1. Introduction
Hyperdimensional imaging microscopy (HDIM) is a powerful method for combining different fluorescence parameters into discernable combinations of biochemical species [
1]. A lack of optimal imaging and computational tools has held back the practical implementations of HDIM, but recent advances in faster computational analyses and imaging techniques have helped advanced this method [
2,
3]. However, HDIM as an imaging technique still needs speed improvements and better automation to boost performance and adapt to a more lab-friendly version. We have re-engineered the HDIM technique with a sample-specific rationalization targeted towards metabolic and rapid imaging contrast. We study the plant model of
Arabidopsis thaliana and the intrinsic fluorescence from the stress-induced pigments anthocyanins using HDIM, enhancing the contrast between cells [
4]. Anthocyanins are autofluorescent vacuolar pigments with reported antioxidant and photoprotection properties [
5,
6]. Previous studies of anthocyanin using fluorescence lifetime imaging microscopy (FLIM) have proven effective in understanding anthocyanin trafficking and chemistry [
7,
8,
9]. Our efforts help move HDIM into a real-time tool that is more robust and extends its comparative and contradistinction abilities into disease diagnostics.
There are essential differences between multiparametric approaches and hyperdimensional imaging. Multiparametric approaches often include methods derived from the fundamental parameters, including those that reflect the molecular size, movement, binding state, and others [
10,
11,
12]. A typical example is in the analysis of membrane dynamics. For example, Heikal [
13] showed that by combining confocal, FLIM, polarization, and fluorescence correlation spectroscopy (FCS) it is possible to decode membrane thermodynamic derivates such as lipid organization, cholesterol content, protein proximity, mechanical forces, and others. Multiparametric methods can help understand the biological system’s function, learn about its structure, and improve the probing technique as the question evolves over the course of the study. Hyperdimensional methods can read the fundamental fluorescence parameters, including the primary five: intensity, spectrum, polarization, lifetime, and spatial-temporal coordinates [
1]. The purpose of HDIM using this direct readout method is to amplify the inherent contrast presented in a biochemical specimen without drawing assumptions on the model of the state or function. Another significant difference is the simultaneous measurement scheme employed in hyperdimensional imaging that can paint ensemble measurements and single-molecule measurements easier to decode. This direct approach gives HDIM an added advantage to pose as a quasi-real-time tool for differentiation in a sample [
14]. It is also interesting to note that term Multimodal imaging describes a wide range of non-fluorescence-based techniques or modes of imaging. This list includes not only macroscopic imaging modalities such as magnetic resonance imaging (MRI), optical coherence tomography (OCT), computerized tomography (C.T.), but also the non-imaging modalities such as metabolic profiling and proliferation rates [
15].
Fluorescence is a ubiquitous research tool in fields such as biophysics research and clinical diagnostics, spanning a large spatial and temporal ranges of activity. Molecules that emit fluorescence photons unveil their location, orientation, yield, selective underlying energy states, and relaxation times. These five properties are widely used as fluorescence techniques under the common names of fluorescence imaging, fluorescence polarization imaging, fluorescence intensity imaging, fluorescence hyperspectral imaging, and fluorescence lifetime imaging, respectively [
12,
16,
17,
18]. A literature review of these methods is outside the scope of this paper, but interested readers are encouraged to pursue these studies [
12,
19,
20,
21]. Briefly, fluorescence intensity imaging reveals the spatial and temporal yield of fluorescence photons from a location at a given time. Fluorescence anisotropy or fluorescence polarization imaging identifies the relative polarization state of the emission photons with respect to the excitation [
22]. Fluorescence spectral imaging or hyperspectral imaging modalities investigate the fluorescence molecules’ emission spectrum [
23]. Fluorescence lifetime imaging calculates the decay rate of a fluorophore [
24].
There is only a limited research data available on HDIM contrast, owing to its technical demands. Esposito and Venkitaraman pointed out current limitations in simultaneous observation of physical properties of fluorescence [
1]. These authors used grating-based time-resolved spectral detectors on two perpendicularly polarized emission channels. These fast spectral detectors are time-resolved spectrographs made of a multi-cathode array in 16 × 1 or 32 × 1 fashion, coupled to fast-timing electronics units to tag every photon with a time of arrival and the wavelength of origin from each cathode. Using two such detector arrays to tag the polarization states produces a photon with three parameters: time of arrival, a narrow spectral channel of emission, and its polarization state. The individual photon-tags are tallied to make arrival histograms of custom width of resolution. For example, the resulting data for an image of 256 × 256 pixels and 64 time bins is organized in a 16 × 2 × 64 × 256 × 256 matrix (S, P, T, X, Y). S is used to denote Spectrum, P to denote Polarization, T to denote Lifetime, X, and Y for spatial pixel coordinate. This data is analyzed and classified using a dimensionality reduction approach to 3 × 256 × 256 image with the top three components of separation (in the case of principal component analysis (PCA)—based separation). The implementation of HDIM successfully separated the biochemical properties of solutions (rhodamine) and fluorescence imaging samples (convallaria) under varying quenching proportions, viscosity, and polarity [
1,
25]. Our research attempts to re-engineer the current hyperdimensional imaging set up to accommodate ease of implementation and reduce the burden on computational requirements while revealing biochemical contrast without compromising critical features, such as dynamic range of an image and speed of acquisition. The computational demands from traditional HDIM analysis includes microscopic image matrix compression (data storage), quick histogram calculation (for time-resolved contrast), classification and feature extraction (for spatial selectivity) [
26]. Multidimensional matrix calculations are conventionally opted as the solution here; for example, projections of selected axes of interest such as peak-wavelength anisotropy images, and frequency split-hyperspectral images. The choice of dimensionality-reducing methods like PCA, LDA, K-Means, and machine learning techniques such as t-SNE, UMAP, and Autoencoders are often more demanding to implement, let alone extend the method to real-time imaging/visualization [
27,
28]. Many approaches lighten this load by choosing analysis frameworks such as phasors and other transformation matrices that simplify the multidimensionality into a configurable range of layers of interest [
2].
In this work, we show for the first time an optical HDIM setup that derives contrast using an optical fiber-based scheme, resolving underlying species without a substantial computational cost. This system that we named hyper-dimensional contrast imaging is achieved by fixing the axis of time-resolved anisotropy data and convolving the spectral contrast on top of the lifetime signature. The data is collected in 256-time channels for 256 × 256 image size that results in an XYT matrix (256 × 256 × 256), and we paint a contrast based on phasors (2 × X × Y) or custom-gated (1 × X × Y) for real-time analysis. We present a re-engineered version of the previously published HDIM work (full-resolution HDIM) that translates a known fluorescence parameter contrast in a sample of choice into fast imaging [
1]. A comparison of full-resolution HDIM and our HDIM-contrast imaging (this paper) is presented in
Figure 1.
This manuscript presents our hardware-based dimension reduction method for applying hyperdimensional contrast on
Arabidopsis thaliana epidermal cells to identify cellular heterogeneity distribution of anthocyanins. Anthocyanins are valuable intrinsic stress-markers, existing in multiple variations and modified forms, including methylated and acetylated forms [
29]. Anthocyanins play a photo-protector role against ultraviolet-B radiations in many vegetative tissues and can also act as a free radical scavenger. However, anthocyanin synthesis and sequestration in plant cells are complex and understanding the localization and functional nature of these pigments at the single-cell level has enormous value. The many endogenous fluorophores in plant cells often present challenges in anthocyanin imaging; however, FLIM has shown promise in separating anthocyanins and localizing subcellular pigmentation [
8]. We chose this important plant model due to our active work in learning anthocyanin trafficking and function using FLIM [
4,
7,
8,
9]. In this research, we present an enhanced plant cell heterogeneity results measured by intrinsic anthocyanins distributions. We demonstrate a fiber-based instrumentation solution for reducing computational complexity associated with hyperdimensional imaging. Our HDIM contrast system can image a biological sample such as
Arabidopsis tissues with maximized contrast derived from a combination of the fluorophores spectrum, lifetime, and anisotropy.
3. Results
HDIM imaging modalities were briefly discussed in the introduction section (
Figure 1). The full-resolution method of collection, as described by Esposito, allows a complete analysis of the photons. The photons analyzed paint a pixel in discriminating fractions of different species, plotting kinetic changes over time, or similar biochemical changes. This contrast in a pixel results from discrimination drawn between photons along n-number of axes (for an n-dim PCA reduction). In some instances where principal axis #1 (PC1) alone brings the amount of variance of the transform to 99% of the original dimensions, the contrast lies mainly in one axis drawn across an n-dim tensor space of spectrum-anisotropy-lifetime space. This axis of separation can be partially simulated using hardware by controlled anisotropy-spectral collection. Otherwise, there exists a set of polarization, spectral, lifetime states that generate a maximum difference between species in a pixel. When extended in an image, these static values help the pixel distribution generate maximum contrast between pixels.
For fiber-HDIM contrast imaging, we find the best emission polarization contrast and fix the polarization state to optimize the anisotropy-spectral contrast. We do this by tuning the excitation polarization instead of emission because the dichroic on emission paths has its polarization preference. Moreover, controlling excitation polarization gives better control of the system. The emission photon’s spectral position is encoded as a delay using fiber optics [
33]. The spectral resolution can be varied by changing the fiber length, and we found 30 m length suitable for our experiments. The convolution of photon’s spectral information on the lifetime-decay curve makes the dynamic information content in the decay curve higher. This decay curve is fit using an exponential-curve fit or transformed using phasors to paint contrast in the image. However, the spectral convolution is selectively biased by the dominating fraction in the pixel. The exponential intensity decay curve can separate multiple species by virtue of fitting; however, the spectral information coded on it is challenging to be separated.
The signal in the lifetime curve is a polarization filtered spectrally convolved intensity decay curve in the order of nanoseconds. The two values that need to be set for a particular unknown sample is the best anisotropy contrast (aka the excitation polarization angle) and appropriate spectral bandpass filter (to restrict the spectral broadening to a smaller range). Once obtained for a sample, these optimal values of polarization and spectral range can be used as a priori information for fiber-HDIM to operate at a full-imaging pace. This step of identification of the tensor axis is derivable even by the sequential approach to formulating the P-S-T matrix and fix the best P-S values that aid the image contrast. In the case of Arabidopsis leaves, we did a sequential collection and set the spectral filter at 630/69 nm and polarization angle combination of HWP: QWP at (160:80°). The polarization optimization was carried out the entire 360°–360° degree matrix for static anisotropy contrast because there was variability in the peak contrast obtained each unit of (90:45°) combination. This variation is possibly due to imperfect collection and delivery of polarized light.
The following results show a PCA analysis of a sequential collection of time-lag, anisotropy, and spectral-peak using the fiber. We also show how a spectral bandpass filter helps in improving the contrast. Finally, when used with the bandpass spectral filter, the direct analysis of the “polarization filtered spectrally convolved intensity decay curve” from the fiber yields a better contrast than the sequential collection PCA analysis.
3.1. Hyperdimensional Imaging Using Derived Parameters
In order to validate the HDIM contrast, we measured the different parameters sequentially. We selected a smaller areas of the cotyledon lower epidermis and (a) calculated the mean lifetime using a bi-exponential decay; (b) the fractional distribution of lifetime species; (c) measured the static anisotropy using two orthogonally polarized detectors and calculating the intensity ratio; (d) calculated the rotational time using anisotropy decay fit into an associated anisotropy decay model using two lifetime values; and finally, (e) imaged using the fiber to measure spectral peaks (encoded as time-shifts). The five resultant images were analyzed using a segmented intensity map to paint single leaf-cells with the respective parameter’s mean value. The images are presented in
Figure 4.
We used the five parameters shown in
Figure 4, along with the intensity data and the fractional anisotropy data, into a sparse-PCA. This reduced them into two components: principal component #1, #2: PC1, PC2) [
39]. Although we could perform PCA by using the 2 (P) × 256 (T) × 256 (S) dataset from a single pixel, the comparison would not be fair because the fiber-based spectral collection is not a full-hyperspectral imaging unit. The [S] dimension has the lifetime and spectral information are convolved on each other. The seven parameters were standardized (as shown in
Figure 5A), and then two axes of separation were derived and colored per leaf-cell (
Figure 5B). The PCA scatter points were further analyzed using a variable k-means clustering algorithm to identify clusters, and no satisfying separation of clusters (measured using silhouette score) was obtained. The two-component k-means separation is shown in
Figure 5C.
This method was a means to understand if we could derive a satisfying PCA axis using sequential measurement. This analysis is not perfect; however, standardizing this way helps us deduce which variables offer the most contrast. Although we present only seven parameters, a total of 25 derived parameters were tested as PCA dimensions. This list includes the fit parameters from both time-resolved anisotropy fit and lifetime fit, their fit-goodness values (by a chi-square value), intensity offset, mean-lifetime (t
m), and intensity weighted-mean lifetime (it
m). The 25 variables were (T: (t
1, t
2, a
1, a
2, f
1%, offset, chi-sq, it
m, t
m, sum), P: (t
1, t
2, a
1, a
2, f
1%, offset, chi-sq, it
m, t
m), P (r-static, r
0, r-infinity), S (peak-shift, peak-width, offset)). These additional parameters did not help the PCA separation; instead affected the noise covariance and lowered the cumulative explained variance ratio of all components [
37].
3.2. Single-Shot HDIM
The spectral information derived using the fiber was further studied for increasing the HDIM contrast. The highest photon emitter often dominates the spectral widening of the lifetime curve in a single pixel. This limits the separation to a small range because most cells default to the anthocyanin spectral peak. We found that the spectral distribution widens when we collect in a smaller spectral range by adding an emission filter. This spectral-band restriction of total fluorescence photons allows more contrast between pixels owing to minor changes in spectral peaks. A comparison of time-shift due to dispersion with and without a spectral bandpass filter is shown in
Figure 6. The time-shift images of leaf-cells in
Figure 6 (A: without filter, B: with filter) is a smaller distribution, while with addition of the 630/69 nm filter, the spectral peaks time-shifts) are broadly distributed to a six-time wider distribution (
Figure 6C,D). Although this causes a reduction in total available photons, the enhancement of cellular contrast is significant.
3.3. Comparing Fiber HDIM vs. Derived Parameters HDIM
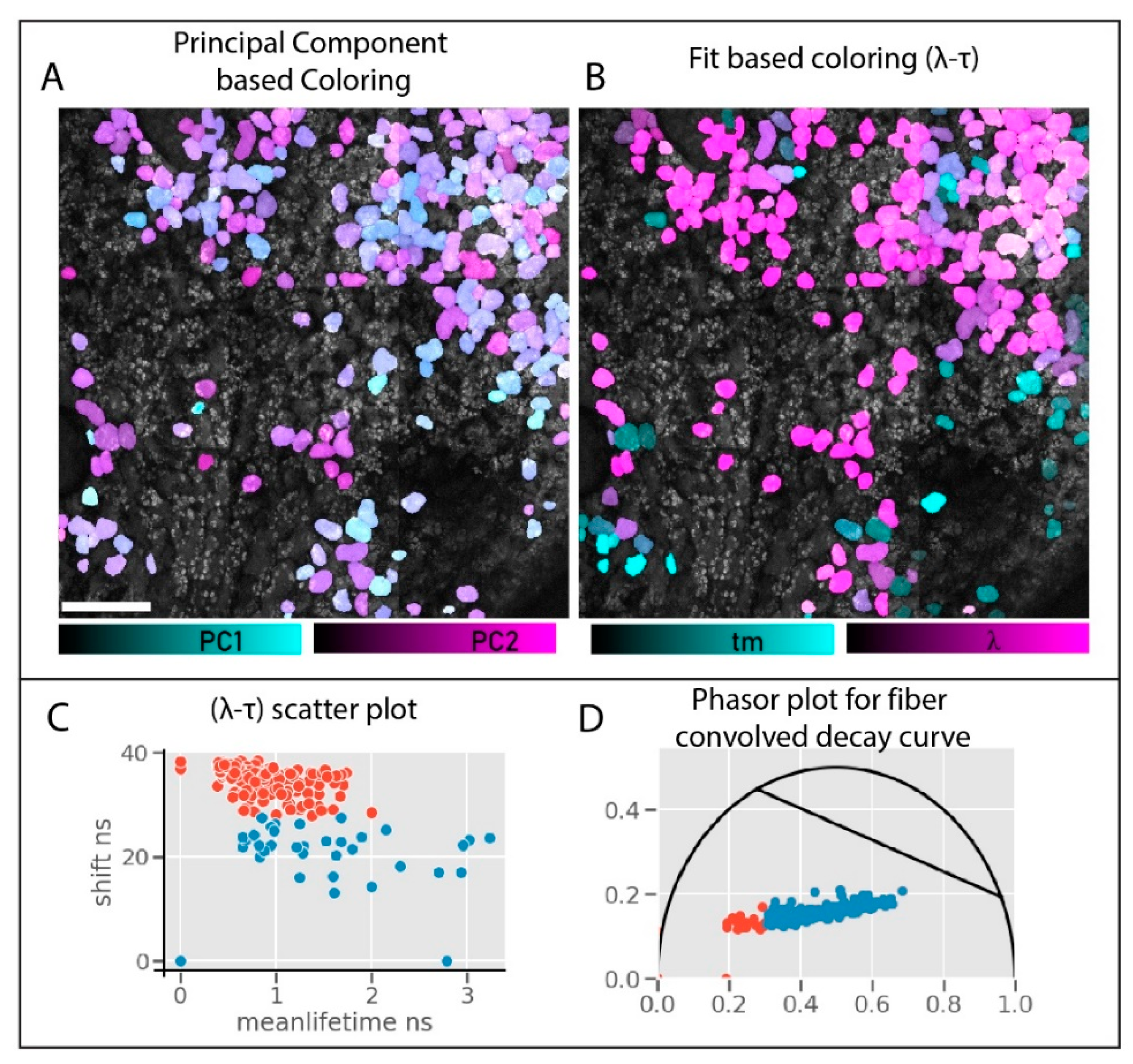
This spectral selection and polarization optimization help us reduce the collection to a single channel. The single detector connected to the fiber is used for fiber-HDIM. We compare the results from the fiber-HDIM with the derived-parameter PCA output in
Figure 7. The intensity decay curve with maximum anisotropy contrast and selected spectral range is analyzed in two different schemes. (1) The time-shift is derived as lambda max and plotted against the fitted mean-lifetime value: this 2D histogram plot is called lambda-tau map (following the naming from previous work) [
33]. (2) The data can be transformed into a phasor plot where both shift and lifetime will significantly affect the transform. These two schemes are shown in
Figure 7C,D. The λ-τ (lambda-tau) values can be color-coded on the leaf cells for maximum contrast. (This can also be done using g-s of phasor transform, not shown). We compare (
Figure 7A,B) the λ-τ (lambda-tau) map contrast against the derived PCA contrast obtained before (
Figure 5). We can identify two distribution and easily separable HDIM contrast using the λ-τ maps. Moreover, this fitting process is fast (in milliseconds [
34]) and does not rely on extensive data processing, making this a real-time contrast modality in imaging.
The enhanced contrast can be compared by comparing the scatter plot from PCA analysis
Figure 5C and λ-τ maps in
Figure 7C. The PCA separation is hardly noticeable, while the scatter plots show a more apparent separation. The phasor plot in
Figure 7D shows a separation with a good silhouette score (>0.5).
4. Discussion
Fluorescence-based studies harness the physical properties of fluorescence that change with microenvironmental changes of the molecule. Different properties enable us to study a chemical event of interest with different perceptions. Anisotropic studies often lead to an understanding of orientation and extend into the rotation, size, and binding state of that molecule. Similarly, spectral and decay rate studies unveil chemical events in relation to temperature, viscosity, and molecular conformation. Most biophysical studies limit their work to one of these fluorescence properties, which often produce contrast in their images and study the problem. In order to learn more details about the molecule, all the molecular properties need to be mapped out and analyzed in a hyper-dimensional space—furthermore, either a dimension reduction method or a multi-dimensional analysis to extract parameters of interest must be employed. These methods, although not difficult to deploy, are often time-consuming and expensive. Here we give an alternate option where a hyperdimensional contrast from multiple fluorescence properties can be encoded into a single observation using a single detector.
Fluorescence can reveal molecular binding, molecular selectivity, and physical behaviors such as rotation, translational restrictions, channeling, and environmental changes such as temperature and viscosity. Most biological system studies often confound in this multidimensional information space because extensive studies require a longer time and additional research to validate the properties. We propose a method by which biological imaging can raise the contrast in any imaging scheme.
Our method has many advantages. First, even after filtering, the photon budget per pixel is large (because of binning spectral-lifetime data). Second, the lack of computation makes the processing faster and a real-time-imaging option. Third, unknown samples can be probed with a quick anisotropy optimization and spectral collection to generate a contrast axis without any information of the sample. Similarly, known samples can be calibrated for the best polarization-spectral range for HDIM contrast. Fourth, the imaging implementation requires a FLIM system and a fiber as opposed to a costly spectral-setup. Our calibration setup uses two PMTs but can be achieved using a single PMT and a rotating emission polarizer. However, there are many challenges involved with the current implementation, and more research and optimization are required. This list includes the lack of a real-time FLIM analysis tool that can integrate the custom phasor or λ-τ based contrast. More research is necessary to integrate this marker into imaging as a contrast metric. Similarly, adding a mechanism to void parameter influence would also be helpful. This parameter cancelation can be achieved using (1) setting a circular polarization for removing anisotropy, (2) replacing the fiber with the free-space collection for removing spectral effects, and (3) summing the time-resolved axis to remove intensity decay information. This switching can be fully automated and can be useful for parameter separation. Another avenue of improvement is the lack of spectral variation in the fiber-based dispersion. Observing only the spectral peak but not the width and number of modes severely limits the spectral information encoded in our imaging scheme. More careful processing of spectral-lifetime analysis is necessary to achieve the exact width of the emission spectra. In this manuscript, we present single-cell analysis from one sample, but we have observed an increased contrast for every leaf sample we tested (n > 6). Additional ongoing experiments to show multiple extrinsic fluorescence markers and biochemical fingerprinting of those markers in a single pixel level would strengthen our method. Future work in these directions to optimize HD-Contrast Imaging are underway, and we hope this can become an unsupervised biomedical tool for fluorescence contrast.
We presented the fiber-based HDIM method that extracts an enhanced fluorescence contrast than lifetime, spectrum, or polarization could generate alone. However, there are challenges in this scheme of separation. Unlike full-resolution HDIM, when we selectively filter and encode information, there can be losses. For example, if the spectral shift contrast increases and lifetime-contrast decreases, they could cancel each other. A more detailed study into the sources of autofluorescence is necessary to address such anomalies. However, in our measurements with anthocyanins, we do not observe this. Future studies can identify and isolate the contrast source using a multiparametric collection like Raman, mass spectrometry, and others. As far as image contrast is considered, this method of encoding information provides improved contrast in biological imaging without losing photons.



 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}






