S.-H.; Chen, Z. A novel in situ electrochemical NMR cell with a palisade gold film electrode. AIP Adv. 2017, 7, 085205. [Google Scholar] [CrossRef] [Green Version]
- Hoult, D.I.; Richards, R.E. The signal-to-noise ratio of the nuclear magnetic resonance experiment. J. Magn. Reson. 1976, 24, 71–85. [Google Scholar] [CrossRef]
- Hoult, D.I. The principle of reciprocity in signal strength calculations–A mathematical guide. Concepts Magn. Reson. 2000, 12, 173–187. [Google Scholar] [CrossRef]
- Nishizaki, Y.; Lankin, D.C.; Chen, S.; Pauli, G.F. Accurate and precise external calibration enhances the versatility of quantitative NMR (qNMR). Anal. Chem. 2021, 93, 2733–2741. [Google Scholar] [CrossRef]
- Burton, I.W.; Quilliam, M.A.; Walter, J.A. Quantitative 1H NMR with external standards: Use in preparation of calibration solutions for algal toxins and other natural products. Anal. Chem. 2005, 77, 3123–3131. [Google Scholar] [CrossRef]
- Chaplin, B.P. Critical review of electrochemical advanced oxidation processes for water treatment applications. Environ. Sci.-Process Impacts 2014, 16, 1182–1203. [Google Scholar] [CrossRef]
- Sires, I.; Brillas, E.; Oturan, M.A.; Rodrigo, M.A.; Panizza, M. Electrochemical advanced oxidation processes: Today and tomorrow. A review. Environ. Sci. Pollut. Res. 2014, 21, 8336–8367. [Google Scholar] [CrossRef] [PubMed]
- Moreira, F.C.; Boaventura, R.A.R.; Brillas, E.; Vilar, V.J.P. Electrochemical advanced oxidation processes: A review on their application to synthetic and real wastewaters. Appl. Catal. B-Environ. 2017, 202, 217–261. [Google Scholar] [CrossRef]
- Comninellis, C. Electrocatalysis in the electrochemical conversion/combustion of organic pollutants for waste water treatment. Electrochim. Acta 1994, 39, 1857–1862. [Google Scholar] [CrossRef]
- Mohammed, A.A.; Mohamed, K.A.; Wan Mohd, A.W.D.; Shaukat, A.M. Effect of adsorption and passivation phenomena on the electrochemical oxidation of phenol and 2-Chlorophenol at carbon black diamond composite electrode. Ind. Eng. Chem. Res. 2017, 56, 1652–1660. [Google Scholar] [CrossRef]
- Seid, L.; Lakhdari, D.; Berkani, M.; Belgherbi, O.; Chouder, D.; Vasseghian, Y.; Lakhdari, N. High-efficiency electrochemical degradation of phenol in aqueous solutions using Ni-PPy and Cu-PPy composite materials. J. Hazard. Mater. 2022, 423, 126986. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, M.; Ni, Z.-R.; Sun, H.-J.; Cao, S.-H.; Chen, Z. In Situ Real-Time Quantitative Determination in Electrochemical Nuclear Magnetic Resonance Spectroscopy. Sensors 2022, 22, 282. https://doi.org/10.3390/s22010282
Liu M, Ni Z-R, Sun H-J, Cao S-H, Chen Z. In Situ Real-Time Quantitative Determination in Electrochemical Nuclear Magnetic Resonance Spectroscopy. Sensors. 2022; 22(1):282. https://doi.org/10.3390/s22010282
Chicago/Turabian StyleLiu, Min, Zu-Rong Ni, Hui-Jun Sun, Shuo-Hui Cao, and Zhong Chen. 2022. "In Situ Real-Time Quantitative Determination in Electrochemical Nuclear Magnetic Resonance Spectroscopy" Sensors 22, no. 1: 282. https://doi.org/10.3390/s22010282
APA StyleLiu, M., Ni, Z.-R., Sun, H.-J., Cao, S.-H., & Chen, Z. (2022). In Situ Real-Time Quantitative Determination in Electrochemical Nuclear Magnetic Resonance Spectroscopy. Sensors, 22(1), 282. https://doi.org/10.3390/s22010282