Fluorescence Polarization-Based Bioassays: New Horizons


 ,
,  and
and 
Abstract
1. Introduction
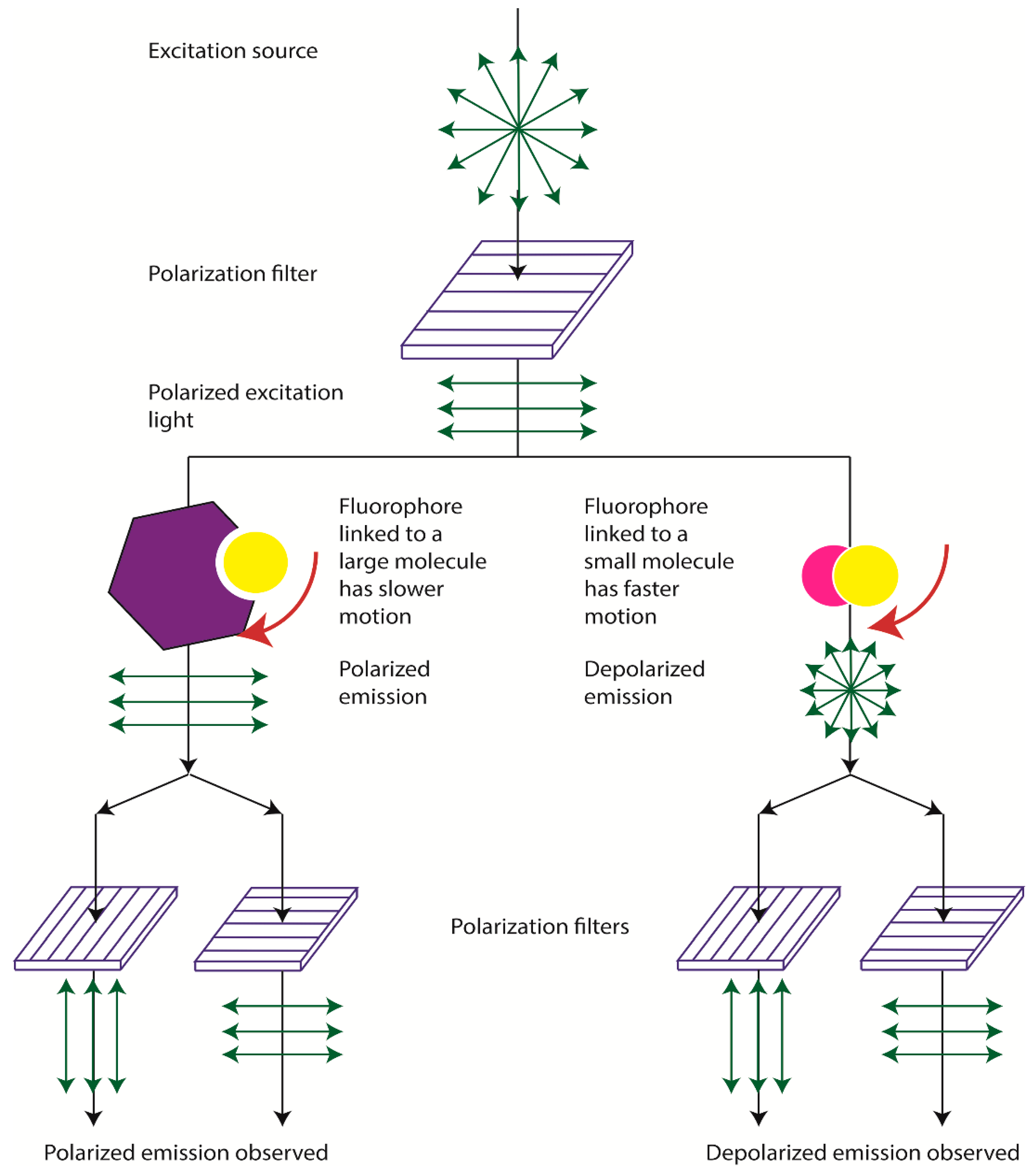
2. Physical Bases of Fluorescence Polarization
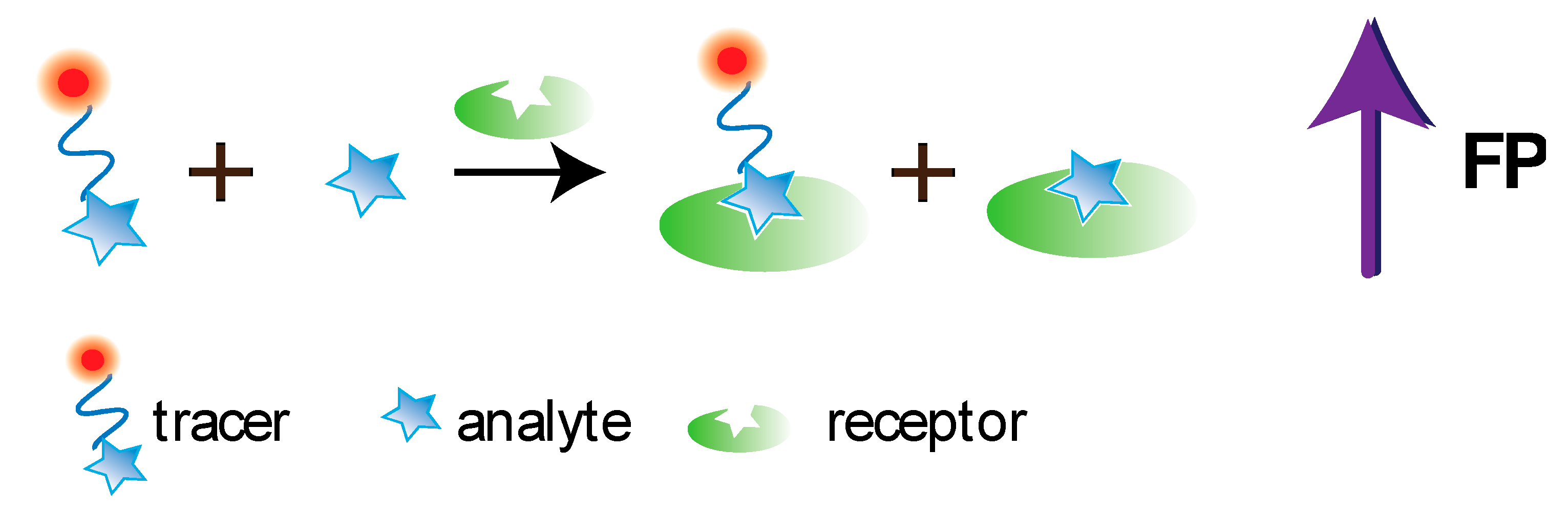
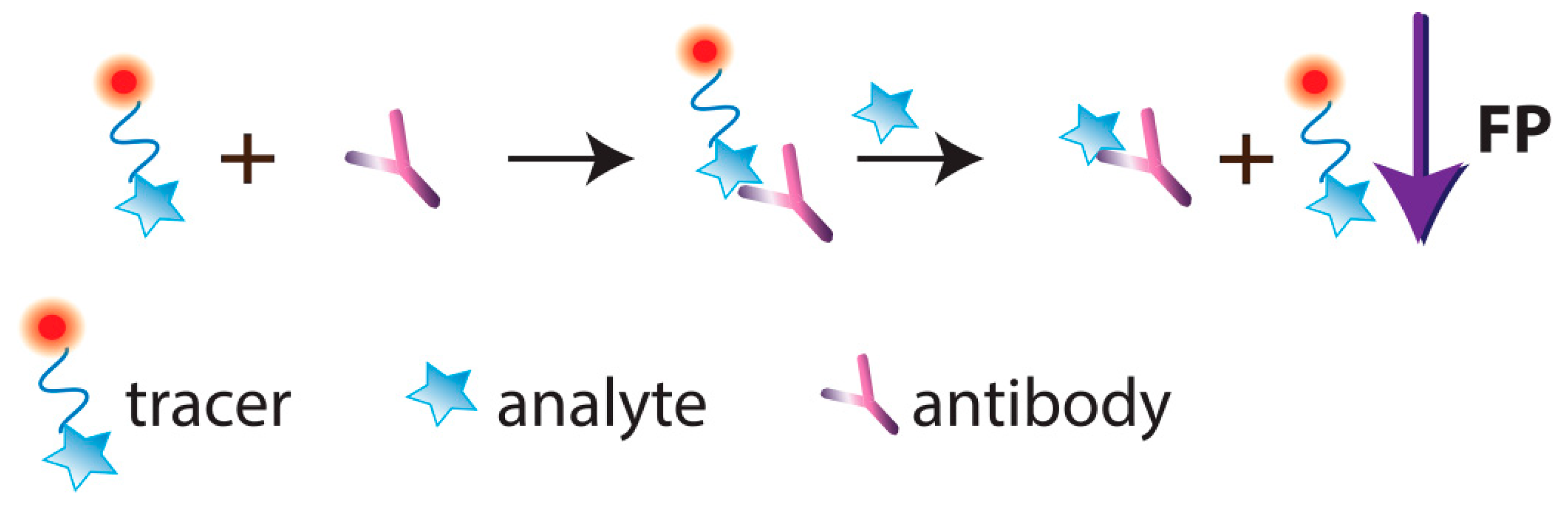
3. Conventional FPIA, Its Advantages and Drawbacks
- It is a simple homogeneous method that requires no washing and separation steps.
- The analysis time is limited only by the pipetting because the kinetic constant of binding of small analyte molecules to antibodies in solution usually varies from 107 to 108 1/M • s.
- The P value is a relative and dimensionless parameter that smooths fluctuations in instrument measurements and leads to very high reproducibility of results. The variation coefficients usually do not exceed 3–5%.
- Fluorescent labels can be synthesized quite easily and remain stable during storage for many years.
- However, the following features of conventional FPIA should be considered as its drawbacks.
- The assay typically needs in high concentration of reactants for reliable measurements of the FP; due to this FPIA is less sensitive as compared with other immunoanalytical techniques.
- Some compounds of the matrix can absorb excitation and emitted light, as well as exhibit their own fluorescence in the same spectral region as the used fluorophore; as a result, measurement results are distorted.
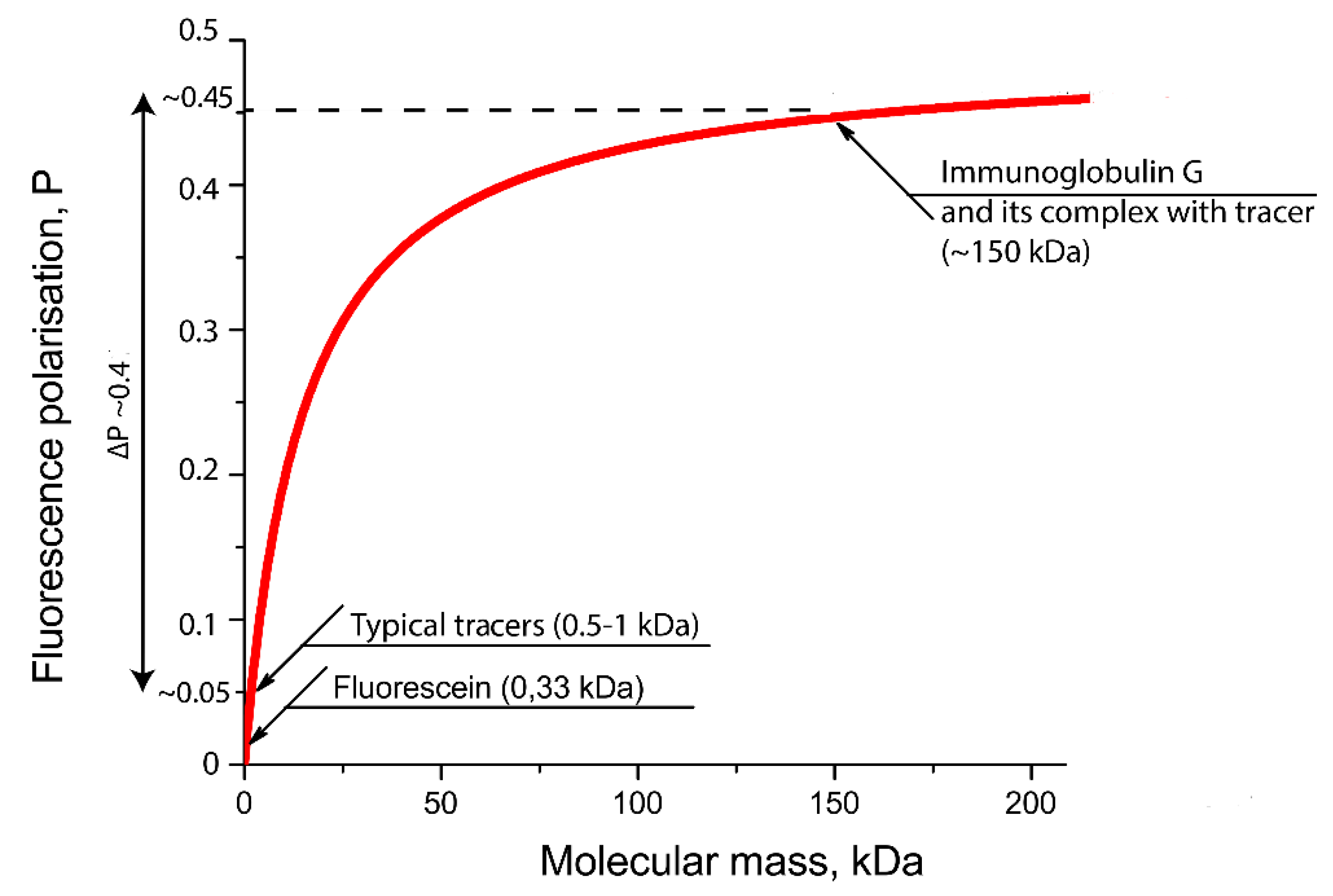
- The technique may be applied only to detect low molecular weight compounds, as well as it is based on significant difference of molecular weights for fluorophore-labeled antigen and its complex with antibody.
4. Modes of FPIA
4.1. Single-Reagent FPIA
4.2. FPIA in the Kinetic Mode
4.3. FPIA in Organic Solvents
4.4. Non-Competitive FPIA
5. FP-Based Assays with Alternate Recognizing Elements
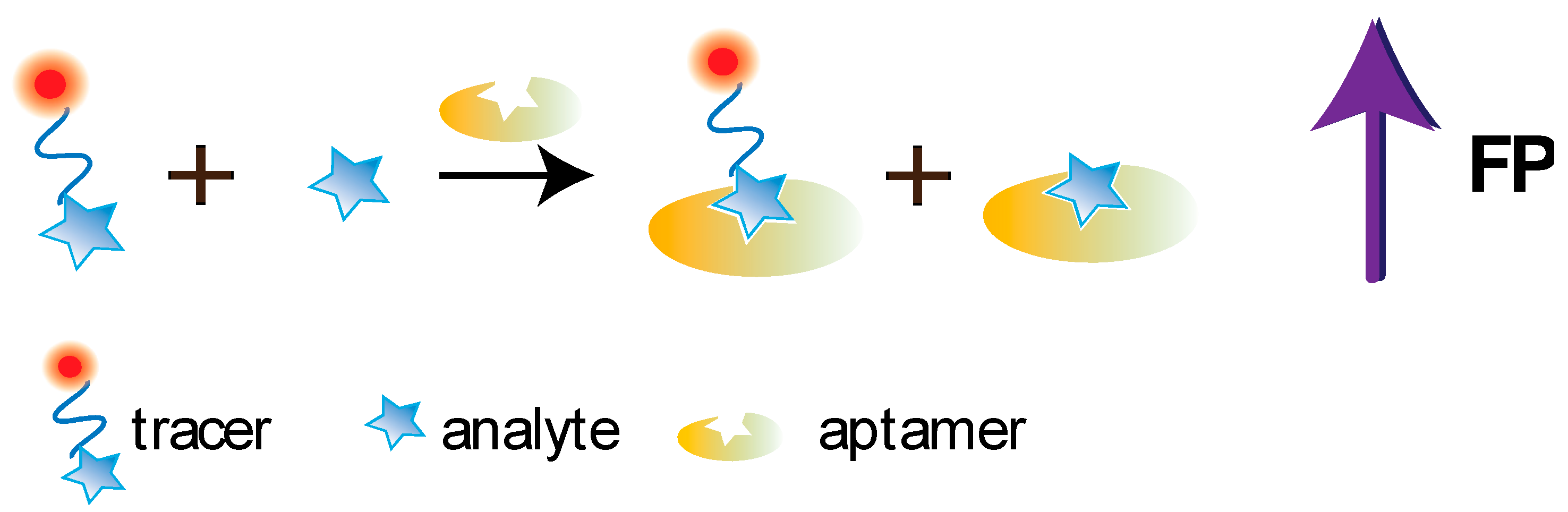
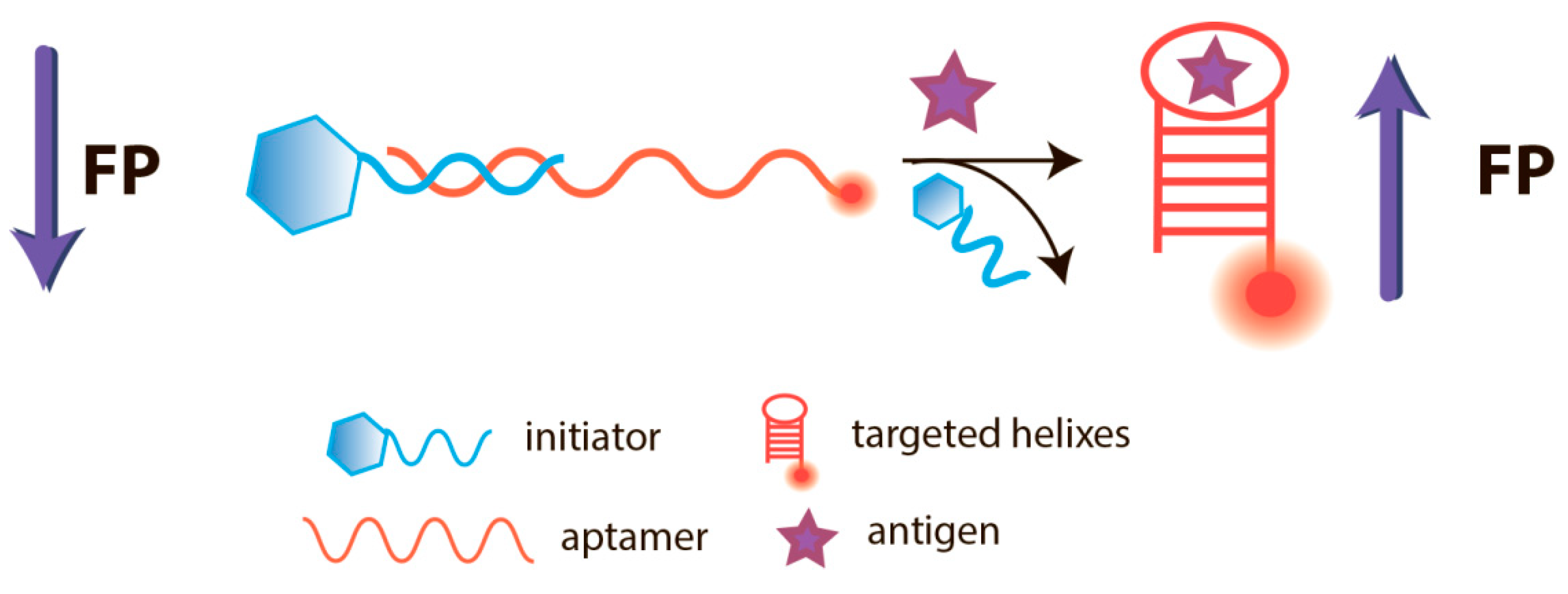
5.1. Aptamer-Based FPA
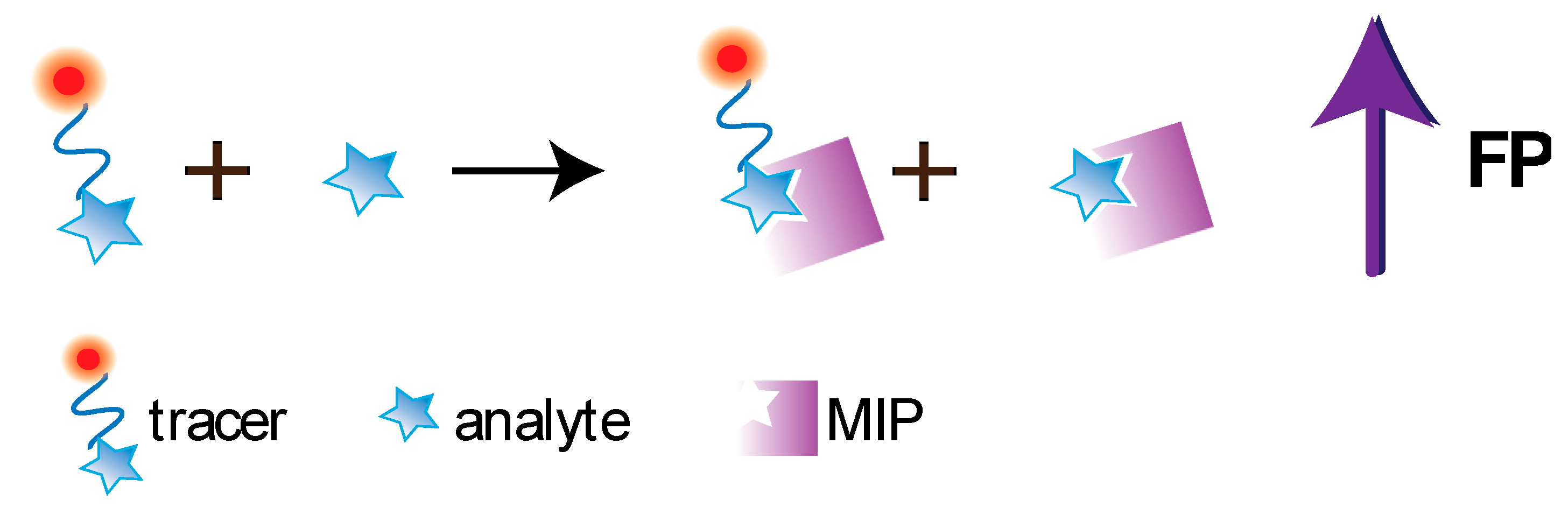
5.2. FPA Based on Molecularly Imprinted Polymers
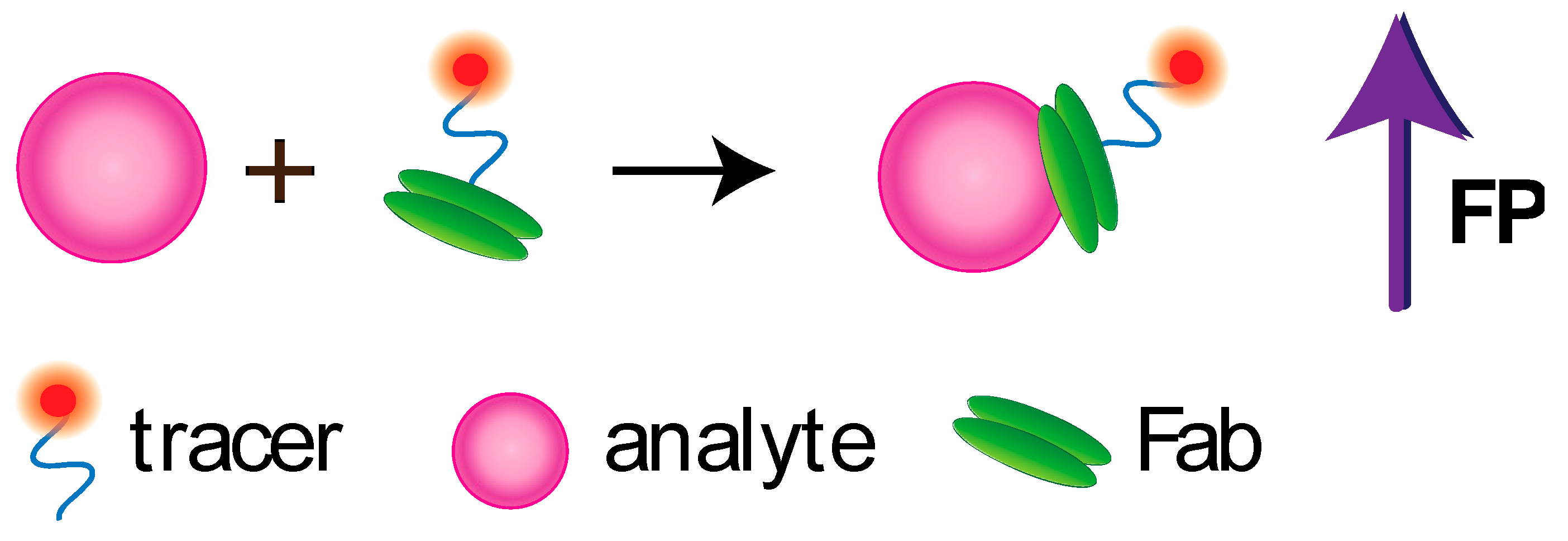
5.3. FPA Based on Other Recognizing Elements
6. Fluorescein and Alternative Fluorophores
7. FPIA for the Determination of Metal Ions
8. FPA of Nucleic Acids
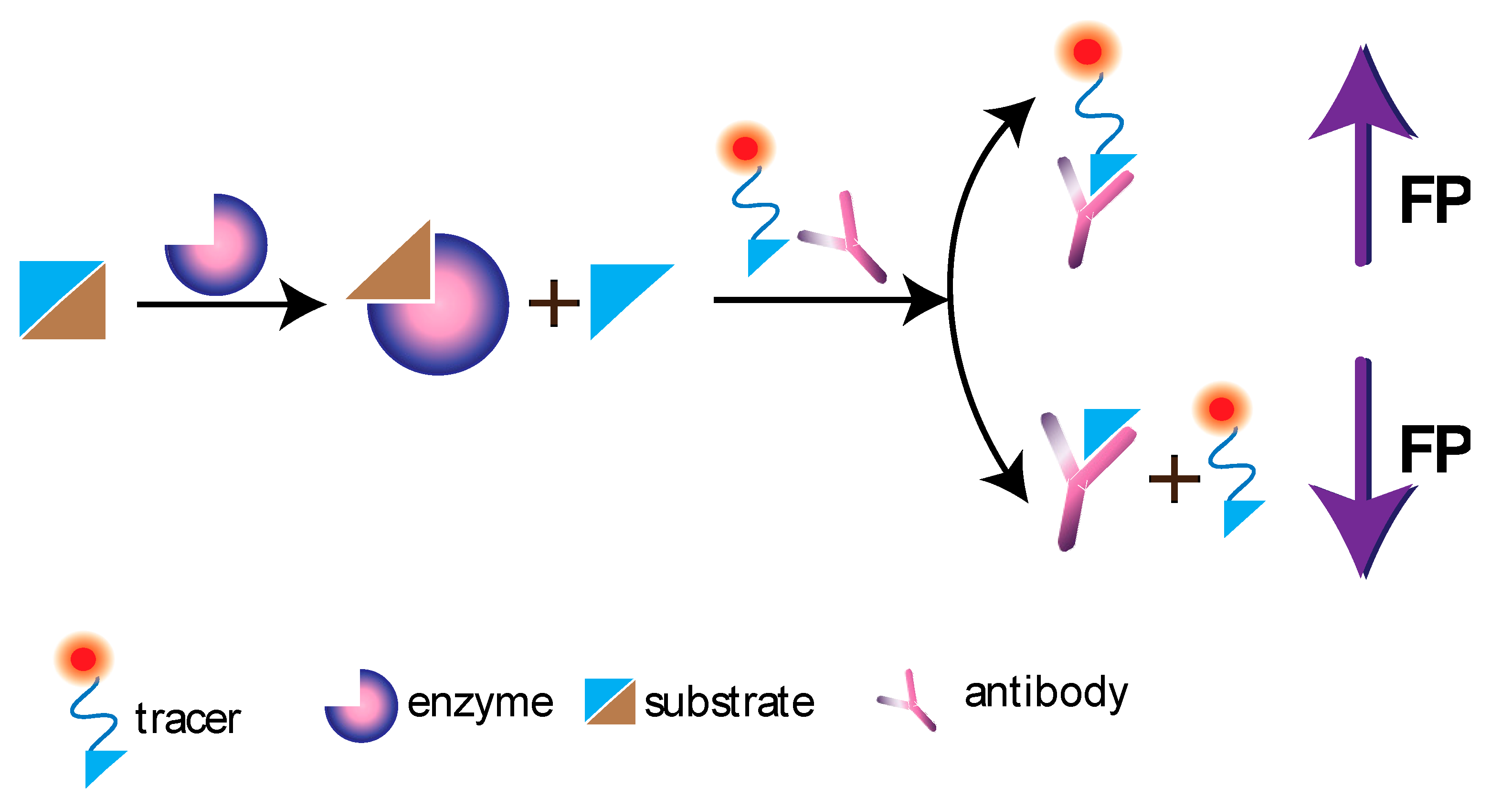
9. FP in the Monitoring of Catalytic Processes
10. Towards Multiplex Analysis
11. Switched FP and its Analytical Use
12. FPA with Signal Enhancement
12.1. Nanoparticles as Signal Enhancers
12.2. Proteins-Based Signal Amplification
13. Overall Estimation and Validation of FP-Based Assays
14. Equipment for FPA
15. Prospects of FPIA
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Lu, C.; Yang, H.; Zhu, C.; Chen, X.; Chen, G. A Graphene Platform for Sensing Biomolecules. Angew. Chem. 2009, 121, 4879–4881. [Google Scholar] [CrossRef]
- Farka, Z.; Juřík, T.; Kovář, D.; Trnková, L.; Skládal, P. Nanoparticle-Based Immunochemical Biosensors and Assays: Recent Advances and Challenges. Chem. Rev. 2017, 117, 9973–10042. [Google Scholar] [CrossRef] [PubMed]
- Tian, J.; Wang, Y.; Chen, S.; Jiang, Y.; Zhao, Y.; Zhao, S. Mass-amplifying quantum dots in a fluorescence polarization-based aptasensor for ATP. Microchim. Acta 2012, 180, 203–209. [Google Scholar] [CrossRef]
- Tõntson, L.; Kopanchuk, S.; Rinken, A. Characterization of 5-HT1A receptors and their complexes with G-proteins in budded baculovirus particles using fluorescence anisotropy of Bodipy-FL-NAN-190. Neurochem. Int. 2014, 67, 32–38. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Yang, S.; De Ruyck, K.; Beloglazova, N.V.; Eremin, S.A.; De Saeger, S.; Zhang, S.; Shen, J.; Wang, Z. Fluorescence polarization assays for chemical contaminants in food and environmental analyses. TrAC Trends Anal. Chem. 2019, 114, 293–313. [Google Scholar] [CrossRef]
- Kaur, A.; Dhakal, S. Recent applications of FRET-based multiplexed techniques. TrAC Trends Anal. Chem. 2020, 123, 115777. [Google Scholar] [CrossRef]
- Gutierrez, M.C.; Gomez-Hens, A.; Perez-Bendito, D. Immunoassay methods based on fluorescence polarization. Talanta 1989, 36, 1187–1201. [Google Scholar] [CrossRef]
- Chen, J.; Liu, J.; Chen, X.; Qiu, H. Recent progress in nanomaterial-enhanced fluorescence polarization/anisotropy sensors. Chin. Chem. Lett. 2019, 30, 1575–1580. [Google Scholar] [CrossRef]
- Lakowicz, J.R. Principles of Fluorescence Spectroscopy; Springer Science & Business Media: Berlin, Germany, 2013. [Google Scholar]
- Dandliker, W.; Kelly, R.; Dandliker, J.; Farquhar, J.; Levin, J. Fluorescence polarization immunoassay. Theory and experimental method. Immunochemistry 1973, 10, 219–227. [Google Scholar] [CrossRef]
- Samokhvalov, A.V.; Safenkova, I.V.; Eremin, S.A.; Zherdev, A.; Dzantiev, B. Use of anchor protein modules in fluorescence polarisation aptamer assay for ochratoxin A determination. Anal. Chim. Acta 2017, 962, 80–87. [Google Scholar] [CrossRef]
- Choi, M.J.; Lee, J.R.; Eremin, S.A. Development of Single Reagent for Fluorescence Polarization Immunoassay of Atrazine. Food Agric. Immunol. 2010, 14, 107–120. [Google Scholar] [CrossRef]
- Lippolis, V.; Porricelli, A.C.R.; Mancini, E.; Ciasca, B.; Lattanzio, V.M.T.; De Girolamo, A.; Maragos, C.M.; McCormick, S.P.; Li, P.; Logrieco, A.F.; et al. Fluorescence Polarization Immunoassay for the Determination of T-2 and HT-2 Toxins and Their Glucosides in Wheat. Toxins 2019, 11, 380. [Google Scholar] [CrossRef] [PubMed]
- Mi, T.; Wang, Z.; Eremin, S.A.; Shen, J.; Zhang, S. Simultaneous Determination of Multiple (Fluoro)quinolone Antibiotics in Food Samples by a One-Step Fluorescence Polarization Immunoassay. J. Agric. Food Chem. 2013, 61, 9347–9355. [Google Scholar] [CrossRef] [PubMed]
- Eremin, S.A.; Knopp, D.; Niessner, R.; Hong, J.Y.; Park, S.-J.; Choi, M.J. High Throughput Determination of BTEX by a One-Step Fluorescence Polarization Immunoassay. Environ. Chem. 2005, 2, 227–234. [Google Scholar] [CrossRef]
- Eremin, S.A.; Matveeva, E.G.; Gómez-Hens, A.; Pérez-Bendito, D. Kinetic Determination of 2,4-Dichlorophenoxyacetic Acid by Stopped-Flow Fluorescence Polarization Immunoassay. Int. J. Environ. Anal. Chem. 1998, 71, 137–146. [Google Scholar] [CrossRef]
- Sendra, B. Kinetic determination of atrazine in foods based on stopped-flow fluorescence polarization immunoassay. Talanta 1998, 47, 153–160. [Google Scholar] [CrossRef]
- Eremin, S.; Smith, D. Fluorescence Polarization Immunoassays for Pesticides. Comb. Chem. High Throughput Screen. 2003, 6, 257–266. [Google Scholar] [CrossRef]
- Reiner, D.; Stark, H. Ligand binding kinetics at histamine H3 receptors by fluorescence-polarization with real-time monitoring. Eur. J. Pharmacol. 2019, 848, 112–120. [Google Scholar] [CrossRef]
- Wolfe, A.J.; Hsueh, Y.-C.; Blanden, A.R.; Mohammad, M.M.; Pham, B.; Thakur, A.K.; Loh, S.N.; Chen, M.; Movileanu, L. Interrogating Detergent Desolvation of Nanopore-Forming Proteins by Fluorescence Polarization Spectroscopy. Anal. Chem. 2017, 89, 8013–8020. [Google Scholar] [CrossRef]
- Yakovleva, J.; Lobanova, A.I.; Panchenko, O.A.; Eremin, S.A. Production of Antibodies and Development of Specific Polarization Fluoroimmunoassay for Acetochlor. Int. J. Environ. Anal. Chem. 2010, 82, 851–863. [Google Scholar] [CrossRef]
- Shim, W.-B.; Kolosova, A.Y.; Kim, Y.-J.; Yang, Z.-Y.; Park, S.-J.; Eremin, S.A.; Lee, I.-S.; Chung, D.-H. Fluorescence polarization immunoassay based on a monoclonal antibody for the detection of ochratoxin A. Int. J. Food Sci. Technol. 2004, 39, 829–837. [Google Scholar] [CrossRef]
- El-Hay, S.S.A.; Belal, F.F. Development of a micelle-enhanced high-throughput fluorometric method for determination of niclosamide using a microplate reader. Luminescence 2019, 34, 48–54. [Google Scholar] [CrossRef] [PubMed]
- Fukuyama, M.; Nakamura, A.; Nishiyama, K.; Imai, A.; Tokeshi, M.; Shigemura, K.; Hibara, A. Noncompetitive Fluorescence Polarization Immunoassay for Protein Determination. Anal. Chem. 2020, 92, 14393–14397. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.-E.; Si, S. Direct fluorescence polarization aptamer-based assay for the determination of adenosine. Anal. Methods 2013, 5, 840. [Google Scholar] [CrossRef]
- Zhao, Q.; Lv, Q.; Yin, J. Identification of Allosteric Nucleotide Sites of Tetramethylrhodamine-Labeled Aptamer for Noncompetitive Aptamer-Based Fluorescence Anisotropy Detection of a Small Molecule, Ochratoxin A. Anal. Chem. 2013, 86, 1238–1245. [Google Scholar] [CrossRef]
- Geng, X.; Zhang, D.; Yin, J.; Zhao, Q. Screening interaction between ochratoxin A and aptamers by fluorescence anisotropy approach. Anal. Bioanal. Chem. 2013, 405, 2443–2449. [Google Scholar] [CrossRef]
- Nishiyama, K.; Fukuyama, M.; Maeki, M.; Ishida, A.; Tani, H.; Hibara, A.; Maeki, M. One-step non-competitive fluorescence polarization immunoassay based on a Fab fragment for C-reactive protein quantification. Sens. Actuators B Chem. 2021, 326, 128982. [Google Scholar] [CrossRef]
- Tarannum, N.; Hendrickson, O.D.; Khatoon, S.; Zherdev, A.; Dzantiev, B.B. Molecularly imprinted polymers as receptors for assays of antibiotics. Crit. Rev. Anal. Chem. 2019, 50, 291–310. [Google Scholar] [CrossRef]
- Chen, C.; Luo, J.; Li, C.; Ma, M.; Yu, W.; Shen, J.; Wang, Z. Molecularly Imprinted Polymer as an Antibody Substitution in Pseudo-immunoassays for Chemical Contaminants in Food and Environmental Samples. J. Agric. Food Chem. 2018, 66, 2561–2571. [Google Scholar] [CrossRef]
- McKeague, M.; McConnell, E.M.; Cruz-Toledo, J.; Bernard, E.D.; Pach, A.; Mastronardi, E.; Zhang, X.; Beking, M.; Francis, T.; Giamberardino, A.; et al. Analysis of In Vitro Aptamer Selection Parameters. J. Mol. Evol. 2015, 81, 150–161. [Google Scholar] [CrossRef]
- McKeague, M.; DeRosa, M.C. Challenges and Opportunities for Small Molecule Aptamer Development. J. Nucleic Acids 2012, 2012, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Klug, S.J.; Famulok, M. All you wanted to know about SELEX. Mol. Biol. Rep. 1994, 20, 97–107. [Google Scholar] [CrossRef] [PubMed]
- Luzi, E.; Minunni, M.; Tombelli, S.; Mascini, M. New trends in affinity sensing. TrAC Trends Anal. Chem. 2003, 22, 810–818. [Google Scholar] [CrossRef]
- Song, K.-M.; Lee, S.; Ban, C. Aptamers and Their Biological Applications. Sensors 2012, 12, 612–631. [Google Scholar] [CrossRef] [PubMed]
- Antipova, O.; Zavyalova, E.; Golovin, A.V.; Pavlova, G.V.; Kopylov, A.; Reshetnikov, R.V. Advances in the Application of Modified Nucleotides in SELEX Technology. Biochemistry (Moscow) 2018, 83, 1161–1172. [Google Scholar] [CrossRef] [PubMed]
- Ilgu, M.; Nilsen-Hamilton, M. Aptamers in analytics. Analyst 2016, 141, 1551–1568. [Google Scholar] [CrossRef]
- Lakhin, A.V.; Tarantul, V.Z.; Gening, L.V. Aptamers: Problems, Solutions and Prospects. Acta Nat. 2013, 5, 34–43. [Google Scholar] [CrossRef]
- Schüling, T.; Eilers, A.; Scheper, T.; Walter, J.-G. Aptamer-based lateral flow assays. AIMS Environ. Sci. 2018, 5, 78–102. [Google Scholar] [CrossRef]
- Bai, Y.; Shu, T.; Su, L.; Zhang, X. Functional nucleic acid-based fluorescence polarization/anisotropy biosensors for detection of biomarkers. Anal. Bioanal. Chem. 2020, 412, 1–11. [Google Scholar] [CrossRef]
- Zhao, Q.; Tao, J.; Uppal, J.S.; Peng, H.; Wang, H.; Le, X.C. Nucleic acid aptamers improving fluorescence anisotropy and fluorescence polarization assays for small molecules. TrAC Trends Anal. Chem. 2019, 110, 401–409. [Google Scholar] [CrossRef]
- Cui, L.; Zou, Y.; Lin, N.; Zhu, Z.; Jenkins, G.; Yang, C.J. Mass Amplifying Probe for Sensitive Fluorescence Anisotropy Detection of Small Molecules in Complex Biological Samples. Anal. Chem. 2012, 84, 5535–5541. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Zhao, Q. A simple fluorescence anisotropy assay for detection of bisphenol A using fluorescently labeled aptamer. J. Environ. Sci. 2020, 97, 19–24. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Zhao, Q. Direct fluorescence anisotropy assay for cocaine using tetramethylrhodamine-labeled aptamer. Anal. Bioanal. Chem. 2017, 237, 1219–4000. [Google Scholar] [CrossRef] [PubMed]
- Chovelon, B.; Fiore, E.; Faure, P.; Peyrin, E.; Ravelet, C. Kissing interactions for the design of a multicolour fluorescence anisotropy chiral aptasensor. Talanta 2019, 205, 120098. [Google Scholar] [CrossRef]
- Kang, L.; Yang, B.; Zhang, X.; Cui, L.; Meng, H.; Mei, L.; Wu, C.; Ren, S.; Tan, W. Enzymatic cleavage and mass amplification strategy for small molecule detection using aptamer-based fluorescence polarization biosensor. Anal. Chim. Acta 2015, 879, 91–96. [Google Scholar] [CrossRef]
- Li, Y.; Sun, Y.; Ye, J.; Pan, F.; Peng, B.; Li, H.; Zhang, M.; Xu, Y. Sensitive and selective detection of microRNA in complex biological samples based on protein-enhanced fluorescence anisotropy. Anal. Methods 2020, 12, 687–692. [Google Scholar] [CrossRef]
- Huang, Y.; Liu, X.; Shi, M.; Zhao, S.; Hu, K.; Chen, Z.-F.; Liang, H. Ultrasensitive Fluorescence Polarization Aptasensors Based on Exonuclease Signal Amplification and Polystyrene Nanoparticle Amplification. Chem. Asian J. 2014, 9, 2755–2760. [Google Scholar] [CrossRef]
- Huang, Y.; Zhao, S.; Chen, Z.-F.; Shi, M.; Liang, H. Amplified fluorescence polarization aptasensors based on structure-switching-triggered nanoparticles enhancement for bioassays. Chem. Commun. 2012, 48, 7480–7482. [Google Scholar] [CrossRef]
- Hunt, C.E.; Pasetto, P.; Ansell, R.J.; Haupt, K. A fluorescence polarisation molecular imprint sorbent assay for 2,4-D: A non-separation pseudo-immunoassay. Chem. Commun. 2006, 16, 1754. [Google Scholar] [CrossRef]
- Hunt, C.E.; Ansell, R.J. Use of fluorescence shift and fluorescence anisotropy to evaluate the re-binding of template to (S)-propranolol imprinted polymers. Analyst 2006, 131, 678. [Google Scholar] [CrossRef]
- Ton, X.-A.; Acha, V.; Haupt, K.; Bui, B.T.S. Direct fluorimetric sensing of UV-excited analytes in biological and environmental samples using molecularly imprinted polymer nanoparticles and fluorescence polarization. Biosens. Bioelectron. 2012, 36, 22–28. [Google Scholar] [CrossRef] [PubMed]
- Murase, N.; Taniguchi, S.-I.; Takano, E.; Kitayama, Y.; Takeuchi, T. A molecularly imprinted nanocavity-based fluorescence polarization assay platform for cortisol sensing. J. Mater. Chem. B 2016, 4, 1770–1777. [Google Scholar] [CrossRef] [PubMed]
- Xiao, P.; Weibel, N.; Dudal, Y.; Corvini, P.F.-X.; Shahgaldian, P. A cyclodextrin-based polymer for sensing diclofenac in water. J. Hazard. Mater. 2015, 299, 412–416. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Li, T.; Zhang, T.; Xue, P.; Guan, T.; Yuan, Y.; Yu, H. Receptor-Based Fluorescence Polarization Assay to Detect Phthalate Esters in Chinese Spirits. Food Anal. Methods 2016, 10, 1293–1300. [Google Scholar] [CrossRef]
- Guan, T.; Sun, Y.; Zhang, T.; Li, T.; Li, Z.; Zhang, Y.; Zhang, J.; Wang, Y. Simultaneous determination of bisphenol A and its halogenated analogues in soil by fluorescence polarization assay. J. Soils Sediments 2017, 18, 845–851. [Google Scholar] [CrossRef]
- Guan, T.; Li, T.; Zhang, T.; Li, Z.; Wang, Y.; Yu, H.; Ruan, P.; Zhang, J.; Wang, Y. Fluorescence polarization assay for the simultaneous determination of bisphenol A, bisphenol F and their diglycidyl ethers in canned tuna. Int. J. Food Prop. 2017, 20, 1–10. [Google Scholar] [CrossRef]
- Zhang, J.; Zhang, T.; Guan, T.; Yu, H.; Li, T. In vitro and in silico assessment of the structure-dependent binding of bisphenol analogues to glucocorticoid receptor. Anal. Bioanal. Chem. 2017, 409, 2239–2246. [Google Scholar] [CrossRef]
- Allikalt, A.; Kopanchuk, S.; Rinken, A. Implementation of fluorescence anisotropy-based assay for the characterization of ligand binding to dopamine D1 receptors. Eur. J. Pharmacol. 2018, 839, 40–46. [Google Scholar] [CrossRef]
- Yao, Y.; Kadam, R.U.; Lee, C.-C.D.; Woehl, J.L.; Wu, N.C.; Zhu, X.; Kitamura, S.; Wilson, I.A.; Wolan, D.W. An influenza A hemagglutinin small-molecule fusion inhibitor identified by a new high-throughput fluorescence polarization screen. Proc. Natl. Acad. Sci. USA 2020, 117, 18431–18438. [Google Scholar] [CrossRef]
- Wang, Z.; Liang, X.; Wen, K.; Zhang, S.; Li, C.; Shen, J. A highly sensitive and class-specific fluorescence polarisation assay for sulphonamides based on dihydropteroate synthase. Biosens. Bioelectron. 2015, 70, 1–4. [Google Scholar] [CrossRef]
- Alfonso, A.; Fernández-Araujo, A.; Carames, B.; Tobío, A.; Louzao, M.C.; Vieytes, M.; Botana, L.M.; Alfonso, C. Palytoxin detection and quantification using the fluorescence polarization technique. Anal. Biochem. 2012, 424, 64–70. [Google Scholar] [CrossRef] [PubMed]
- Hall, M.D.; Yasgar, A.; Peryea, T.; Braisted, J.C.; Jadhav, A.; Simeonov, A.; Coussens, N.P. Fluorescence polarization assays in high-throughput screening and drug discovery: A review. Methods Appl. Fluoresc. 2016, 4, 022001. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zhang, N.; Yin, J.; Zhao, Q. Fluorescence Anisotropy-Based Signal-Off and Signal-On Aptamer Assays Using Lissamine Rhodamine B as a Label for Ochratoxin A. J. Agric. Food Chem. 2020, 68, 4277–4283. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Zhao, Q. Direct fluorescence anisotropy approach for aflatoxin B1 detection and affinity binding study by using single tetramethylrhodamine labeled aptamer. Talanta 2018, 189, 442–450. [Google Scholar] [CrossRef] [PubMed]
- Ren, D.; Ren, D.; You, Z.; Yalikun, Y.; Tanaka, Y. Ultrasensitive detection of nucleic acids based on dually enhanced fluorescence polarization. Analyst 2018, 143, 3560–3569. [Google Scholar] [CrossRef]
- Choi, J.-W.; Jo, B.-G.; Demello, A.J.; Choo, J.; Kim, H.-Y. Streptavidin-triggered signal amplified fluorescence polarization for analysis of DNA–protein interactions. Analyst 2016, 141, 6499–6502. [Google Scholar] [CrossRef]
- Rusinova, E.; Tretyachenko-Ladokhina, V.; Vele, O.E.; Senear, D.F.; Ross, J.A. Alexa and Oregon Green dyes as fluorescence anisotropy probes for measuring protein-protein and protein-nucleic acid interactions. Anal. Biochem. 2002, 308, 18–25. [Google Scholar] [CrossRef]
- Zhang, X.; Eremin, S.A.; Wen, K.; Yu, X.; Li, C.; Ke, Y.; Jiang, H.; Shen, J.; Wang, Z. Fluorescence Polarization Immunoassay Based on a New Monoclonal Antibody for the Detection of the Zearalenone Class of Mycotoxins in Maize. J. Agric. Food Chem. 2017, 65, 2240–2247. [Google Scholar] [CrossRef]
- Tereshchenkov, A.; Shishkina, A.V.; Karpenko, V.V.; Chertkov, V.A.; Konevega, A.L.; Kasatsky, P.S.; Bogdanov, A.A.; Sumbatyan, N.V. New fluorescent macrolide derivatives for studying interactions of antibiotics and their analogs with the ribosomal exit tunnel. Biochemistry (Moscow) 2016, 81, 1163–1172. [Google Scholar] [CrossRef]
- Jurewicz, M.M.; Willis, R.A.; Ramachandiran, V.; Altman, J.D.; Stern, L.J. MHC-I peptide binding activity assessed by exchange after cleavage of peptide covalently linked to β2-microglobulin. Anal. Biochem. 2019, 584, 113328. [Google Scholar] [CrossRef]
- Laursen, B.W.; Bogh, S.A.; Sørensen, T.J. Chapter Twelve—Long fluorescence lifetime triangulenium dyes in imaging and fluorescence polarization assay. In Methods in Enzymology; Chenoweth, D.M., Ed.; Academic Press: Cambridge, MA, USA, 2020; pp. 249–265. [Google Scholar]
- Ohashi, H.; Matsumoto, T.; Jeong, H.-J.; Dong, J.; Abe, R.; Ueda, H. Insight into the Working Mechanism of Quenchbody: Transition of the Dye around Antibody Variable Region That Fluoresces upon Antigen Binding. Bioconjugate Chem. 2016, 27, 2248–2253. [Google Scholar] [CrossRef] [PubMed]
- Okada, T.; Minoura, N. Fluorescence emission and polarization analyses for evaluating binding of ruthenium metalloglycoclusters to lectins and tetanus toxin C-fragment. J. Biomed. Opt. 2011, 16, 037001. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Martínez, M.L.; Aguilar-Caballos, A.M.P.; Gómez-Hens, A. Long-Wavelength Fluorescence Polarization Immunoassay: Determination of Amikacin on Solid Surface and Gliadins in Solution. Anal. Chem. 2007, 79, 7424–7430. [Google Scholar] [CrossRef] [PubMed]
- Lo, K.K.-W.; Hui, W.-K.; Ng, D.C.-M.; Cheung, K.-K. Synthesis, Characterization, Photophysical Properties, and Biological Labeling Studies of a Series of Luminescent Rhenium(I) Polypyridine Maleimide Complexes. Inorg. Chem. 2002, 41, 40–46. [Google Scholar] [CrossRef] [PubMed]
- Tian, J.; Zhou, L.; Zhao, Y.; Wang, Y.; Peng, Y.; Hong, X.; Zhao, S. The Application of CdTe/CdS in the Detection of Carcinoembryonic Antigen by Fluorescence Polarization Immunoassay. J. Fluoresc. 2012, 22, 1571–1579. [Google Scholar] [CrossRef] [PubMed]
- Tian, J.; Zhou, L.; Zhao, Y.; Wang, Y.; Peng, Y.; Zhao, S. Multiplexed detection of tumor markers with multicolor quantum dots based on fluorescence polarization immunoassay. Talanta 2012, 92, 72–77. [Google Scholar] [CrossRef] [PubMed]
- Shao, M.; Wang, H.-F. Heparin-mediated fluorescence anisotropy assay of antithrombin based on polyethyleneimine capped Mn-doped ZnS quantum dots. Analyst 2013, 138, 4618. [Google Scholar] [CrossRef]
- He, Y.; Tian, J.; Hu, K.; Zhang, J.; Chen, S.; Jiang, Y.; Zhao, Y.; Zhao, S. An ultrasensitive quantum dots fluorescent polarization immunoassay based on the antibody modified Au nanoparticles amplifying for the detection of adenosine triphosphate. Anal. Chim. Acta 2013, 802, 67–73. [Google Scholar] [CrossRef]
- Shokri, E.; Hosseini, M.; Sadeghan, A.A.; Bahmani, A.; Nasiri, N.; Hosseinkhani, S. Virus-directed synthesis of emitting copper nanoclusters as an approach to simple tracer preparation for the detection of Citrus Tristeza Virus through the fluorescence anisotropy immunoassay. Sens. Actuators B Chem. 2020, 321, 128634. [Google Scholar] [CrossRef]
- Yue, Q.; Shen, T.; Wang, L.; Xu, S.; Li, H.; Xue, Q.; Zhang, Y.; Gu, X.; Zhang, S.; Liu, J. A convenient sandwich assay of thrombin in biological media using nanoparticle-enhanced fluorescence polarization. Biosens. Bioelectron. 2014, 56, 231–236. [Google Scholar] [CrossRef]
- Liu, J.; Yu, J.; Chen, J.; Yang, R.; Shih, K. Signal-amplification and real-time fluorescence anisotropy detection of apyrase by carbon nanoparticle. Mater. Sci. Eng. C 2014, 38, 206–211. [Google Scholar] [CrossRef] [PubMed]
- El Kojok, H.; El Darra, N.; Khalil, M.; Capo, A.; Pennacchio, A.; Staiano, M.; Camarca, A.; D’Auria, S.; Varriale, A. Fluorescence polarization assay to detect the presence of traces of ciprofloxacin. Sci. Rep. 2020, 10, 4550. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Yin, J. Fluorescence Anisotropy Reduction of An Allosteric G-Rich Oligonucleotide for Specific Silver Ion and Cysteine Detection Based on the G-Ag+-G Base Pair. Anal. Chem. 2019, 91, 14538–14544. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Shao, C.; Yan, C.; Li, D.; Liu, Y. Fluorescence polarization sensor platform based on gold nanoparticles for the efficient detection of Ag (І). J. Lumin. 2019, 210, 21–27. [Google Scholar] [CrossRef]
- Wang, G.; Wang, S.; Yan, C.; Bai, G.; Liu, Y. DNA-functionalized gold nanoparticle-based fluorescence polarization for the sensitive detection of silver ions. Colloids Surf. B Biointerfaces 2018, 167, 150–155. [Google Scholar] [CrossRef]
- Qi, L.; Yan, Z.; Huo, Y.; Hai, X.-M.; Zhang, Z.-Q. MnO2 nanosheet-assisted ligand-DNA interaction-based fluorescence polarization biosensor for the detection of Ag+ ions. Biosens. Bioelectron. 2017, 87, 566–571. [Google Scholar] [CrossRef]
- Jiang, Y.; Tian, J.; Hu, K.; Zhao, Y.; Zhao, S. Sensitive aptamer-based fluorescence polarization assay for mercury(II) ions and cysteine using silver nanoparticles as a signal amplifier. Microchim. Acta 2014, 181, 1423–1430. [Google Scholar] [CrossRef]
- Mccord, B.R.; Gauthier, Q.; Cho, S.; Roig, M.N.; Gibson-Daw, G.C.; Young, B.; Taglia, F.; Zapico, S.C.; Mariot, R.F.; Lee, S.B.; et al. Forensic DNA Analysis. Anal. Chem. 2018, 91, 673–688. [Google Scholar] [CrossRef]
- McGinn, S.; Bauer, D.L.V.; Brefort, T.; Dong, L.; El-Sagheer, A.; ElSharawy, A.; Evans, G.; Falk-Sörqvist, E.; Forster, M.; Fredriksson, S.; et al. New technologies for DNA analysis—A review of the READNA Project. New Biotechnol. 2016, 33, 311–330. [Google Scholar] [CrossRef]
- Nikiforov, T.T.; Jeong, S. Detection of Nucleic Acid Hybridization by Fluorescence Polarization. European Patent EP1297179, 17 August 2004. [Google Scholar]
- Latif, S.; Bauer-Sardina, I.; Ranade, K.; Livak, K.J.; Kwok, P.-Y. Fluorescence Polarization in Homogeneous Nucleic Acid Analysis II: 5’-Nuclease Assay. Genome Res. 2001, 11, 436–440. [Google Scholar] [CrossRef]
- Walker, G.T.; Linn, C.P.; Nadeau, J.G. DNA detection by strand displacement amplification and fluorescence polarization with signal enhancement using a DNA binding protein. Nucleic Acids Res. 1996, 24, 348–353. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.-Y.; Li, D.-H.; Yang, H.-H.; Zhu, Q.-Z.; Xu, J.-G.; Zhao, Y. Interaction of a novel red-region fluorescent probe, Nile Blue, with DNA and its application to nucleic acids assay. Analyst 1999, 124, 901–906. [Google Scholar] [CrossRef] [PubMed]
- Kareiva, P.; Parker, I.M.; Pascual, M. Can We Use Experiments and Models in Predicting the Invasiveness of Genetically Engineered Organisms? Ecology 1996, 77, 1670–1675. [Google Scholar] [CrossRef]
- Pajukanta, P.; Nuotio, I.; Terwilliger, J.D.; Porkka, K.V.; Ylitalo, K.; Pihlajamäki, J.; Suomalainen, A.J.; Syvänen, A.-C.; Lehtimäki, T.; Viikari, J.S.; et al. Linkage of familial combined hyperlipidaemia to chromosome 1q21–q23. Nat. Genet. 1998, 18, 369–373. [Google Scholar] [CrossRef]
- Zhu, Z.; Ravelet, C.; Perrier, S.; Guieu, V.; Fiore, E.; Peyrin, E. Single-Stranded DNA Binding Protein-Assisted Fluorescence Polarization Aptamer Assay for Detection of Small Molecules. Anal. Chem. 2012, 84, 7203–7211. [Google Scholar] [CrossRef]
- Gaus, H.J.; Gupta, R.; Chappell, A.E.; Østergaard, M.E.; Swayze, E.E.; Seth, P.P. Characterization of the interactions of chemically-modified therapeutic nucleic acids with plasma proteins using a fluorescence polarization assay. Nucleic Acids Res. 2019, 47, 1110–1122. [Google Scholar] [CrossRef]
- Oleksy, J.; Boeckman, F.; Hungate, E.; Rencs, E. Fluorescence Polarization Detection of Nucleic Acids. European Patent EP1509622A4, 27 November 2003. [Google Scholar]
- Wu, X.; Pei, X.-J.; Lin, R.-Y.; Liu, F.; Li, N. Fluorescence Anisotropy and Applications Based on Functional Nucleic Acid Recognition. Guang Pu Xue Yu Guang Pu Fen Xi 2017, 37, 13–20. [Google Scholar]
- Miura, H. Application of fluorescence polarization to the determination of urinary lysozyme activity. Clin. Biochem. 1985, 18, 40–47. [Google Scholar] [CrossRef]
- Fiene, A.; Baqi, Y.; Lecka, J.; Sévigny, J.; Müller, C.E. Fluorescence polarization immunoassays for monitoring nucleoside triphosphate diphosphohydrolase (NTPDase) activity. Analyst 2015, 140, 140–148. [Google Scholar] [CrossRef]
- Kleman-Leyer, K.M.; Klink, T.A.; Kopp, A.L.; Westermeyer, T.A.; Koeff, M.D.; Larson, B.R.; Worzella, T.J.; Pinchard, C.A.; Van De Kar, S.A.; Zaman, G.J.; et al. Characterization and Optimization of a Red-Shifted Fluorescence Polarization ADP Detection Assay. ASSAY Drug Dev. Technol. 2009, 7, 56–67. [Google Scholar] [CrossRef]
- Beebe, J.A.; Wiepz, G.J.; Guadarrama, A.G.; Bertics, P.J.; Burke, T.J. A Carboxyl-terminal Mutation of the Epidermal Growth Factor Receptor Alters Tyrosine Kinase Activity and Substrate Specificity as Measured by a Fluorescence Polarization Assay. J. Biol. Chem. 2003, 278, 26810–26816. [Google Scholar] [CrossRef] [PubMed]
- Su, Y.; Hickey, S.F.; Keyser, S.G.L.; Hammond, M.C. In VitroandIn VivoEnzyme Activity Screening via RNA-Based Fluorescent Biosensors forS-Adenosyl-l-homocysteine (SAH). J. Am. Chem. Soc. 2016, 138, 7040–7047. [Google Scholar] [CrossRef] [PubMed]
- Graves, T.L.; Zhang, Y.; Scott, J.E. A universal competitive fluorescence polarization activity assay for S-adenosylmethionine utilizing methyltransferases. Anal. Biochem. 2008, 373, 296–306. [Google Scholar] [CrossRef] [PubMed]
- Mestas, S.P.; Sholders, A.J.; Peersen, O. A fluorescence polarization-based screening assay for nucleic acid polymerase elongation activity. Anal. Biochem. 2007, 365, 194–200. [Google Scholar] [CrossRef]
- Boroduleva, A.Y.; Wu, J.; Yang, Q.; Li, H.; Zhang, Q.; Li, P.; Eremin, S.A. Development of fluorescence polarization immunoassays for parallel detection of pesticides carbaryl and triazophos in wheat grains. Anal. Methods 2017, 9, 6814–6822. [Google Scholar] [CrossRef]
- Zhang, C.; Jiang, Z.; Jin, M.; Du, P.; Chen, G.; Cui, X.; Zhang, Y.; Qin, G.; Yan, F.; El-Aty, A.M.A.; et al. Fluorescence immunoassay for multiplex detection of organophosphate pesticides in agro-products based on signal amplification of gold nanoparticles and oligonucleotides. Food Chem. 2020, 326, 126813. [Google Scholar] [CrossRef]
- Li, C.; Wen, K.; Mi, T.; Zhang, X.; Zhang, H.; Zhang, S.; Shen, J.; Wang, Z. A universal multi-wavelength fluorescence polarization immunoassay for multiplexed detection of mycotoxins in maize. Biosens. Bioelectron. 2016, 79, 258–265. [Google Scholar] [CrossRef]
- Ma, P.; Sun, Y.; Khan, I.M.; Gu, Q.; Yue, L.; Wang, Z. Structure-switching fluorescence aptasensor for sensitive detection of chloramphenicol. Microchim. Acta 2020, 187, 1–10. [Google Scholar] [CrossRef]
- Perrier, S.; Bouilloud, P.; Coelho, G.D.O.; Henry, M.; Peyrin, E. Small molecule aptamer assays based on fluorescence anisotropy signal-enhancer oligonucleotides. Biosens. Bioelectron. 2016, 82, 155–161. [Google Scholar] [CrossRef]
- Li, Y.; Zhao, Q. Aptamer structure switch fluorescence anisotropy assay for aflatoxin B1 using tetramethylrhodamine-guanine interaction to enhance signal change. Chin. Chem. Lett. 2020, 31, 1982–1985. [Google Scholar] [CrossRef]
- Li, Y.; Zhao, Q. Aptamer Structure Switch Fluorescence Anisotropy Assay for Small Molecules Using Streptavidin as an Effective Signal Amplifier Based on Proximity Effect. Anal. Chem. 2019, 91, 7379–7384. [Google Scholar] [CrossRef] [PubMed]
- Goux, E.; Lespinasse, Q.; Guieu, V.; Perrier, S.; Ravelet, C.; Fiore, E.; Peyrin, E. Fluorescence anisotropy-based structure-switching aptamer assay using a peptide nucleic acid (PNA) probe. Methods 2016, 97, 69–74. [Google Scholar] [CrossRef] [PubMed]
- Perrier, S.; Guieu, V.; Chovelon, B.; Ravelet, C.; Peyrin, E. Panoply of Fluorescence Polarization/Anisotropy Signaling Mechanisms for Functional Nucleic Acid-Based Sensing Platforms. Anal. Chem. 2018, 90, 4236–4248. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Mazouzi, Y.; Salmain, M.; Liedberg, B.; Boujday, S. Antibody-Gold Nanoparticle Bioconjugates for Biosensors: Synthesis, Characterization and Selected Applications. Biosens. Bioelectron. 2020, 165, 112370. [Google Scholar] [CrossRef] [PubMed]
- Yan, S.-R.; Foroughi, M.; Safaei, M.; Jahani, S.; Ebrahimpour, N.; Borhani, F.; Baravati, N.R.Z.; Aramesh-Boroujeni, Z.; Foong, L.K. A review: Recent advances in ultrasensitive and highly specific recognition aptasensors with various detection strategies. Int. J. Biol. Macromol. 2020, 155, 184–207. [Google Scholar] [CrossRef]
- Samokhvalov, A.V.; Safenkova, I.V.; Eremin, S.A.; Zherdev, A.V.; Dzantiev, B. Application of Gold Nanoparticles for High-Sensitivity Fluorescence Polarization Aptamer Assay for Ochratoxin A. Nanotechnol. Russ. 2019, 14, 397–404. [Google Scholar] [CrossRef]
- Ye, H.; Duan, N.; Gu, H.; Wang, H.; Wang, Z. Fluorometric determination of lipopolysaccharides via changes of the graphene oxide-enhanced fluorescence polarization caused by truncated aptamers. Microchim. Acta 2019, 186, 173. [Google Scholar] [CrossRef]
- Huang, Y.; Liu, X.; Zhang, L.; Hu, K.; Zhao, S.; Fang, B.; Chen, Z.-F.; Liang, H. Nicking enzyme and graphene oxide-based dual signal amplification for ultrasensitive aptamer-based fluorescence polarization assays. Biosens. Bioelectron. 2015, 63, 178–184. [Google Scholar] [CrossRef]
- Chen, Z.; Li, H.; Zhao, Y.; Xu, M.; Xu, D. Magnetic nanoparticles and polydopamine amplified FP aptasensor for the highly sensitive detection of rHuEPO-α. Talanta 2018, 189, 143–149. [Google Scholar] [CrossRef]
- Ma, P.; Ye, H.; Deng, J.; Khan, I.M.; Yue, L.; Wang, Z. A fluorescence polarization aptasensor coupled with polymerase chain reaction and streptavidin for chloramphenicol detection. Talanta 2019, 205, 120119. [Google Scholar] [CrossRef]
- Liu, Y.X.; Xiao, X.; Li, C.H.; Men, C.; Ye, Q.C.; Lv, W.Y.; Li, Y.F.; Huang, C.Z.; Zhen, S.J. DNA nanosheet as an excellent fluorescence anisotropy amplification platform for accurate and sensitive biosensing. Talanta 2020, 211, 120730. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Wan, B.; Du, W.; Hu, H.; Zeng, L.; Duan, X.; Liu, J.; Wei, Z.; Tang, L.; Peng, Y. A ligation-triggered and protein-assisted fluorescence anisotropy amplification platform for sensitive and selective detection of small molecules in a biological matrix. RSC Adv. 2020, 10, 21789–21794. [Google Scholar] [CrossRef]
- Liu, G.; Nash, P.J.; Johnson, B.; Pietzsch, C.; Ilagan, M.X.G.; Bukreyev, A.; Basler, C.F.; Bowlin, T.L.; Moir, D.T.; Leung, D.W.; et al. A Sensitive in Vitro High-Throughput Screen to Identify Pan-filoviral Replication Inhibitors Targeting the VP35–NP Interface. ACS Infect. Dis. 2017, 3, 190–198. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Liu, R.; Boroduleva, A.; Eremin, S.; Guo, Y.; Zhu, G. A highly specific and sensitive fluorescence polarization immunoassay for the rapid detection of triazophos residue in agricultural products. Anal. Methods 2016, 8, 6636–6644. [Google Scholar] [CrossRef]
- Chen, J.; Shanin, I.A.; Lv, S.; Wang, Q.; Mao, C.; Xu, Z.; Sun, Y.; Wu, Q.; Eremin, S.A.; Lei, H. Heterologous strategy enhancing the sensitivity of the fluorescence polarization immunoassay of clinafloxacin in goat milk. J. Sci. Food Agric. 2015, 96, 1341–1346. [Google Scholar] [CrossRef]
- Lippolis, V.; Porricelli, A.C.R.; Cortese, M.; Suman, M.; Zanardi, S.; Pascale, M. Determination of Ochratoxin A in Rye and Rye-Based Products by Fluorescence Polarization Immunoassay. Toxins 2017, 9, 305. [Google Scholar] [CrossRef]
- Ansideri, F.; Lange, A.; El-Gokha, A.; Boeckler, F.M.; Koch, P. Fluorescence polarization-based assays for detecting compounds binding to inactive c-Jun N-terminal kinase 3 and p38α mitogen-activated protein kinase. Anal. Biochem. 2016, 503, 28–40. [Google Scholar] [CrossRef]
- Kalleshamurthy, T.; Yaranna, C.; Shekar, R.; Natesan, K.; Sahay, S.; Shome, B.R.; Rahman, H.; Barbuddhe, S.B.; Barman, N.N.; Das, S.K.; et al. Fluorescence polarization assay: Diagnostic evaluation for porcine brucellosis. J. Microbiol. Methods 2019, 156, 46–51. [Google Scholar] [CrossRef]
- Beloglazova, N.; Shmelin, P.; Eremin, S. Sensitive immunochemical approaches for quantitative (FPIA) and qualitative (lateral flow tests) determination of gentamicin in milk. Talanta 2016, 149, 217–224. [Google Scholar] [CrossRef]
- Porricelli, A.C.R.; Lippolis, V.; Valenzano, S.; Cortese, M.; Suman, M.; Zanardi, S.; Pascale, M. Optimization and Validation of a Fluorescence Polarization Immunoassay for Rapid Detection of T-2 and HT-2 Toxins in Cereals and Cereal-Based Products. Food Anal. Methods 2016, 9, 3310–3318. [Google Scholar] [CrossRef]
- Boroduleva, A.Y.; Manclús, J.J.; Montoya, Á.; Eremin, S.A. Fluorescence polarization immunoassay for rapid screening of the pesticides thiabendazole and tetraconazole in wheat. Anal. Bioanal. Chem. 2018, 410, 6923–6934. [Google Scholar] [CrossRef] [PubMed]
- Boroduleva, A.Y.; Eremin, S.A. Determination of 2,4-dichlorophenoxyacetic acid in cereals by fluorescence polarization immunoassay. J. Anal. Chem. 2016, 71, 949–954. [Google Scholar] [CrossRef]
- Zhao, Z.; Wei, L.; Cao, M.; Lu, M. A smartphone-based system for fluorescence polarization assays. Biosens. Bioelectron. 2019, 128, 91–96. [Google Scholar] [CrossRef] [PubMed]
- Wargocki, P.; Deng, W.; Anwer, A.G.; Goldys, E.M. Medically Relevant Assays with a Simple Smartphone and Tablet Based Fluorescence Detection System. Sensors 2015, 15, 11653–11664. [Google Scholar] [CrossRef]
- Wakao, O.; Satou, K.; Nakamura, A.; Galkina, P.A.; Nishiyama, K.; Sumiyoshi, K.; Kurosawa, F.; Maeki, M.; Ishida, A.; Tani, H.; et al. High-throughput fluorescence polarization immunoassay by using a portable fluorescence polarization imaging analyzer. Lab Chip 2019, 19, 2581–2588. [Google Scholar] [CrossRef]
- Nishiyama, K.; Takeda, Y.; Maeki, M.; Ishida, A.; Tani, H.; Shigemura, K.; Hibara, A.; Yonezawa, Y.; Imai, K.; Ogawa, H.; et al. Rapid detection of anti-H5 avian influenza virus antibody by fluorescence polarization immunoassay using a portable fluorescence polarization analyzer. Sens. Actuators B Chem. 2020, 316, 128160. [Google Scholar] [CrossRef]
- Choi, J.-W.; Vasamsetti, B.M.K.; Choo, J.; Kim, H.-Y. Analysis of deoxyribonuclease activity by conjugation-free fluorescence polarisation in sub-nanolitre droplets. Analyst 2020, 145, 3222–3228. [Google Scholar] [CrossRef]
- Schrell, A.M.; Mukhitov, N.; Yi, L.; Adablah, J.E.; Menezes, J.; Roper, M.G. Online fluorescence anisotropy immunoassay for monitoring insulin secretion from islets of Langerhans. Anal. Methods 2017, 9, 38–45. [Google Scholar] [CrossRef]
- Zvereva, E.A.; Zherdev, A.V.; Formanovsky, A.A.; Abuknesha, R.A.; Eremin, S.A.; Dzantiev, B.B. Fluorescence polarization immunoassay of colchicine. J. Pharm. Biomed. Anal. 2018, 159, 326–330. [Google Scholar] [CrossRef]
- Zvereva, E.A.; Shpakova, N.A.; Zherdev, A.; Liu, L.; Xu, C.; Eremin, S.A.; Dzantiev, B. Fluorescence polarization immunoassay of ractopamine. Appl. Biochem. Microbiol. 2016, 52, 673–678. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Analyte/Medium | Aptamer Used | Limit of Detection | References |
|---|---|---|---|
| Bisphenol A/tap water | 5′-CCG CCG TTG GTG TGG TGG GCC TAG GGC CGG CGG-(TMR)-3′ | 0.5 µM | [44] |
| Cocaine/serum and urine | 5′-GAC AAG GAA AAT CCT TCA ATG AAG T(TMR)GG GTC-3′ | 5 µM | [45] |
| D- and L-arginine vasopressin / serum | 5′-TCA CGT GCA TGA TAA CAC TCC CAT TCT GAG TTG CTG TGT GCC GAT GCA CGT GA-fluorescein(or Texas-Red)-3′ | 40 nM; 20 nM | [46] |
| Adenosine/cell culture medium | 5′-fluorescein-AGdT GAA CCT GGG GGA GTA TTG CGG AGG AAG GT-biotin-3′ | 500 nM | [47] |
| MicroRNA/tumor cell lysates | 5′-GGT TGG TGT GGT TGG TCA ACA TCA GTC TGA TAA GCTA-fluorescein-3′ | 3.4 pM | [48] |
| Ochratoxin A/buffer | 5′-fluorescein-GGG AGC ATC GGA CTT TTT TT-biotin-3′ | 25.2 fM | [49] |
| Ochratoxin A/white wine | 5′-GAT CGG GTG TGG GTG GCG TAA AGG GAG CAT CGG ACA-biotin-3′ | 2.8 nM | [11] |
| Adenosine/cell culture medium | 5′-GGT TGG TGT GGT TGG ACC TGG GGG AGT ATT GCG GAG GAA GGT-fluorescein-3′ | 0.5 μM | [43] |
| Cocaine/urine | 5′-fluorescein-AGAC AAG GAA AAT CCT TCA ATG AAG TGG GTC G GGT TGG TGT GGT TGG-3′ | 0.8 μM | [43] |
| Adenosine/serum | 5’-TTG TTA CCT GGG TTT TTT TTTT-SH-3’ | 20 pM | [50] |
| Parameter | Comparative Characterization |
|---|---|
| Rapidity | High (5–10 min for typical protocols) |
| Labor intensity | Low (one-step assays with minimal manipulations) |
| LoD | Comparable with other immunotechniques (individual for analyte) |
| Working range | Variable (for competitive assay—about an order of magnitude) |
| Reproducibility | High (due to dimensionless registered FP and FA values) |
| Accuracy | Depends on the reaction volume |
| Analyte | Reference Method | R2 | Reference |
|---|---|---|---|
| other immune techniques | |||
| N-terminal kinase 3 | ELISA | 0.9555 | [131] |
| p38a mitogen-activated protein kinase | ELISA | 0.8447 | [131] |
| Brucellosis | ELISA (two formats) | 0.8564/0.9116 | [132] |
| Gentamicin | immunochromatography | 0.976 | [133] |
| other non- immune techniques | |||
| Bisphenol A | HPLC | 0.9636 | [56] |
| Fluoroquinolones | HPLC | 0.9665 | [132] |
| T-2 and HT-2 toxins | UHPLC | 0.953–0.998 depending on matrix | [134] |
| Triazophos | Gas chromatography | 0.9680 | [128] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hendrickson, O.D.; Taranova, N.A.; Zherdev, A.V.; Dzantiev, B.B.; Eremin, S.A. Fluorescence Polarization-Based Bioassays: New Horizons. Sensors 2020, 20, 7132. https://doi.org/10.3390/s20247132
Hendrickson OD, Taranova NA, Zherdev AV, Dzantiev BB, Eremin SA. Fluorescence Polarization-Based Bioassays: New Horizons. Sensors. 2020; 20(24):7132. https://doi.org/10.3390/s20247132
Chicago/Turabian StyleHendrickson, Olga D., Nadezhda A. Taranova, Anatoly V. Zherdev, Boris B. Dzantiev, and Sergei A. Eremin. 2020. "Fluorescence Polarization-Based Bioassays: New Horizons" Sensors 20, no. 24: 7132. https://doi.org/10.3390/s20247132
APA StyleHendrickson, O. D., Taranova, N. A., Zherdev, A. V., Dzantiev, B. B., & Eremin, S. A. (2020). Fluorescence Polarization-Based Bioassays: New Horizons. Sensors, 20(24), 7132. https://doi.org/10.3390/s20247132