
Computational Strategies for the Identification of a Transcriptional Biomarker Panel to Sense Cellular Growth States in Bacillus subtilis


Abstract
1. Introduction
2. Materials and Methods
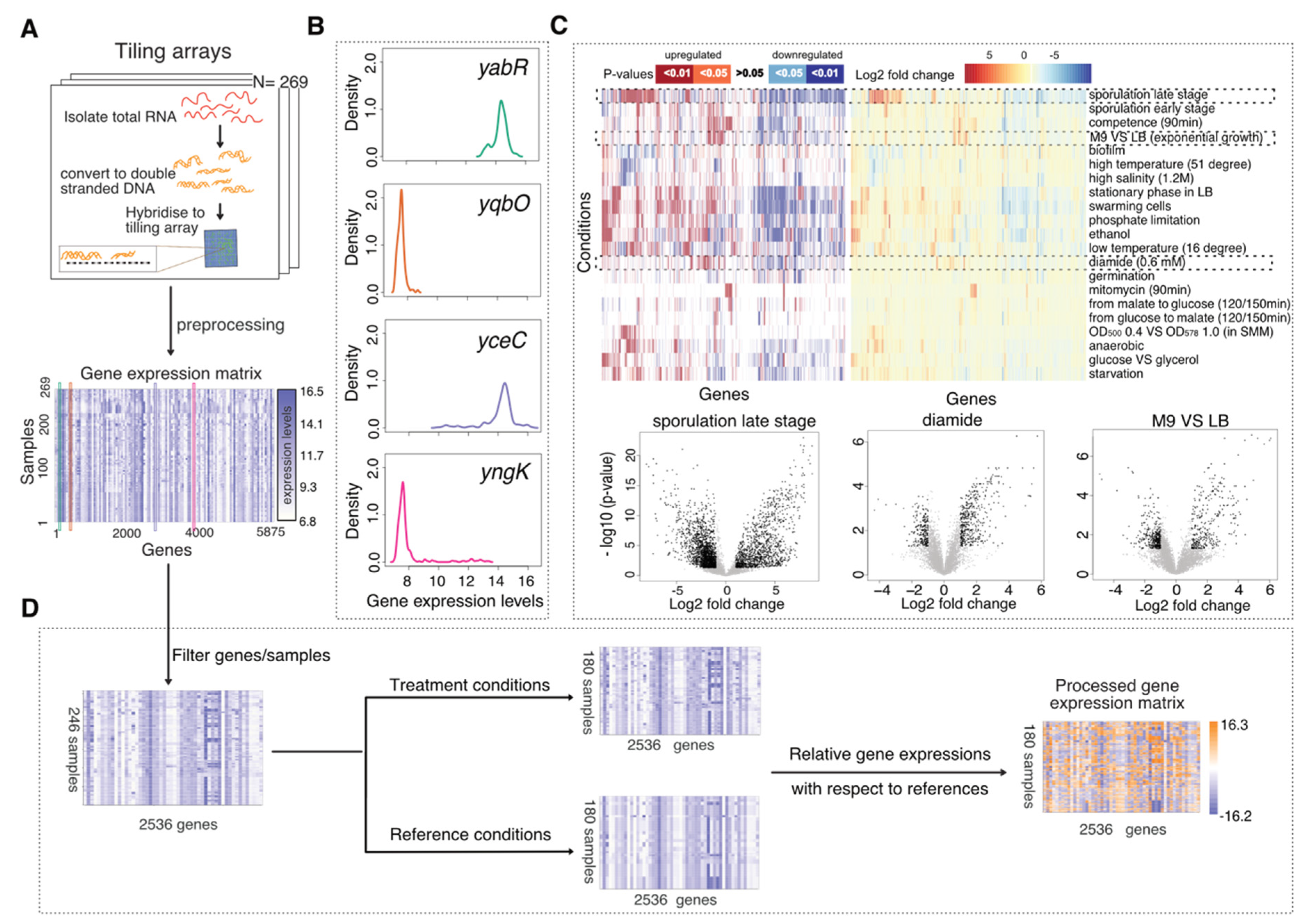
2.1. Tiling Array Data and Data Pre-Processing
2.2. Data Processing
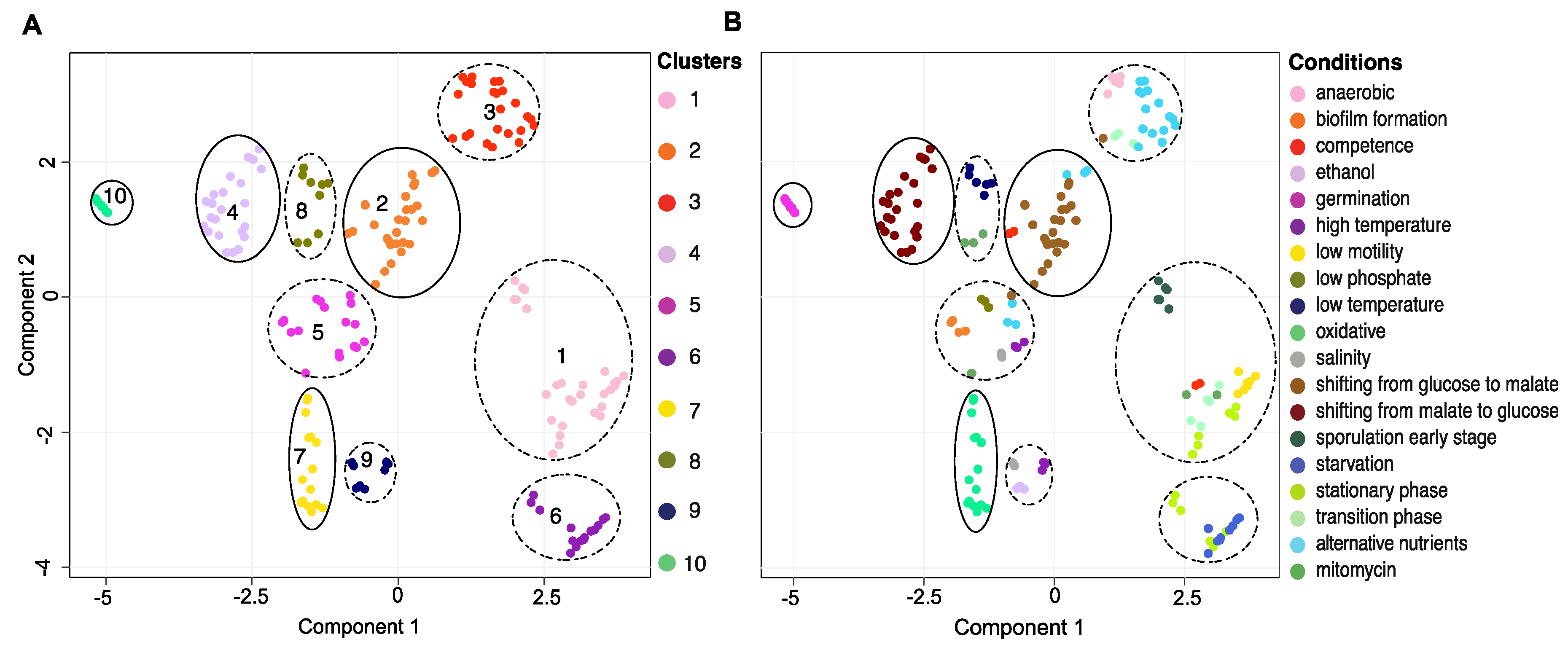
2.3. Dimension Reduction and Clustering
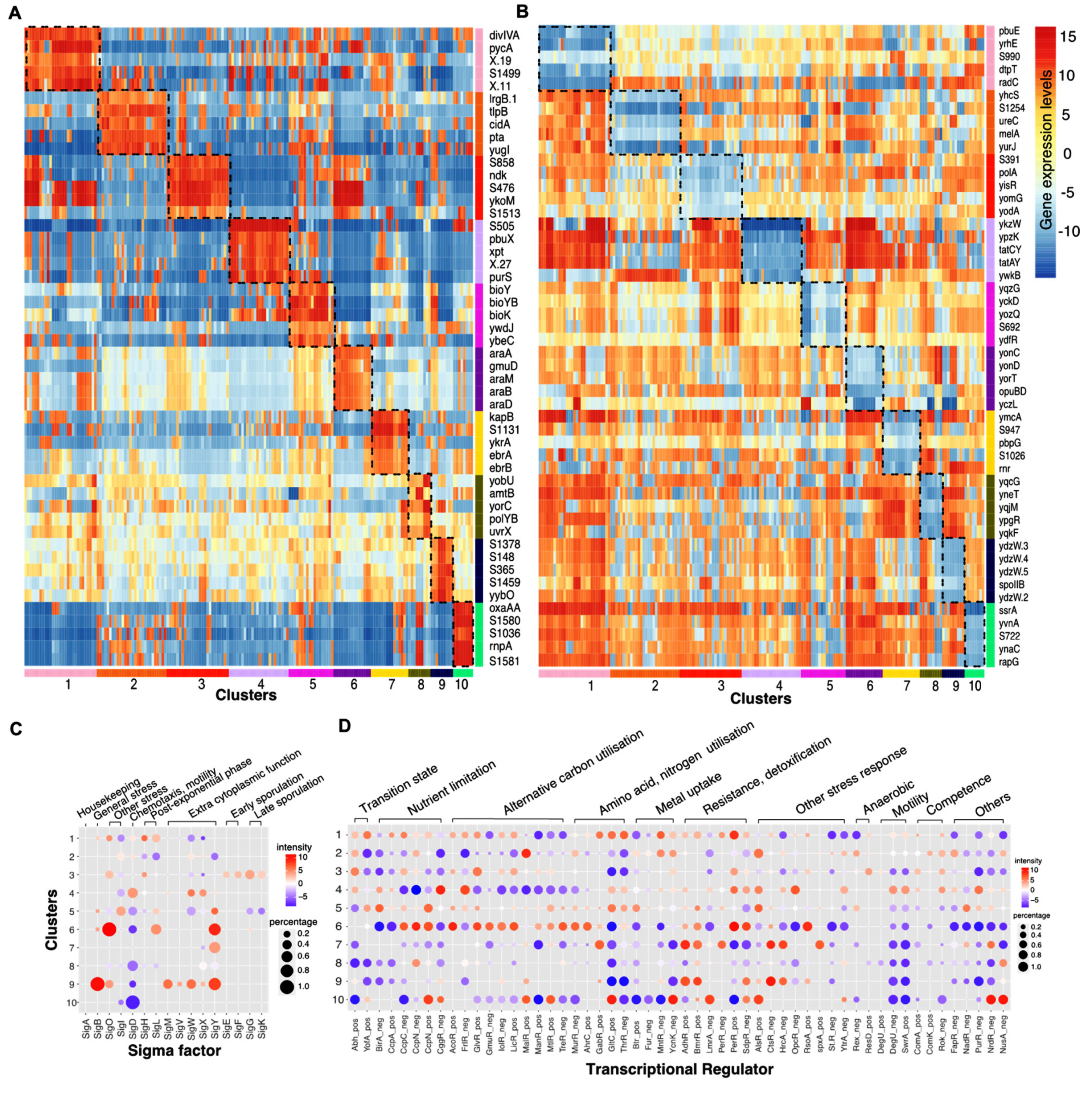
2.4. Feature Selection Method
2.5. Statistical Tests: Differential and Enrichment Analysis
2.6. Knowledgebase: Gene Annotations, Transcriptional Regulatory Network, Gene Ontology, KEGG Pathways
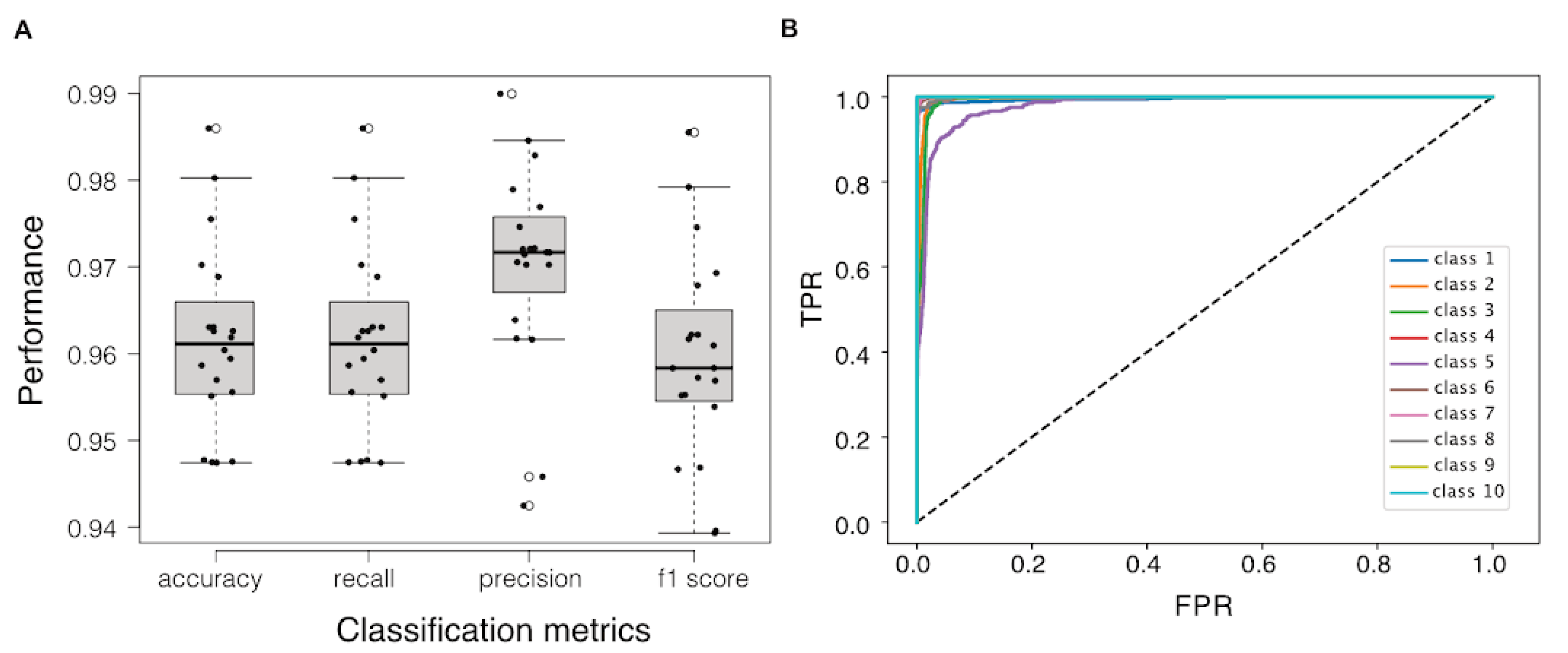
2.7. Validation of the Biomarker Panel on Independent Datasets
3. Results
3.1. The Transcriptional Landscape Constructed by Condition-Dependent Transcriptomes Reveals Major Cellular States of B. subtilis
3.2. A Small Subset of Genes Can Be Used as the Biomarker Panel to Indicate the Cellular State of B. Subtilis
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hoe, C.H.; Raabe, C.A.; Rozhdestvensky, T.S.; Tang, T.H. Bacterial sRNAs: Regulation in stress. Int. J. Med. Microbiol. 2013, 303, 217–229. [Google Scholar] [CrossRef]
- Hecker, M.; Völker, U. General stress response of Bacillus subtilis and other bacteria. Adv. Microb. Physiol. 2001, 44, 35–91. [Google Scholar] [CrossRef] [PubMed]
- Boor, K.J. Bacterial stress responses: What doesn’t kill them can make them stronger. PLoS Biol. 2006, 4, 0018–0020. [Google Scholar] [CrossRef] [PubMed]
- Bonilla, C.Y. Generally stressed out bacteria: Environmental stress response mechanisms in gram-positive bacteria. Integr. Comp. Biol. 2020, 60, 126–133. [Google Scholar] [CrossRef] [PubMed]
- Steil, L.; Hoffmann, T.; Budde, I.; Völker, U.; Bremer, E. Genome-Wide Transcriptional Profiling Analysis of Adaptation of Bacillus subtilis to High Salinity. J. Bacteriol. 2003, 185, 6358–6370. [Google Scholar] [CrossRef] [PubMed]
- Poole, K. Bacterial stress responses as determinants of antimicrobial resistance. J. Antimicrob. Chemother. 2012, 67, 2069–2089. [Google Scholar] [CrossRef]
- Jordan, S.; Hutchings, M.I.; Mascher, T. Cell envelope stress response in Gram-positive bacteria. FEMS Microbiol. Rev. 2008, 32, 107–146. [Google Scholar] [CrossRef]
- Gruber, T.M.; Gross, C.A. Multiple Sigma Subunits and the Partitioning of Bacterial Transcription Space. Annu. Rev. Microbiol. 2003, 57, 441–466. [Google Scholar] [CrossRef]
- Schmidt, F.R. Optimization and scale up of industrial fermentation processes. Appl. Microbiol. Biotechnol. 2005, 68, 425–435. [Google Scholar] [CrossRef]
- Veening, J.W.; Smits, W.K.; Kuipers, O.P. Bistability, epigenetics, and bet-hedging in bacteria. Annu. Rev. Microbiol. 2008, 62, 193–210. [Google Scholar] [CrossRef]
- Gefen, O.; Balaban, N.Q. The importance of being persistent: Heterogeneity of bacterial populations under antibiotic stress: Review article. FEMS Microbiol. Rev. 2009, 33, 704–717. [Google Scholar] [CrossRef] [PubMed]
- Ge, H.; Walhout, A.J.M.; Vidal, M. Integrating “omic” information: A bridge between genomics and systems biology. Trends Genet. 2003, 19, 551–560. [Google Scholar] [CrossRef] [PubMed]
- Bervoets, I.; Charlier, D. Diversity, versatility and complexity of bacterial gene regulation mechanisms: Opportunities and drawbacks for applications in synthetic biology. FEMS Microbiol. Rev. 2019, 43, 304–339. [Google Scholar] [CrossRef] [PubMed]
- Scott, M.; Gunderson, C.W.; Mateescu, E.M.; Zhang, Z.; Hwa, T. Interdependence of Cell Growth and Gene Expression: Origins and Consequences. Science 2010, 330, 1099–1102. [Google Scholar] [CrossRef]
- Dahl, R.H.; Zhang, F.; Alonso-Gutierrez, J.; Baidoo, E.; Batth, T.S.; Redding-Johanson, A.M.; Petzold, C.J.; Mukhopadhyay, A.; Lee, T.S.; Adams, P.D.; et al. Engineering dynamic pathway regulation using stress-response promoters. Nat. Biotechnol. 2013, 31, 1039–1046. [Google Scholar] [CrossRef]
- Anderson, J.C.; Voigt, C.A.; Arkin, A.P. Environmental signal integration by a modular and gate. Mol. Syst. Biol. 2007, 3, 133. [Google Scholar] [CrossRef]
- Gohl, D.M.; Vangay, P.; Garbe, J.; MacLean, A.; Hauge, A.; Becker, A.; Gould, T.J.; Clayton, J.B.; Johnson, T.J.; Hunter, R.; et al. Systematic improvement of amplicon marker gene methods for increased accuracy in microbiome studies. Nat. Biotechnol. 2016, 34, 942–949. [Google Scholar] [CrossRef]
- Schöler, A.; Jacquiod, S.; Vestergaard, G.; Schulz, S.; Schloter, M. Analysis of soil microbial communities based on amplicon sequencing of marker genes. Biol. Fertil. Soils 2017, 53, 485–489. [Google Scholar] [CrossRef]
- Zubakov, D.; Boersma, A.W.M.; Choi, Y.; Van Kuijk, P.F.; Wiemer, E.A.C.; Kayser, M. MicroRNA markers for forensic body fluid identification obtained from microarray screening and quantitative RT-PCR confirmation. Int. J. Legal Med. 2010, 124, 217–226. [Google Scholar] [CrossRef]
- Tanga, F.Y.; Raghavendra, V.; DeLeo, J.A. Quantitative real-time RT-PCR assessment of spinal microglial and astrocytic activation markers in a rat model of neuropathic pain. Neurochem. Int. 2004, 45, 397–407. [Google Scholar] [CrossRef]
- Glick, B.R. Metabolic load and heterologous gene expression. Biotechnol. Adv. 1995, 13, 247–261. [Google Scholar] [CrossRef]
- Silva, F.; Queiroz, J.A.; Domingues, F.C. Evaluating metabolic stress and plasmid stability in plasmid DNA production by Escherichia coli. Biotechnol. Adv. 2012, 30, 691–708. [Google Scholar] [CrossRef]
- Carrera, J.; Rodrigo, G.; Singh, V.; Kirov, B.; Jaramillo, A. Empirical model and in vivo characterization of the bacterial response to synthetic gene expression show that ribosome allocation limits growth rate. Biotechnol. J. 2011, 6, 773–783. [Google Scholar] [CrossRef]
- Kurland, C.G.; Dong, H. Bacterial growth inhibition by overproduction of protein. Mol. Microbiol. 1996, 21, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Hamadeh, A.; Del Vecchio, D. Mitigation of resource competition in synthetic genetic circuits through feedback regulation. In Proceedings of the 53rd IEEE Conference on Decision and Control, Los Angeles, CA, USA, 15–17 December 2014; pp. 3829–3834. [Google Scholar]
- Borkowski, O.; Ceroni, F.; Stan, G.B.; Ellis, T. Overloaded and stressed: Whole-cell considerations for bacterial synthetic biology. Curr. Opin. Microbiol. 2016, 33, 123–130. [Google Scholar] [CrossRef]
- Gorochowski, T.E.; Avcilar-Kucukgoze, I.; Bovenberg, R.A.L.; Roubos, J.A.; Ignatova, Z. A Minimal Model of Ribosome Allocation Dynamics Captures Trade-offs in Expression between Endogenous and Synthetic Genes. ACS Synth. Biol. 2016, 5, 710–720. [Google Scholar] [CrossRef] [PubMed]
- Ceroni, F.; Furini, S.; Stefan, A.; Hochkoeppler, A.; Giordano, E. A synthetic Post-transcriptional controller to explore the modular design of gene circuits. ACS Synth. Biol. 2012, 1, 163–171. [Google Scholar] [CrossRef]
- Brown, D.R.; Barton, G.; Pan, Z.; Buck, M.; Wigneshweraraj, S. Nitrogen stress response and stringent response are coupled in Escherichia coli. Nat. Commun. 2014, 5, 1–8. [Google Scholar] [CrossRef]
- Chiang, S.M.; Schellhorn, H.E. Regulators of oxidative stress response genes in Escherichia coli and their functional conservation in bacteria. Arch. Biochem. Biophys. 2012, 525, 161–169. [Google Scholar] [CrossRef]
- Bojanovic, K.; Arrigo, I.D.; Long, K.S. Global Transcriptional Responses to Osmotic, Oxidative, and Imipenem Stress Conditions in Pseudomonas putida. Appl. Environ. Microbiol. 2017, 83, 1–18. [Google Scholar] [CrossRef]
- den Besten, H.M.W.; Arvind, A.; Gaballo, H.M.S.; Moezelaar, R.; Zwietering, M.H.; Abee, T. Short- and long-term biomarkers for bacterial robustness: A framework for quantifying correlations between cellular indicators and adaptive behavior. PLoS ONE 2010, 5, e13746. [Google Scholar] [CrossRef]
- Rau, M.H.; Bojanovič, K.; Nielsen, A.T.; Long, K.S. Differential expression of small RNAs under chemical stress and fed-batch fermentation in E. coli. BMC Genom. 2015, 16, 1–16. [Google Scholar] [CrossRef]
- Utaida, S.; Dunman, P.M.; Macapagal, D.; Murphy, E.; Projan, S.J.; Singh, V.K.; Jayaswal, R.K.; Wilkinson, B.J. Genome-wide transcriptional profiling of the response of Staphylococcus aureus to cell-wall-active antibiotics reveals a cell-wall-stress stimulon. Microbiology 2003, 149, 2719–2732. [Google Scholar] [CrossRef]
- Alsaker, K.V.; Paredes, C.; Papoutsakis, E.T. Metabolite stress and tolerance in the production of biofuels and chemicals: Gene-expression-based systems analysis of butanol, butyrate, and acetate stresses in the anaerobe Clostridium acetobutylicum. Biotechnol. Bioeng. 2010, 105, 1131–1147. [Google Scholar] [CrossRef]
- Avican, K.; Aldahdooh, J.; Togninalli, M.; Tang, J.; Borgwardt, K.; Rhen, M.; Fällman, M. RNA Atlas of Human Bacterial Pathogens Uncovers Stress Dynamics Linked to Infection. bioRxiv 2020. [Google Scholar] [CrossRef]
- Kim, M.; Rai, N.; Zorraquino, V.; Tagkopoulos, I. Multi-omics integration accurately predicts cellular state in unexplored conditions for Escherichia coli. Nat. Commun. 2016, 7, 1–12. [Google Scholar] [CrossRef]
- Skelly, D.A.; Squiers, G.T.; McLellan, M.A.; Bolisetty, M.T.; Robson, P.; Rosenthal, N.A.; Pinto, A.R. Single-Cell Transcriptional Profiling Reveals Cellular Diversity and Intercommunication in the Mouse Heart. Cell Rep. 2018, 22, 600–610. [Google Scholar] [CrossRef]
- Tepe, B.; Hill, M.C.; Pekarek, B.T.; Hunt, P.J.; Martin, T.J.; Martin, J.F.; Arenkiel, B.R. Single-Cell RNA-Seq of Mouse Olfactory Bulb Reveals Cellular Heterogeneity and Activity-Dependent Molecular Census of Adult-Born Neurons. Cell Rep. 2018, 25, 2689–2703.e3. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.; Spielmann, M.; Qiu, X.; Huang, X.; Ibrahim, D.M.; Hill, A.J.; Zhang, F.; Mundlos, S.; Christiansen, L.; Steemers, F.J.; et al. The single-cell transcriptional landscape of mammalian organogenesis. Nature 2019, 566, 496–502. [Google Scholar] [CrossRef] [PubMed]
- Alkhateeb, A.; Rezaeian, I.; Singireddy, S.; Cavallo-Medved, D.; Porter, L.A.; Rueda, L. Transcriptomics Signature from Next-Generation Sequencing Data Reveals New Transcriptomic Biomarkers Related to Prostate Cancer. Cancer Inform. 2019, 18. [Google Scholar] [CrossRef] [PubMed]
- Smith, B.P.; Auvil, L.S.; Welge, M.; Bushell, C.B.; Bhargava, R.; Elango, N.; Johnson, K.; Madak-Erdogan, Z. Identification of early liver toxicity gene biomarkers using comparative supervised machine learning. Sci. Rep. 2020, 10, 1–27. [Google Scholar] [CrossRef] [PubMed]
- Peng, J.; Sun, B.F.; Chen, C.Y.; Zhou, J.Y.; Chen, Y.S.; Chen, H.; Liu, L.; Huang, D.; Jiang, J.; Cui, G.S.; et al. Single-cell RNA-seq highlights intra-tumoral heterogeneity and malignant progression in pancreatic ductal adenocarcinoma. Cell Res. 2019, 29, 725–738. [Google Scholar] [CrossRef] [PubMed]
- Tabl, A.A.; Alkhateeb, A.; ElMaraghy, W.; Rueda, L.; Ngom, A. A machine learning approach for identifying gene biomarkers guiding the treatment of breast cancer. Front. Genet. 2019, 10, 1–13. [Google Scholar] [CrossRef]
- Nicolas, P.; Mäder, U.; Dervyn, E.; Rochat, T.; Leduc, A.; Pigeonneau, N.; Bidnenko, E.; Marchadier, E.; Hoebeke, M.; Aymerich, S.; et al. Condition-dependent transcriptome reveals high-level regulatory architecture in Bacillus subtilis. Science 2012. [Google Scholar] [CrossRef] [PubMed]
- McInnes, L.; Healy, J.; Melville, J. UMAP: Uniform Manifold Approximation and Projection for Dimension Reduction. arXiv 2018, arXiv:1802.03426. [Google Scholar]
- Traag, V.A.; Waltman, L.; van Eck, N.J. From Louvain to Leiden: Guaranteeing well-connected communities. Sci. Rep. 2019, 9, 1–12. [Google Scholar] [CrossRef]
- Lazzarini, N.; Bacardit, J. RGIFE: A ranked guided iterative feature elimination heuristic for the identification of biomarkers. BMC Bioinform. 2017, 18, 1–22. [Google Scholar] [CrossRef]
- Bolstad, B.M.; Irizarry, R.A.; Åstrand, M.; Speed, T.P. A comparison of normalization methods for high density oligonucleotide array data based on variance and bias. Bioinformatics 2003, 19, 185–193. [Google Scholar] [CrossRef]
- Smyth, G.K. Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Stat. Appl. Genet. Mol. Biol. 2004, 3, 1–26. [Google Scholar] [CrossRef]
- Qiu, X.; Hill, A.; Packer, J.; Lin, D.; Ma, Y.A.; Trapnell, C. Single-cell mRNA quantification and differential analysis with Census. Nat. Methods 2017, 14, 309–315. [Google Scholar] [CrossRef]
- Stuart, T.; Butler, A.; Hoffman, P.; Hafemeister, C.; Papalexi, E.; Mauck, W.M.; Hao, Y.; Stoeckius, M.; Smibert, P.; Satija, R. Comprehensive Integration of Single-Cell Data. Cell 2019, 177, 1888–1902.e21. [Google Scholar] [CrossRef]
- Andrews, T.S.; Hemberg, M. Identifying cell populations with scRNASeq. Mol. Aspects Med. 2018, 59, 114–122. [Google Scholar] [CrossRef] [PubMed]
- Swan, A.L.; Stekel, D.J.; Hodgman, C.; Allaway, D.; Alqahtani, M.H.; Mobasheri, A.; Bacardit, J. A machine learning heuristic to identify biologically relevant and minimal biomarker panels from omics data. BMC Genomics 2015, 16, S2. [Google Scholar] [CrossRef] [PubMed]
- Lazzarini, N.; Runhaar, J.; Bay-Jensen, A.C.; Thudium, C.S.; Bierma-Zeinstra, S.M.A.; Henrotin, Y.; Bacardit, J. A machine learning approach for the identification of new biomarkers for knee osteoarthritis development in overweight and obese women. Osteoarthr. Cartil. 2017, 25, 2014–2021. [Google Scholar] [CrossRef] [PubMed]
- Bindea, G.; Mlecnik, B.; Hackl, H.; Charoentong, P.; Tosolini, M.; Kirilovsky, A.; Fridman, W.H.; Pagès, F.; Trajanoski, Z.; Galon, J. ClueGO: A Cytoscape plug-in to decipher functionally grouped gene ontology and pathway annotation networks. Bioinformatics 2009, 25, 1091–1093. [Google Scholar] [CrossRef] [PubMed]
- Faria, J.P.; Overbeek, R.; Taylor, R.C.; Conrad, N.; Vonstein, V.; Goelzer, A.; Fromion, V.; Rocha, M.; Rocha, I.; Henry, C.S. Reconstruction of the regulatory network for Bacillus subtilis and reconciliation with gene expression data. Front. Microbiol. 2016, 7. [Google Scholar] [CrossRef] [PubMed]
- Zhu, B.; Stülke, J. SubtiWiki in 2018: From genes and proteins to functional network annotation of the model organism Bacillus subtilis. Nucleic Acids Res. 2018, 46, D743–D748. [Google Scholar] [CrossRef]
- Mansourifar, H.; Shi, W. Deep synthetic minority over-sampling technique. arXiv 2020, arXiv:2003.09788. [Google Scholar]
- Kaiser, J.C.; Heinrichs, D.E. Branching out: Alterations in bacterial physiology and virulence due to branched-chain amino acid deprivation. MBio 2018, 9, 1–17. [Google Scholar] [CrossRef]
- Lenhart, J.S.; Schroeder, J.W.; Walsh, B.W.; Simmons, L.A. DNA Repair and Genome Maintenance in Bacillus subtilis. Microbiol. Mol. Biol. Rev. 2012, 76, 530–564. [Google Scholar] [CrossRef]
- Benoist, C.; Guérin, C.; Noirot, P.; Dervyn, E. Constitutive stringent response restores viability of Bacillus subtilis lacking structural maintenance of chromosome protein. PLoS ONE 2015, 10, e0142308. [Google Scholar] [CrossRef] [PubMed]
- Fang, M.; Bauer, C.E. Regulation of stringent factor by branched-chain amino acids. Proc. Natl. Acad. Sci. USA 2018, 115, 6446–6451. [Google Scholar] [CrossRef]
- Burby, P.E.; Simmons, L.A. A bacterial DNA repair pathway specific to a natural antibiotic. Mol. Microbiol. 2019, 111, 338–353. [Google Scholar] [CrossRef] [PubMed]
- Krispin, O.; Allmansberger, R. Changes in DNA supertwist as a response of Bacillus subtilis towards different kinds of stress. FEMS Microbiol. Lett. 1995, 134, 129–135. [Google Scholar] [CrossRef] [PubMed]
- Budde, I.; Steil, L.; Scharf, C.; Völker, U.; Bremer, E. Adaptation of Bacillus subtilis to growth at low temperature: A combined transcriptomic and proteomic appraisal. Microbiology 2006, 152, 831–853. [Google Scholar] [CrossRef]
- Johnson, W.E.; Li, C.; Rabinovic, A. Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics 2007, 8, 118–127. [Google Scholar] [CrossRef]
- Leek, J.T.; Storey, J.D. Capturing heterogeneity in gene expression studies by surrogate variable analysis. PLoS Genet. 2007, 3, e161. [Google Scholar] [CrossRef]
- Fei, T.; Yu, T. Batch Effect Correction of RNA-seq Data through Sample Distance Matrix Adjustment. bioRxiv 2019, 669739. [Google Scholar] [CrossRef]
- Haghverdi, L.; Lun, A.T.L.; Morgan, M.D.; Marioni, J.C. Batch effects in single-cell RNA-sequencing data are corrected by matching mutual nearest neighbors. Nat. Biotechnol. 2018, 36, 421–427. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
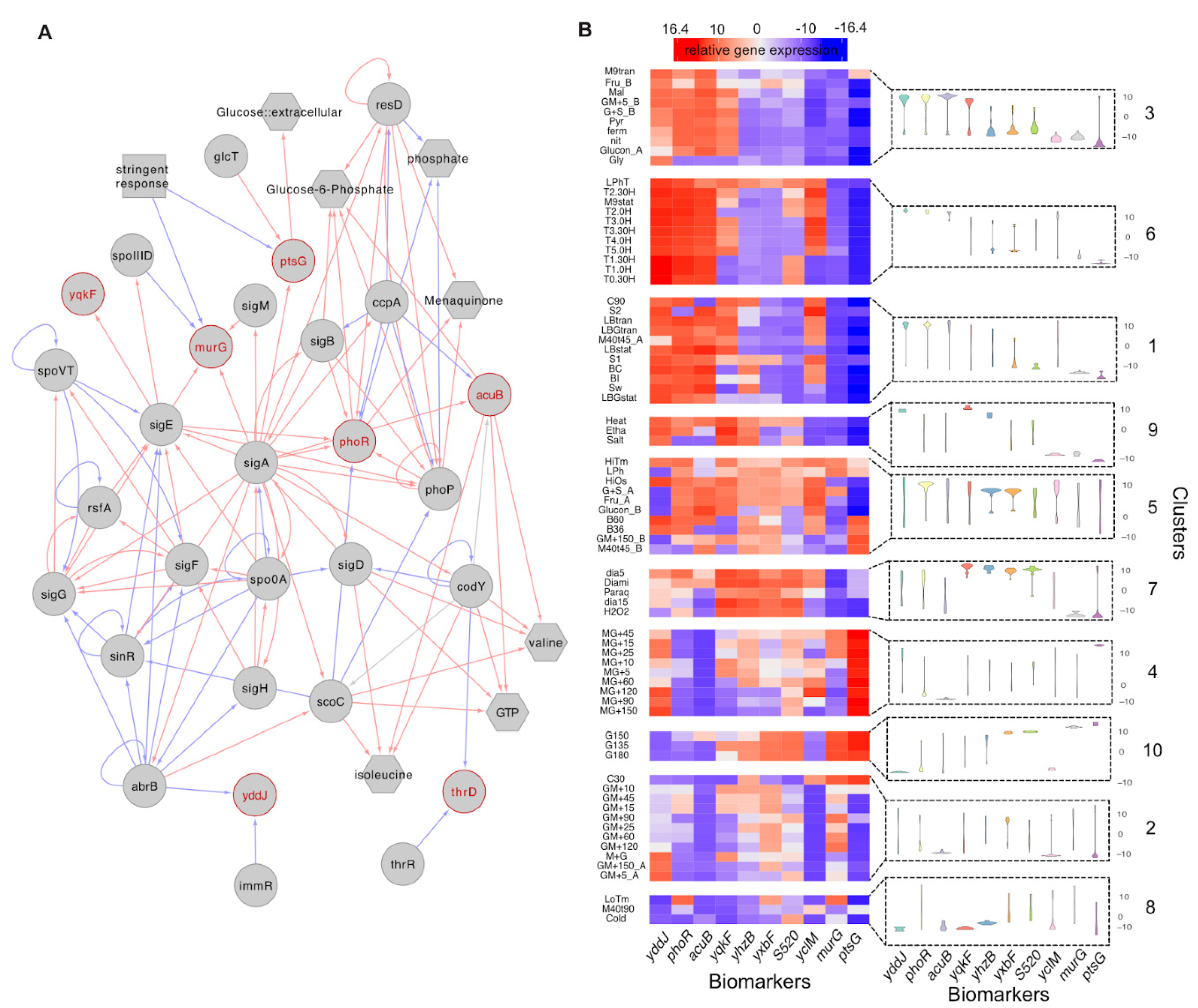{kind=link}
| Biomarker | Function | Product |
|---|---|---|
| yddJ | prevention of redundant transfer of ICEBs1 into host cells | ICEBs1 exclusion factor, putative lipoprotein |
| phoR | regulation of phosphate metabolism | two-component sensor kinase |
| acuB | unknown | unknown |
| yqkF | unknown | NADPH-dependent 4-Hydroxy-2,3-trans-nonenal reductase |
| yhzB | unknown | conserved hypothetical protein |
| yclM (thrD) | unknown | aspartokinase III |
| yxbF | similar to transcriptional regulator (TetR family) | unknown |
| S520 | The antisense RNA of ygxB | 5’UTR of mobA |
| murG | peptidoglycan precursor biosynthesis | UDP-N-acetylglucosamine-N-acetylmuramyl-(pentapeptide)pyrophosphoryl-undecaprenol N-acetylglucosamine transferase |
| ptsG | glucose transport and phosphorylation, control of GlcT activity, phosphotransferase system (PTS) glucose-specific enzyme IICBA component | glucose permease, trigger enzyme |
| Test Condition | Sample Size | Accuracy | Recall | Precision | F1-Score | KS p-Value | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Treatment | Control | Mean | std | Mean | std | Mean | std | Mean | std | ||
| Salinity stress | 6 | 6 | 0.98 | 0.03 | 0.99 | 0.03 | 0.98 | 0.04 | 0.98 | 0.03 | 1.84 × 10−7 |
| Glycine betaine | 6 | 6 | 0.96 | 0.05 | 0.98 | 0.05 | 0.95 | 0.07 | 0.96 | 0.05 | 6.39 × 10−3 |
| Heat stroke | 12 | 9 | 0.89 | 0.04 | 0.92 | 0.05 | 0.88 | 0.05 | 0.90 | 0.04 | <1 × 10−10 |
| Cold shock | 5 | 4 | 0.90 | 0.04 | 0.82 | 0.07 | 1.00 | 0.00 | 0.90 | 0.04 | 2.42 × 10−9 |
| Oxidative stress | 5 | 5 | 0.92 | 0.07 | 0.86 | 0.01 | 0.99 | 0.01 | 0.91 | 0.09 | <1 × 10−10 |
| Pressure stress | 11 | 5 | 0.99 | 0.01 | 0.99 | 0.02 | 1.00 | 0.00 | 0.99 | 0.01 | <1 × 10−10 |
| Stationary phase | 9 | 9 | 1.00 | 0.00 | 1.00 | 0.00 | 1.00 | 0.00 | 1.00 | 0.00 | 4.64 × 10−4 |
| Antibiotic | 9 | 9 | 0.78 | 0.06 | 0.89 | 0.09 | 0.79 | 0.07 | 0.78 | 0.07 | 1.89 × 10−9 |
| Deep starvation | 3 | 4 | 0.89 | 0.12 | 0.99 | 0.03 | 0.84 | 0.16 | 0.90 | 0.10 | <1 × 10−10 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, Y.; Smith, W.; Harwood, C.; Wipat, A.; Bacardit, J. Computational Strategies for the Identification of a Transcriptional Biomarker Panel to Sense Cellular Growth States in Bacillus subtilis. Sensors 2021, 21, 2436. https://doi.org/10.3390/s21072436
Huang Y, Smith W, Harwood C, Wipat A, Bacardit J. Computational Strategies for the Identification of a Transcriptional Biomarker Panel to Sense Cellular Growth States in Bacillus subtilis. Sensors. 2021; 21(7):2436. https://doi.org/10.3390/s21072436
Chicago/Turabian StyleHuang, Yiming, Wendy Smith, Colin Harwood, Anil Wipat, and Jaume Bacardit. 2021. "Computational Strategies for the Identification of a Transcriptional Biomarker Panel to Sense Cellular Growth States in Bacillus subtilis" Sensors 21, no. 7: 2436. https://doi.org/10.3390/s21072436
APA StyleHuang, Y., Smith, W., Harwood, C., Wipat, A., & Bacardit, J. (2021). Computational Strategies for the Identification of a Transcriptional Biomarker Panel to Sense Cellular Growth States in Bacillus subtilis. Sensors, 21(7), 2436. https://doi.org/10.3390/s21072436