
Hierarchical Porous Carbon Electrodes with Sponge-Like Edge Structures for the Sensitive Electrochemical Detection of Heavy Metals


Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
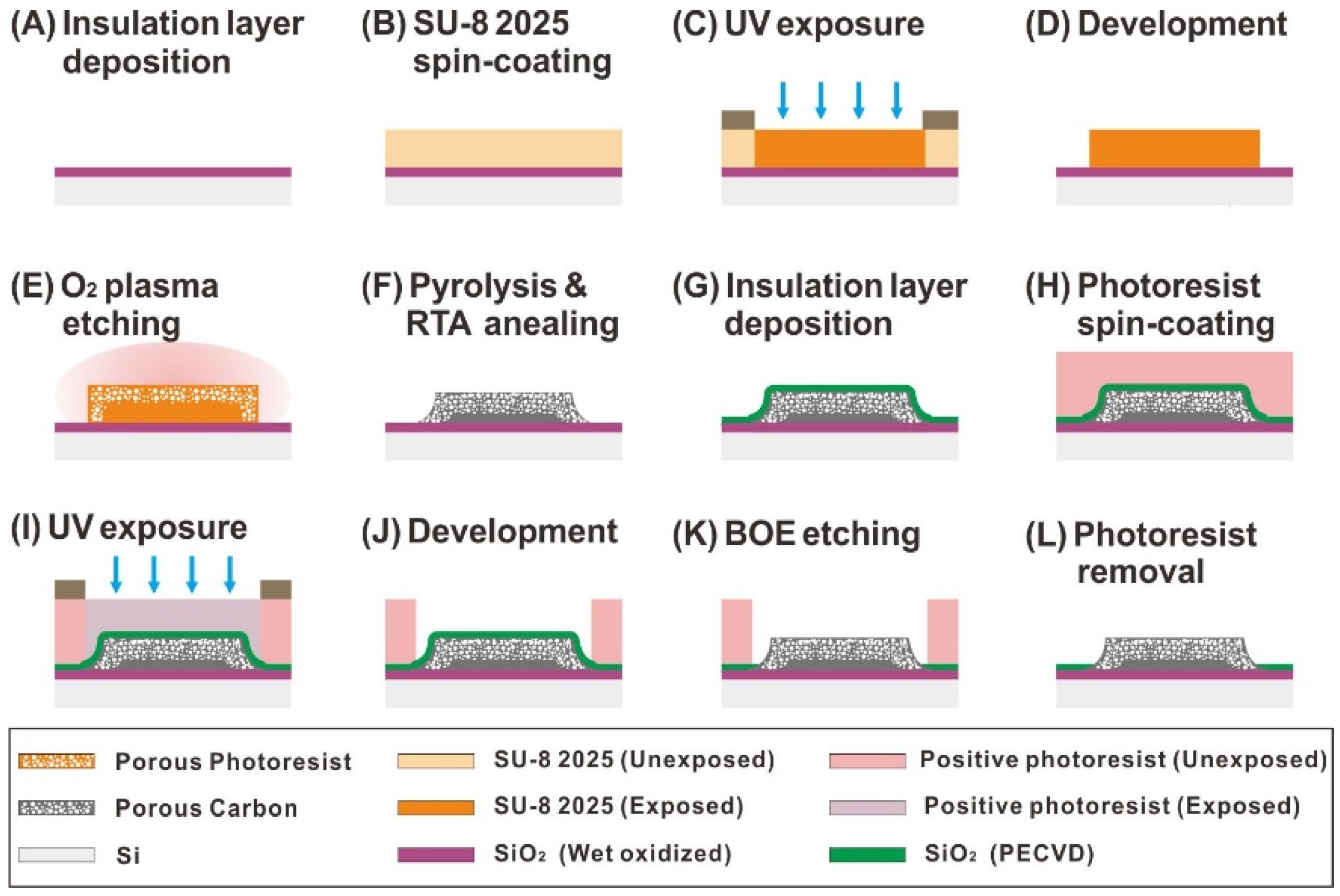
2.2. Fabrication of the Hierarchical Nanoporous Carbon Electrode with Sponge-Like Edge Structures
2.3. Electrochemical Characterization of Hierarchical Nanoporous Carbon Electrodes
3. Results and Discussion
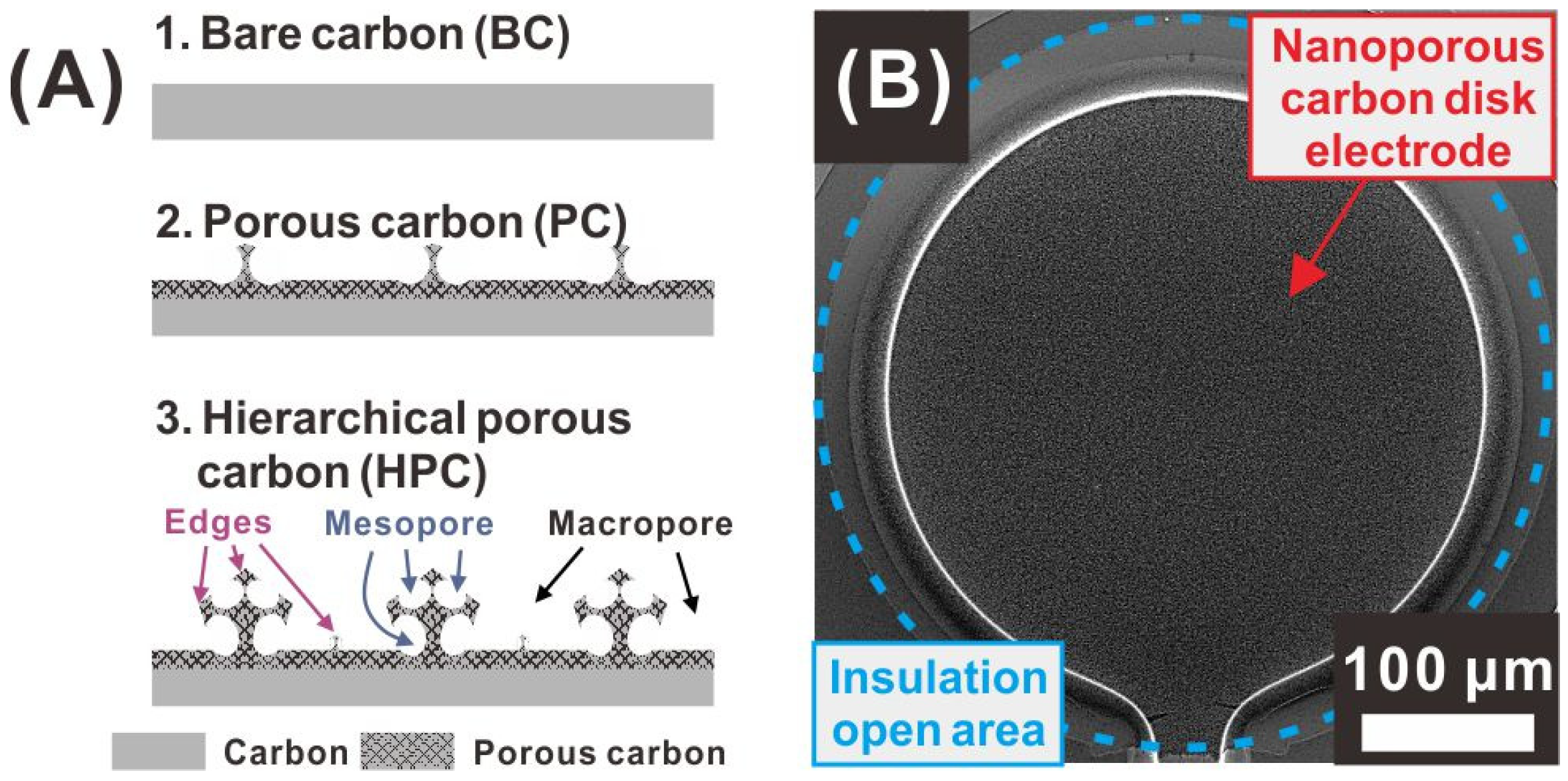
3.1. Morphology of the Nanoporous Carbon Electrode with a Sponge-Like Edge Structure
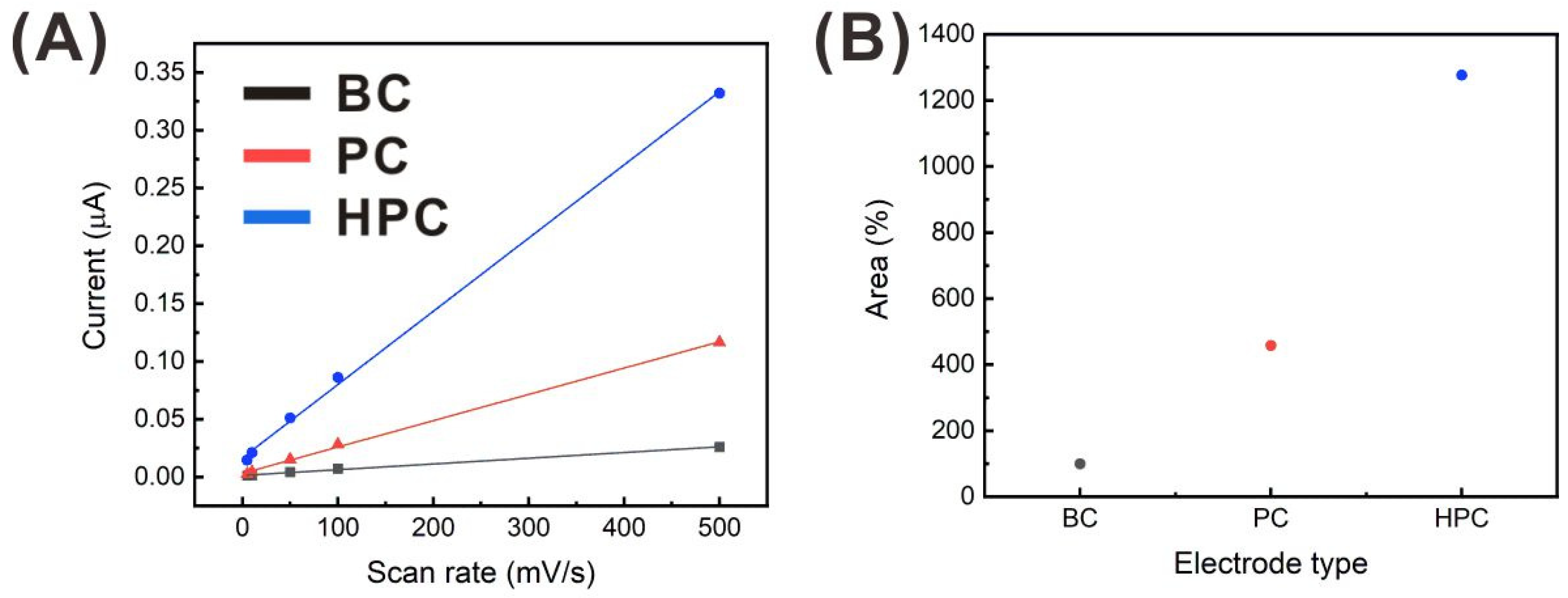
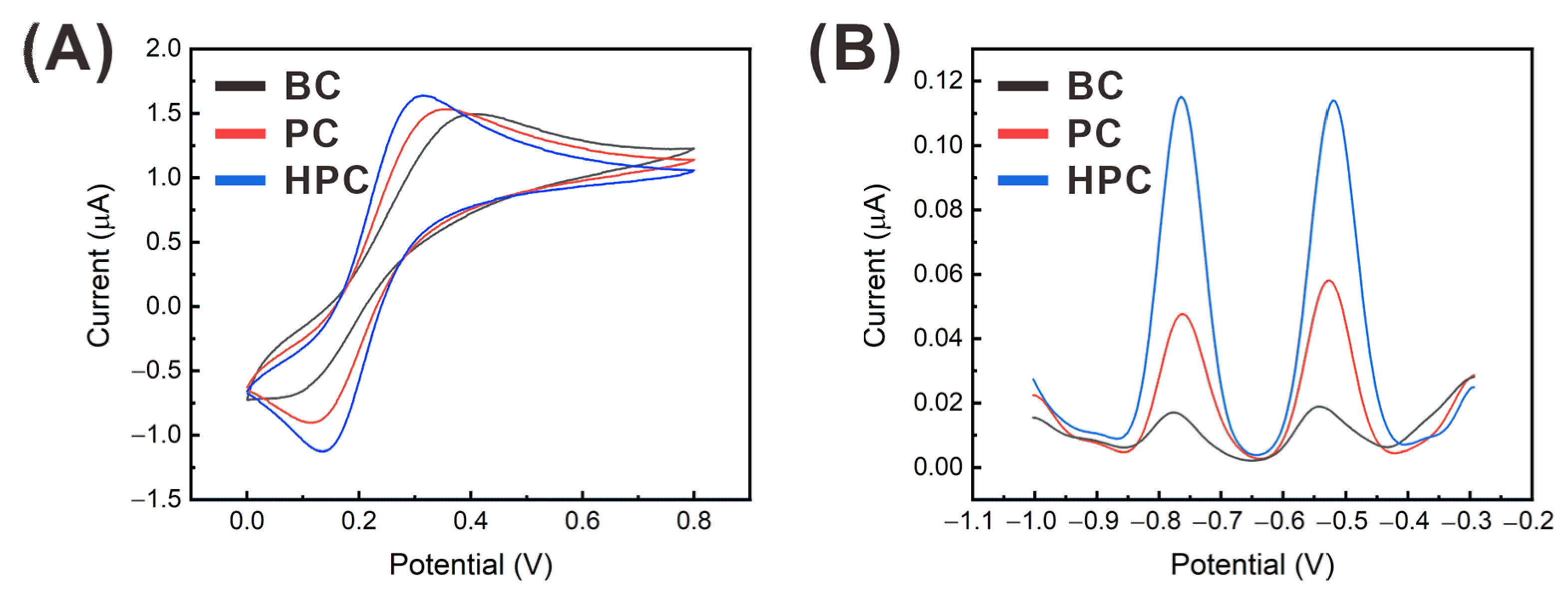
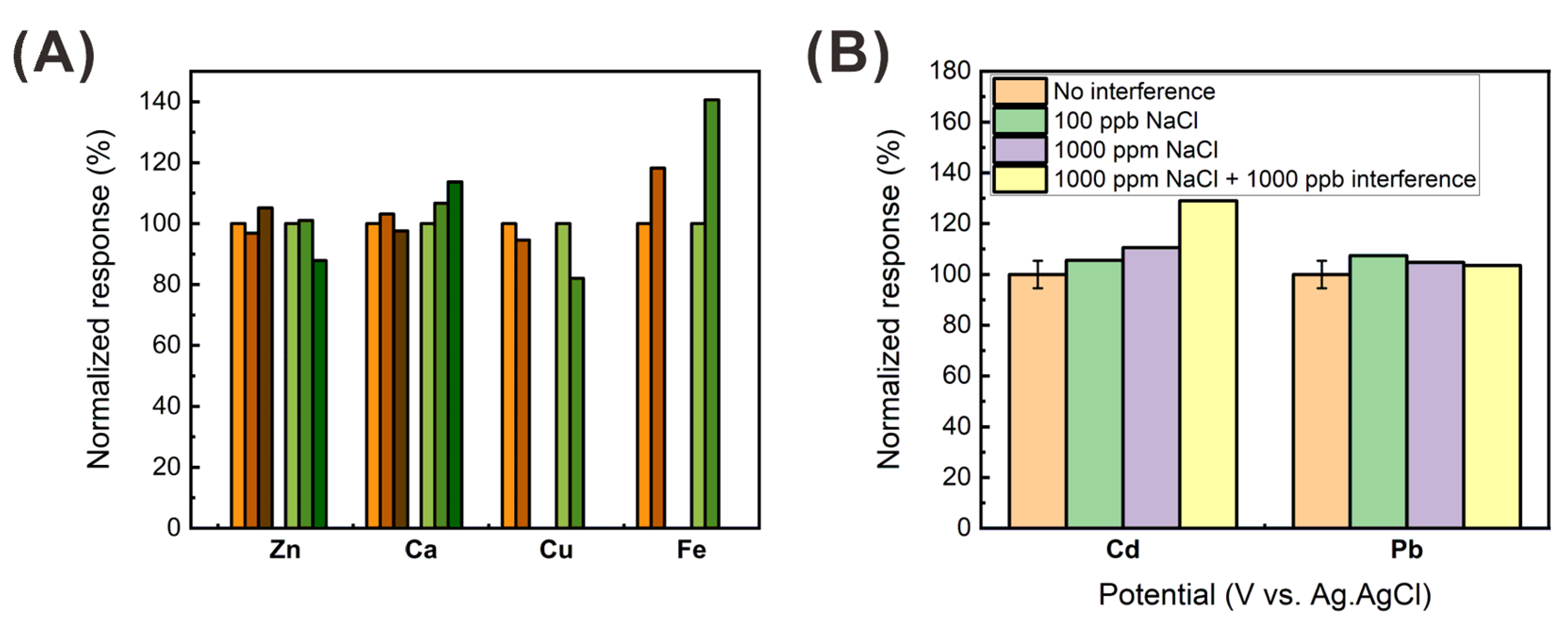
3.2. Effect of the Electrode Surface Properties on the Heavy Metal Detection
3.3. Optimization of the Preconcentration Conditions
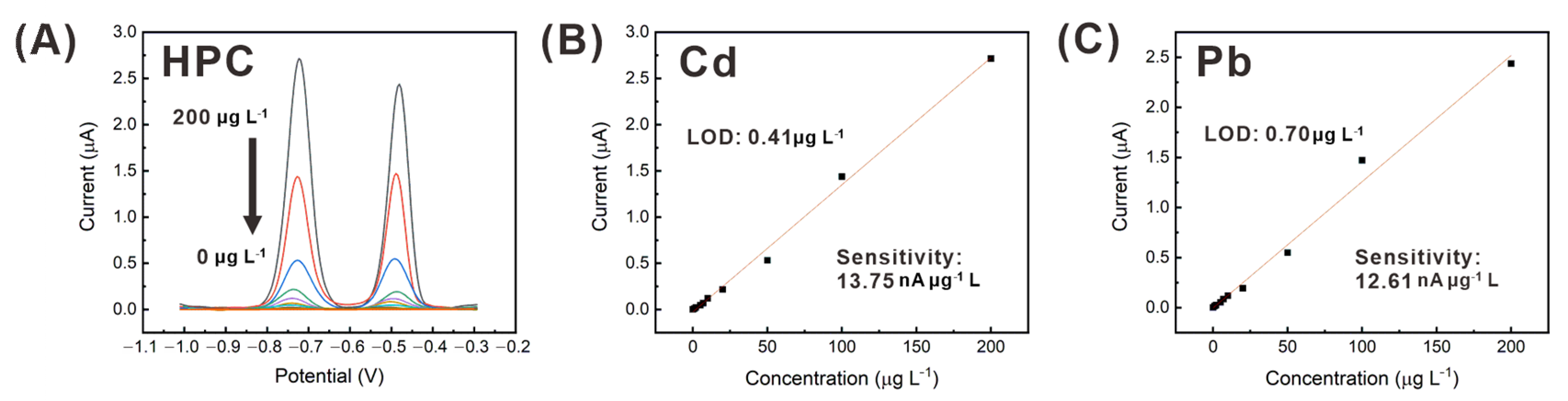
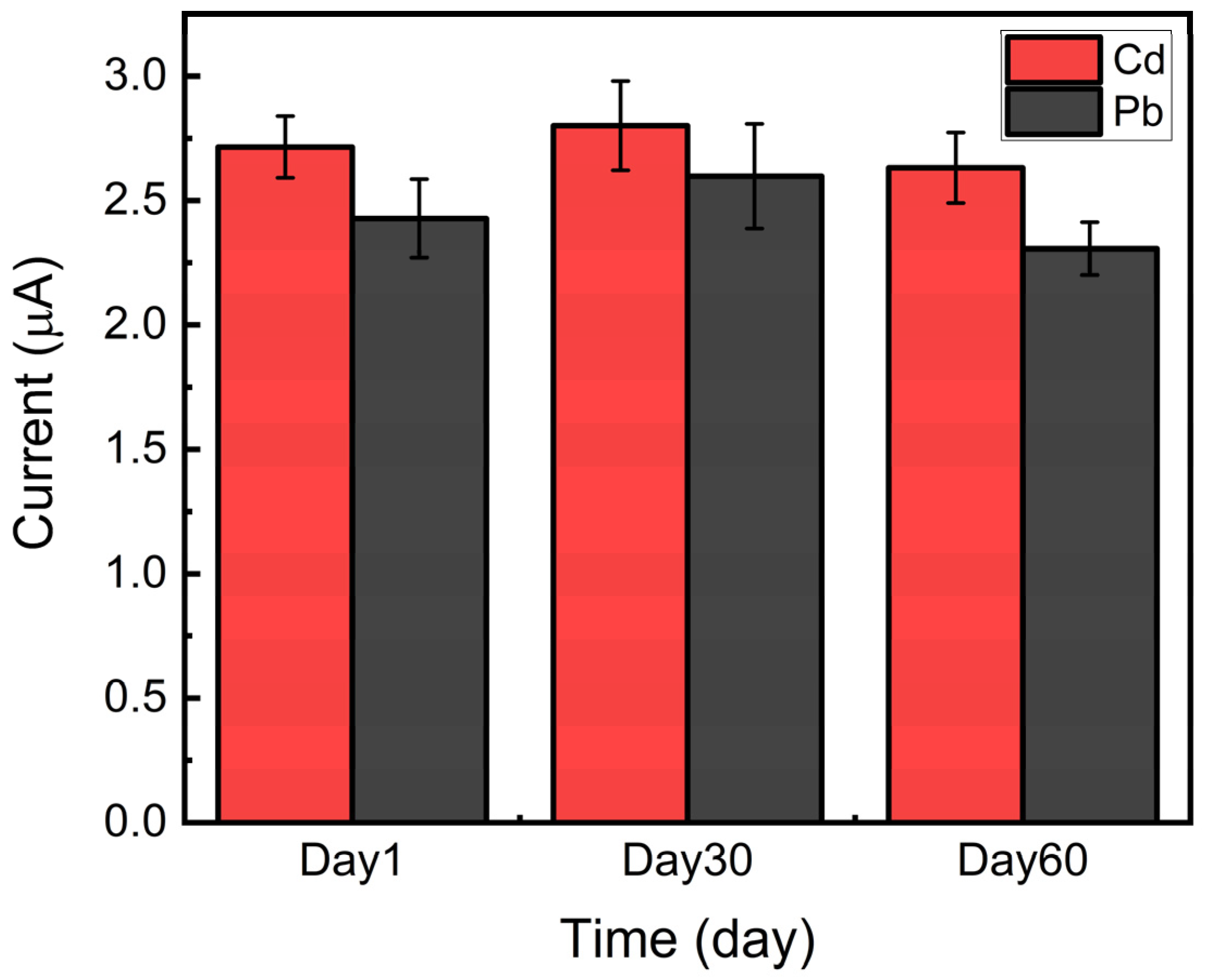
3.4. Simultaneous Detection of Cadmium and Lead, Using the HPC Electrode
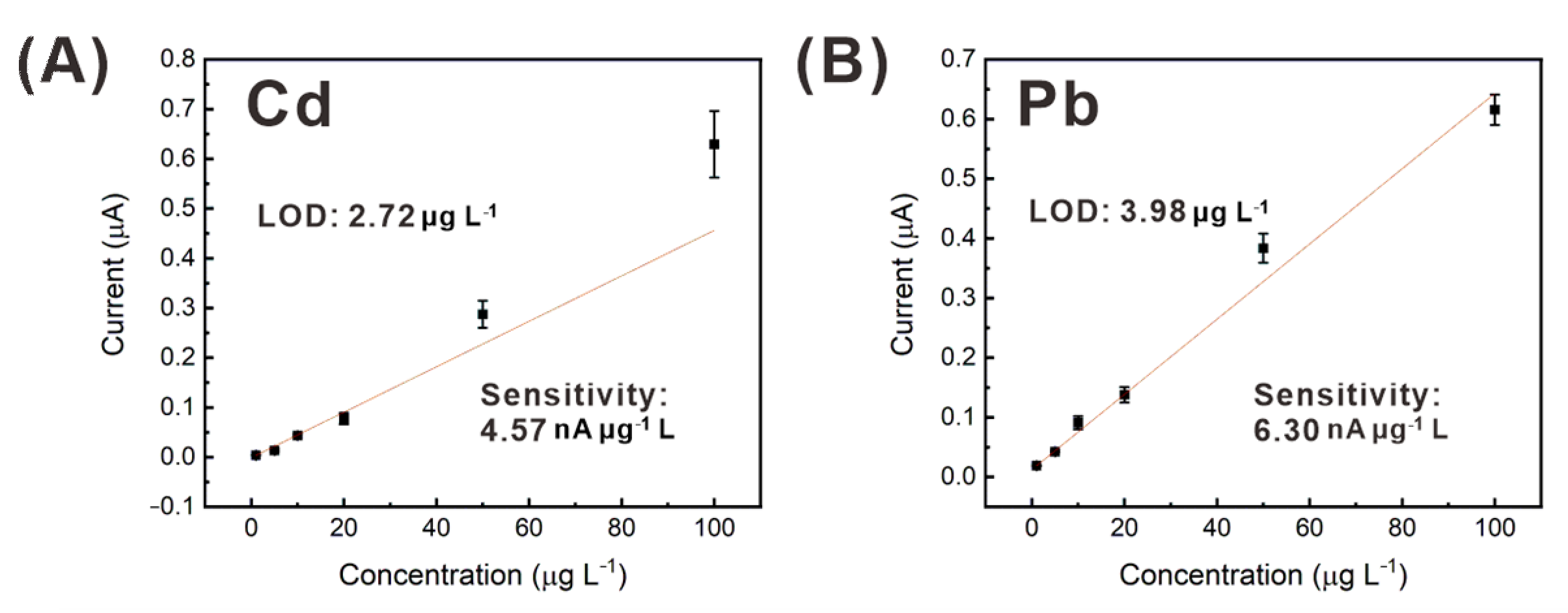
3.5. Heavy Metal Detection in a Real Water Sample
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Aragay, G.; Pons, J.; Merkoci, A. Recent trends in macro-, micro-, and nanomaterial-based tools and strategies for heavy-metal detection. Chem. Rev. 2011, 111, 3433–3458. [Google Scholar] [CrossRef]
- US Environmental Protection Agency. Seminar Publication: Risk Assessment, Management and Communication of Drinking Water Contamination; National Service Center for Environmental Publications: Cincinnati, OH, USA, 1989.
- Habila, M.A.; ALOthman, Z.A.; El-Toni, A.M.; Labis, J.P.; Soylak, M. Synthesis and application of Fe3O4@ SiO2@ TiO2 for photocatalytic decomposition of organic matrix simultaneously with magnetic solid phase extraction of heavy metals prior to ICP-MS analysis. Talanta 2016, 154, 539–547. [Google Scholar] [CrossRef]
- Narin, İ.; Soylak, M.; Elçi, L.; Doğan, M. Determination of trace metal ions by AAS in natural water samples after preconcentration of pyrocatechol violet complexes on an activated carbon column. Talanta 2000, 52, 1041–1046. [Google Scholar] [CrossRef]
- Marguí, E.; Zawisza, B.; Sitko, R. Trace and ultratrace analysis of liquid samples by X-ray fluorescence spectrometry. TrAC Trends Anal. Chem. 2014, 53, 73–83. [Google Scholar] [CrossRef]
- Rex, M.; Hernandez, F.E.; Campiglia, A.D. Pushing the limits of mercury sensors with gold nanorods. Anal. Chem. 2006, 78, 445–451. [Google Scholar] [CrossRef]
- Wagner, K.A.; Levine, K.E.; Jones, B.T. A simple, low cost, multielement atomic absorption spectrometer with a tungsten coil atomizer. Spectrochim. Acta Part B 1998, 53, 1507–1516. [Google Scholar] [CrossRef]
- Waggoner, J.W.; Milstein, L.S.; Belkin, M.; Sutton, K.L.; Caruso, J.A.; Fannin, H.B. Application of a low power/reduced pressure helium ICP ionization source for mass spectrometric detection of organobromine compounds and derivatized organotin compounds. J. Anal. At. Spectrom. 2000, 15, 13–18. [Google Scholar] [CrossRef]
- Zhou, S.; Yuan, Z.; Cheng, Q.; Zhang, Z.; Yang, J. Rapid in situ determination of heavy metal concentrations in polluted water via portable XRF: Using Cu and Pb as example. Environ. Pollut. 2018, 243, 1325–1333. [Google Scholar] [CrossRef] [PubMed]
- Barton, J.; García, M.B.G.; Santos, D.H.; Fanjul-Bolado, P.; Ribotti, A.; McCaul, M.; Diamond, D.; Magni, P. Screen-printed electrodes for environmental monitoring of heavy metal ions: A review. Microchim. Acta 2016, 183, 503–517. [Google Scholar] [CrossRef]
- Braga, M.S.; Jaimes, R.F.; Borysow, W.; Gomes, O.F.; Salcedo, W.J. Portable multispectral colorimeter for metallic ion detection and classification. Sensors 2017, 17, 1730. [Google Scholar] [CrossRef] [PubMed]
- Sriram, G.; Bhat, M.P.; Patil, P.; Uthappa, U.T.; Jung, H.Y.; Altalhi, T.; Kurkuri, M.D. Paper-based microfluidic analytical devices for colorimetric detection of toxic ions: A review. TrAC Trends Anal. Chem. 2017, 93, 212–227. [Google Scholar] [CrossRef]
- Gan, Y.; Liang, T.; Hu, Q.; Zhong, L.; Wang, X.; Wan, H.; Wang, P. In-situ detection of cadmium with aptamer functionalized gold nanoparticles based on smartphone-based colorimetric system. Talanta 2020, 208, 120231. [Google Scholar] [CrossRef]
- Li, M.; Li, D.W.; Li, Y.T.; Xu, D.K.; Long, Y.T. Highly selective in situ metal ion determination by hybrid electrochemical “adsorption–desorption” and colorimetric methods. Anal. Chim. Acta 2011, 701, 157–163. [Google Scholar] [CrossRef] [PubMed]
- Xu, G.; Li, X.; Cheng, C.; Yang, J.; Liu, Z.; Shi, Z.; Liu, Q. Fully integrated battery-free and flexible electrochemical tag for on-demand wireless in situ monitoring of heavy metals. Sens. Actuators B Chem. 2020, 310, 127809. [Google Scholar] [CrossRef]
- Yi, W.J.; Li, Y.; Ran, G.; Luo, H.Q.; Li, N.B. Determination of cadmium (II) by square wave anodic stripping voltammetry using bismuth–antimony film electrode. Sens. Actuators B Chem. 2012, 166, 544–548. [Google Scholar] [CrossRef]
- Li, M.; Gou, H.; Al-Ogaidi, I.; Wu, N. Nanostructured sensors for detection of heavy metals: A review. ACS Sustain. Chem. Eng. 2013, 1, 713–723. [Google Scholar] [CrossRef]
- Vyskočil, V.; Barek, J. Mercury electrodes–possibilities and limitations in environmental electroanalysis. Crit. Rev. Anal. Chem. 2009, 39, 173–188. [Google Scholar] [CrossRef]
- Daniele, S.; Bragato, C.; Baldo, M.A.; Ciani, I. Application of thin-shielded mercury microelectrodes in anodic stripping voltammetry. Talanta 2008, 77, 235–240. [Google Scholar] [CrossRef]
- Wang, J. Stripping analysis at bismuth electrodes: A review. Electroanalysis 2005, 17, 1341–1346. [Google Scholar] [CrossRef]
- Zou, Z.; Jang, A.; Macknight, E.; Wu, P.M.; Do, J.; Bishop, P.L.; Ahn, C.H. Environmentally friendly disposable sensors with microfabricated on-chip planar bismuth electrode for in situ heavy metal ions measurement. Sens. Actuators B Chem. 2008, 134, 18–24. [Google Scholar] [CrossRef]
- Zhang, C.H.; Li, G.-J.; Wang, J.-H. Application of nanopore and porous materials for heavy metal ion detection. Chin. J. Anal. Chem. 2014, 42, 607–615. [Google Scholar] [CrossRef]
- Kim, S.; Dong, W.J.; Gim, S.; Sohn, W.; Park, J.Y.; Yoo, C.J.; Jang, H.W.; Lee, J.L. Shape-controlled bismuth nanoflakes as highly selective catalysts for electrochemical carbon dioxide reduction to formate. Nano Energy 2017, 39, 44–52. [Google Scholar] [CrossRef]
- Zhu, C.; Li, H.; Fu, S.; Du, D.; Lin, Y. Highly efficient nonprecious metal catalysts towards oxygen reduction reaction based on three-dimensional porous carbon nanostructures. Chem. Soc. Rev. 2016, 45, 517–531. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Li, M.; Zhang, Z.; Guo, J. Unravelling the adsorption disparity mechanism of heavy-metal ions on the biomass-derived hierarchically porous carbon. Appl. Surf. Sci. 2019, 471, 615–620. [Google Scholar] [CrossRef]
- Yuan, X.; An, N.; Zhu, Z.; Sun, H.; Zheng, J.; Jia, M.; Liu, N. Hierarchically porous nitrogen-doped carbon materials as efficient adsorbents for removal of heavy metal ions. Process. Saf. Environ. Prot. 2018, 119, 320–329. [Google Scholar] [CrossRef]
- Li, J.; Liu, K.; Gao, X.; Yao, B.; Huo, K.; Cheng, Y.; Ding, D. Oxygen-and nitrogen-enriched 3D porous carbon for supercapacitors of high volumetric capacity. ACS Appl. Mater. Interfaces 2015, 7, 24622–24628. [Google Scholar] [CrossRef]
- Guan, J.; Fang, Y.; Zhang, T.; Wang, L.; Zhu, H.; Du, M.; Zhang, M. Kelp-Derived Activated Porous Carbon for the Detection of Heavy Metal Ions via Square Wave Anodic Stripping Voltammetry. Electrocatalysis 2020, 11, 59–67. [Google Scholar] [CrossRef]
- Wang, C.; Madou, M. From MEMS to NEMS with carbon. Biosens. Bioelectron. 2005, 20, 2181–2187. [Google Scholar] [CrossRef]
- Kostecki, R.; Schnyder, B.; Alliata, D.; Song, X.; Kinoshita, K.; Kötz, R. Surface studies of carbon films from pyrolyzed photoresist. Thin Solid Films 2001, 396, 36–43. [Google Scholar] [CrossRef]
- Sharma, S. Glassy carbon: A promising material for micro-and nanomanufacturing. Materials 2018, 11, 1857. [Google Scholar] [CrossRef]
- VanDersarl, J.J.; Mercanzini, A.; Renaud, P. Integration of 2D and 3D thin film glassy carbon electrode arrays for electrochemical dopamine sensing in flexible neuroelectronic implants. Adv. Funct. Mater. 2015, 25, 78–84. [Google Scholar] [CrossRef]
- Sharma, D.; Lee, J.; Seo, J.; Shin, H. Development of a Sensitive Electrochemical Enzymatic Reaction-Based Cholesterol Biosensor Using Nano-Sized Carbon Interdigitated Electrodes Decorated with Gold Nanoparticles. Sensors 2017, 17, 2128. [Google Scholar] [CrossRef] [PubMed]
- Mohamed Hassan, Y.; Caviglia, C.; Hemanth, S.; Mackenzie, D.M.A.; Petersen, D.H.; Keller, S.S. Pyrolytic carbon microelectrodes for impedance based cell sensing. ECS Trans. 2016, 72, 35–44. [Google Scholar] [CrossRef]
- Thiha, A.; Ibrahim, F.; Muniandy, S.; Dinshaw, I.J.; Teh, S.J.; Thong, K.L.; Leo, B.F.; Madou, M. All-carbon suspended nanowire sensors as a rapid highly-sensitive label-free chemiresistive biosensing platform. Biosens. Bioelectron. 2018, 107, 145–152. [Google Scholar] [CrossRef]
- Mardegan, A.; Kamath, R.; Sharma, S.; Scopece, P.; Ugo, P.; Madou, M. Optimization of Carbon Electrodes Derived from Epoxy-based Photoresist. J. Electrochem. Soc. 2013, 160, B132–B137. [Google Scholar] [CrossRef]
- Du, K.; Wathuthanthri, I.; Liu, Y.; Kang, Y.T.; Choi, C. Fabrication of polymer nanowires via maskless O2 plasma etching. Nanotechnology 2014, 25, 165301. [Google Scholar] [CrossRef]
- Lim, Y.; Woo, J.; Joo, S.H.; Shin, H. Patternable nanoporous carbon electrodes for use as supercapacitors. J. Electrochem. Soc. 2016, 163, A1886. [Google Scholar] [CrossRef]
- Lim, Y.; Chu, J.H.; Kwon, S.Y.; Shin, H. Increase in graphitization and electrical conductivity of glassy carbon nanowires by rapid thermal annealing. J. Alloys Compd. 2017, 702, 465–471. [Google Scholar] [CrossRef]
- Tsougeni, K.; Vourdas, N.; Tserepi, A.; Gogolides, E. Mechanisms of Oxygen Plasma Nanotexturing of Organic Polymer Surfaces: From Stable Super Hydrophilic to Super Hydrophobic Surfaces. Langmuir 2009, 25, 11748–11759. [Google Scholar] [CrossRef]
- Deng, H.; Zhang, C.; Xie, Y.; Tumlin, T.; Giri, L.; Karna, S.P.; Lin, J. Laser induced MoS 2/carbon hybrids for hydrogen evolution reaction catalysts. J. Mater. Chem. A 2016, 4, 6824–6830. [Google Scholar] [CrossRef]
- Łukaszewski, M.; Soszko, M.; Czerwiński, A. Electrochemical methods of real surface area determination of noble metal electrodes—An overview. Int. J. Electrochem. Sci. 2016, 11, 4442–4469. [Google Scholar] [CrossRef]
- Park, S.; Song, Y.J.; Han, J.H.; Boo, H.; Chung, T.D. Structural and electrochemical features of 3D nanoporous platinum electrodes. Electrochim. Acta 2010, 55, 2029–2035. [Google Scholar] [CrossRef]
- Walther, F.; Davydovskaya, P.; Zürcher, S.; Kaiser, M.; Herberg, H.; Gigler, A.M.; Stark, R.W. Stability of the hydrophilic behavior of oxygen plasma activated SU-8. J. Micromech. Microeng. 2007, 17, 524. [Google Scholar] [CrossRef]
- Stone, D.; Ellis, J. Calibration and Linear Regression Analysis: A Self-Guided Tutorial. In CHM314 Instrumental Analysis; Department of Chemistry, University of Toronto: Toronto, ON, Canada, 2003. [Google Scholar]
- Deshmukh, S.; Kandasamy, G.; Upadhyay, R.K.; Bhattacharya, G.; Banerjee, D.; Maity, D.; Roy, S.S. Terephthalic acid capped iron oxide nanoparticles for sensitive electrochemical detection of heavy metal ions in water. J. Electroanal. Chem. 2017, 788, 91–98. [Google Scholar] [CrossRef]
- Liao, J.; Yang, F.; Wang, C.Z.; Lin, S. The crystal facet-dependent electrochemical performance of TiO2 nanocrystals for heavy metal detection: Theoretical prediction and experimental proof. Sens. Actuators B Chem. 2018, 271, 195–202. [Google Scholar] [CrossRef]
- Xuan, X.; Park, J.Y. A miniaturized and flexible cadmium and lead ion detection sensor based on micro-patterned reduced graphene oxide/carbon nanotube/bismuth composite electrodes. Sens. Actuators B Chem. 2018, 255, 1220–1227. [Google Scholar] [CrossRef]
- Yao, Y.; Wu, H.; Ping, J. Simultaneous determination of Cd (II) and Pb (II) ions in honey and milk samples using a single-walled carbon nanohorns modified screen-printed electrochemical sensor. Food Chem. 2019, 274, 8–15. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Zhao, W.; Shen, Z.; Sun, S.; Dai, H.; Ma, H.; Lin, M. Sensitive and selective detection of Pb (II) and Cu (II) using a metal-organic framework/polypyrrole nanocomposite functionalized electrode. Sens. Actuators B Chem. 2020, 304, 127286. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ICP-MS | HPC Electrode | Relative Error (%) | ||||||
|---|---|---|---|---|---|---|---|---|
| Analyte | Added (μg L−1) | Found (μg L−1) | Recovery | Analyte | Added (μg L−1) | Found (μg L−1) | Recovery | |
| Cd | 5 | 6.11 ± 0.14 | 122.2 | Cd | 5 | 5.70 ± 0.68 | 114 | 6.7 |
| Pb | 5 | 4.53 ± 0.19 | 90.6 | Pb | 5 | 4.26 ± 0.20 | 85.2 | 6.0 |
| Cd | 50 | 51.94 ± 0.19 | 103.9 | Cd | 50 | 48.36 ± 4.59 | 96.7 | 6.9 |
| Pb | 50 | 46.09 ± 0.173 | 92.2 | Pb | 50 | 58.42 ± 3.68 | 116.8 | 6.3 |
| Electrode Material | Heavy Metal | LOD (ppb) | Sensing Range (ppb) | * Sensitivity per Unit Sensing Area (nA ppb−1 mm−2) | Electrode Size (mm2) | Real Sample | Reference |
|---|---|---|---|---|---|---|---|
| (Fe3O4/TA)NPs modified GCE | Cd | 22.48 | 44.96–123.7 | 5.508 | 19.625 | River water | [46] |
| Pb | 8.29 | 82.88–227.9 | 2.105 | ||||
| TiO2 nanocrystals modified GCE | Cd | - | 22.48–112.4 | 21.646 | - | River water | [47] |
| Pb | - | 41.44–207.2 | 20.131 | ||||
| rGO/CNT/ Bismuth composite | Cd | 0.6 | 20–200 | 2.62 | 1.5 | Drinking water | [48] |
| Pb | 0.2 | 20–200 | 9.26 | ||||
| SWCNHs modified SPE | Cd | 0.2 | 1–60 | 0.114 | 7 | Milk, Honey | [49] |
| Pb | 0.4 | 1–60 | 0.041 | ||||
| AuNPs/BiNPsmodified CE | Cd | 1.51 | 100–300 | 18.338 | 10 | Solution in cup | [15] |
| Pb | 2.20 | 50–300 | 15.31 | ||||
| Functionalizedgold electrode | Pb | 0.315 | 1–400 | - | 12.56 | Tap water | [50] |
| Kelp-derived porous carbon electrode | Cd | 2.62 | 1.12–56.2 | 33.37 | 7.065 | Tap water | [28] |
| Pb | 2.36 | 2.07–103.6 | 36.48 | 7.065 | |||
| HPC | Cd | 0.41 | 0.5–200 | 109.45 | 0.1256 | Tap water | This work |
| Pb | 0.7 | 0.5–200 | 100.37 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, J.; Kim, S.; Shin, H. Hierarchical Porous Carbon Electrodes with Sponge-Like Edge Structures for the Sensitive Electrochemical Detection of Heavy Metals. Sensors 2021, 21, 1346. https://doi.org/10.3390/s21041346
Lee J, Kim S, Shin H. Hierarchical Porous Carbon Electrodes with Sponge-Like Edge Structures for the Sensitive Electrochemical Detection of Heavy Metals. Sensors. 2021; 21(4):1346. https://doi.org/10.3390/s21041346
Chicago/Turabian StyleLee, Jongmin, Soosung Kim, and Heungjoo Shin. 2021. "Hierarchical Porous Carbon Electrodes with Sponge-Like Edge Structures for the Sensitive Electrochemical Detection of Heavy Metals" Sensors 21, no. 4: 1346. https://doi.org/10.3390/s21041346
APA StyleLee, J., Kim, S., & Shin, H. (2021). Hierarchical Porous Carbon Electrodes with Sponge-Like Edge Structures for the Sensitive Electrochemical Detection of Heavy Metals. Sensors, 21(4), 1346. https://doi.org/10.3390/s21041346