An Advanced Statistical Approach Using Weighted Linear Regression in Electroanalytical Method Development for Epinephrine, Uric Acid and Ascorbic Acid Determination




Abstract
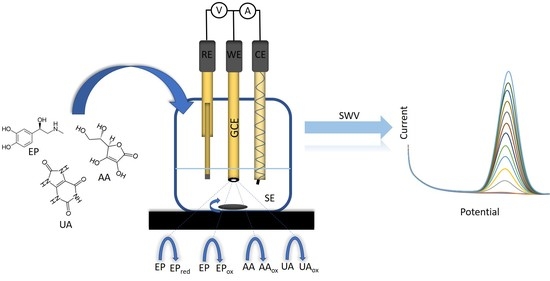
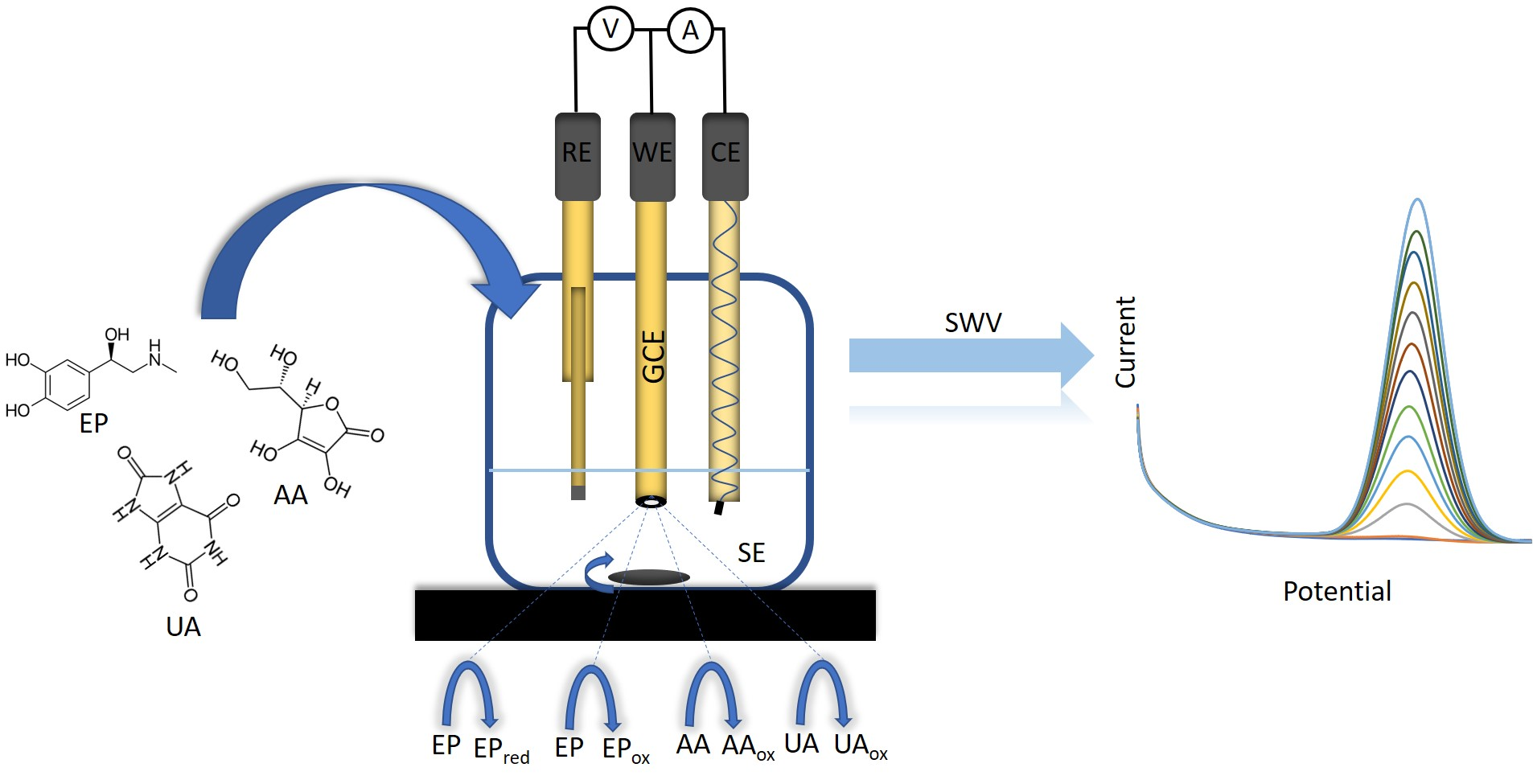
1. Introduction
2. Experimental
2.1. Solutions and Reagents
2.2. Apparatus
2.3. Cyclic Voltammetry
2.4. Square-Wave Voltammetry
2.5. Partial Method Validation
2.6. Real Sample Analysis
3. Results and Discussion
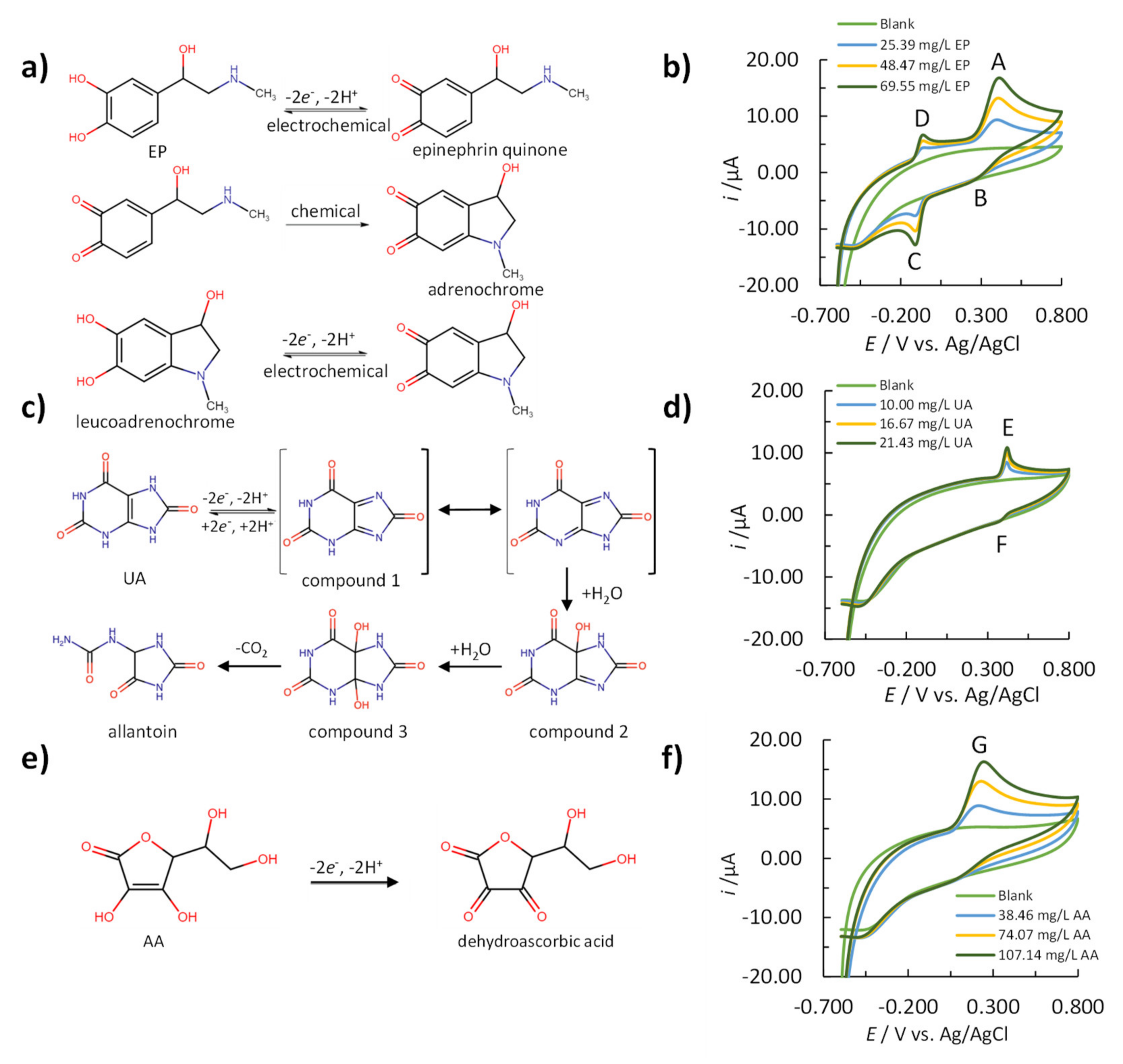
3.1. Electrochemical Reaction Mechanisms of EP, UA, and AA
3.2. Partial Method Validation
3.2.1. LOD and LOQ Determination
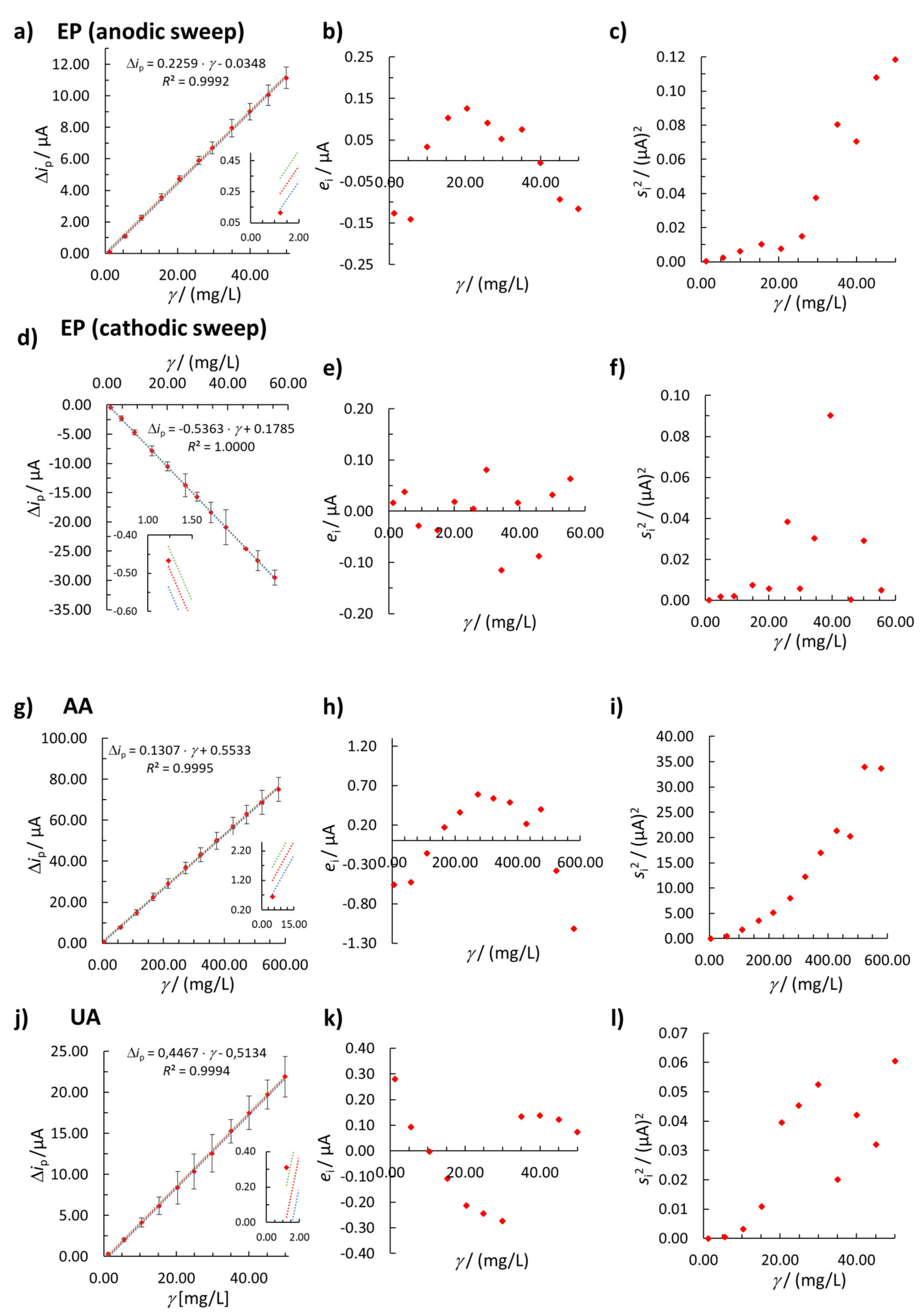
3.2.2. Linearity
3.2.3. Weighted Linear Regression
3.2.4. Accuracy and Precision of the Method
3.3. Real Sample Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ellis, L.; Farrington, D.; Hoskin, A. Handbook of Crime Correlates, 2nd ed.; Academic Press: Cambridge, MA, USA, 2019. [Google Scholar]
- Oyama, M.A. Mechanisms of heart failure. In Textbook of Small Animal Emergency Medicine; Drobatz, K.J., Rozanski, K.H.E., Silverstein, D.C., Eds.; John Wiley and Sons: Hoboken, NJ, USA, 2018; pp. 319–324. [Google Scholar]
- Tipton, M.J.; Harper, A.; Paton, J.F.R.; Costello, J.T. The human ventilatory response to stress: Rate or depth? J. Physiol. 2017, 595, 5729–5752. [Google Scholar] [CrossRef]
- Hoek, T.L.V.; Morrison, L.J.; Shuster, M.; Donnino, M.; Sinz, E.; Lavonas, E.J.; Jeejeebhoy, F.M.; Gabrielli, A. Part 12: Cardiac arrest in special situations. Circulation 2010, 122 (Suppl. S3), S829–S861. [Google Scholar] [CrossRef]
- Vincent, J.-L.; De Backer, D. Circulatory shock. N. Engl. J. Med. 2013, 369, 1726–1734. [Google Scholar] [CrossRef]
- Buleandră, M.; Popa, D.E.; David, I.G.; Ciucu, A.A. A simple and efficient cyclic square wave voltametric method for simultaneous determination of epinephrine and norepinephrine using an activated pencil graphite electrode. Microchem. J. 2020, 160, 105621. [Google Scholar] [CrossRef]
- Freires, A.S.; dos Reis Lima, F.M.; Yotsumoto-Neto, S.; Silva, S.M.; Damos, F.S.; de Cássia Silva Luz, R. Exploiting CdSe/ZnS core-shell photocatalyst modified with cytochrome c for epinephrine determination in drugs utilized in cardiopulmonary resuscitation. Microchem. J. 2018, 139, 18–23. [Google Scholar] [CrossRef]
- Thivya, P.; Wilson, J. Electron rays irradiated polyaniline anchored over bovine serum albumin for simultaneous detection of epinephrine and uric acid. Microchem. J. 2019, 145, 883–891. [Google Scholar] [CrossRef]
- Da Silva, L.V.; Lopes, C.B.; da Silva, W.C.; de Paiva, Y.G.; Silva, F.d.A.d.S.; Lima, P.R.; Kubota, L.T.; Goulart, M.O.F. Electropolymerization of ferulic acid on multi-walled carbon nanotubes modified glassy carbon electrode as a versatile platform for NADH, dopamine and epinephrine separate detection. Microchem. J. 2017, 133, 460–467. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, B.; Xiong, H.; Wen, W.; Cheng, N. A radiometric fluorometric epinephrine and norepinephrine assay based on carbon dot and CdTe quantum dots nanocomposites. Microchem. J. 2019, 146, 66–72. [Google Scholar] [CrossRef]
- Maiuolo, J.; Oppedisano, F.; Gratteri, S.; Muscoli, C.; Mollace, V. Regulation of uric acid metabolism and excretion. Int. J. Cardiol. 2016, 213, 8–14. [Google Scholar] [CrossRef]
- Martillo, M.A.; Nazzal, L.; Crittenden, D.B. The crystallization of monosodium urate. Curr. Rheumatol. Rep. 2013, 16, 400. [Google Scholar] [CrossRef]
- Amjadi, M.; Hallaj, T.; Nasirloo, E. In situ formation of Ag/Au nanorods as a platform to design a non-aggregation colorimetric assay for uric acid detection in biological fluids. Microchem. J. 2020, 154, 104642. [Google Scholar] [CrossRef]
- Hallaj, T.; Amjadi, M.; Mirbirang, F. S, N-doped carbon quantum dots enhanced Luminol-Mn(IV) chemiluminescence reaction for detection of uric acid in biological fluids. Microchem. J. 2020, 156, 104841. [Google Scholar] [CrossRef]
- Krishnan, R.G.; Rejithamol, R.; Saraswathyamma, B. Non-enzymatic electrochemical sensor for the simultaneous determination of adenosine, adenine and uric acid in whole blood and urine. Microchem. J. 2020, 155, 104745. [Google Scholar] [CrossRef]
- Soleh, A.; Kanatharana, P.; Thavarungkul, P.; Limbut, W. Novel electrochemical sensor using a dual-working electrode system for the simultaneous determination of glucose, uric acid and dopamine. Microchem. J. 2020, 153, 104379. [Google Scholar] [CrossRef]
- May, J.M. Vitamin C transport and its role in the central nervous system. In Water Soluble Vitamins: Clinical Research and Future Application; Stanger, O., Ed.; Springer: Dordrecht, The Netherlands, 2012; pp. 85–103. [Google Scholar]
- Abdelwahab, A.A.; Elseman, A.M.; Alotaibi, N.F.; Nassar, A.M. Simultaneous voltammetric determination of ascorbic acid, dopamine, acetaminophen and tryptophan based on hybrid trimetallic nanoparticles-capped electropretreated graphene. Microchem. J. 2020, 156, 104927. [Google Scholar] [CrossRef]
- Musarraf Hussain, M.; Asiri, A.M.; Rahman, M.M. Non-enzymatic simultaneous detection of acetylcholine and ascorbic acid using ZnO·CuO nanoleaves: Real sample analysis. Microchem. J. 2020, 159, 105534. [Google Scholar] [CrossRef]
- Saleh Mohammadnia, M.; Marzi Khosrowshahi, E.; Naghian, E.; Homayoun Keihan, A.; Sohouli, E.; Plonska-Brzezinska, M.E.; Ali Sobhani, N.; Rahimi-Nasrabadi, M.; Ahmadi, F. Application of carbon nanoonion-NiMoO4-MnWO4 nanocomposite for modification of glassy carbon electrode: Electrochemical determination of ascorbic acid. Microchem. J. 2020, 159, 105470. [Google Scholar] [CrossRef]
- He, M.; Wang, C.; Wei, Y. Selective enrichment and determination of monoamine neurotransmitters by CU(II) immobilized magnetic solid phase extraction coupled with high-performance liquid chromatography-fluorescence detection. Talanta 2016, 147, 437–444. [Google Scholar] [CrossRef]
- Aragon, A.; Legradi, J.; Ballesteros-Gómez, A.; Legler, J.; van Velzen, M.; de Boer, J.; Leonards, P. Determination of monoamine neurotransmitters in zebrafish (Danio rerio) by gas chromatography coupled to mass spectrometry with a two-step derivatization. Anal. Bioanal. Chem. 2017, 409, 2931–2939. [Google Scholar] [CrossRef]
- Vladislavić, N.; Rončević Škugor, I.; Buzuk, M.; Buljac, M.; Drventić, I. Electrochemical/chemical synthesis of hydroxyapatite on glassy carbon electrode for electroanalytical determination of cysteine. J. Solid State Electrochem. 2020, 1–17. [Google Scholar] [CrossRef]
- Vladislavić, N.; Rončević Škugor, I.; Buljac, M.; Brinić, S.; Krivič, D.; Buzuk, M. Electroanalytical determination of cysteine using the electrodes based on ternary silver-copper sulfides. Sensors 2018, 18, 3753. [Google Scholar] [CrossRef] [PubMed]
- Moosavi, M.; Ghassabian, S. Linearity of calibration curves for analytical methods: A review of criteria for assessment of method reliability. In Calibration and Validation of Analytical Methods—A Sampling of Current Approaches; IntechOpen Limited: London, UK, 2018. [Google Scholar]
- Sonawane, S.S.; Chhajed, S.S.; Attar, S.S.; Kshirsagar, S.J. An approach to select linear regression model in bioanalytical method validation. J. Anal. Sci. Technol. 2019, 10, 1. [Google Scholar] [CrossRef]
- Almeida, A.M.; Castel-Branco, M.M.; Falcão, A.C. Linear regression for calibration lines revisited: Weighting schemes for bioanalytical methods. J. Chromatogr. B 2002, 774, 215–222. [Google Scholar] [CrossRef]
- Vignaduzzo, S.E.; Maggio, R.M.; Olivieri, A.C. Why should the pharmaceutical industry claim for the implementation of second order chemometric models—A critical review. J. Pharm. Biomed. Anal. 2020, 179, 112965. [Google Scholar] [CrossRef]
- Alcaraz, M.R.; Culzoni, M.J.; Ibañez, G.A.; Lozano, V.A.; Olivieri, A.C. On second-order calibration based on multivariate curve resolution in the presence of highly overlapped profiles. Anal. Chim. Acta 2020, 1096, 53–60. [Google Scholar] [CrossRef]
- Pellegrino Vidal, R.B.; Olivieri, A.C. Contribution to second-order calibration based on multivariate curve resolution with and without previous chromatographic synchronization. Anal. Chim. Acta 2019, 1078, 8–15. [Google Scholar] [CrossRef]
- Finšgar, M.; Kovačec, L. Copper-bismuth-film in situ electrodes for heavy metal detection. Microchem. J. 2020, 154, 104635. [Google Scholar] [CrossRef]
- Finšgar, M.; Xhanari, K.; Otmačić Ćurković, H. Cyclic voltammetry as an electroanalytical tool for analysing the reaction mechanisms of copper in chloride solution containing different azole compounds. Curr. Anal. Chem. 2020, 16, 465–474. [Google Scholar] [CrossRef]
- Massart, D.L.; Vandeginste, B.G.M.; Buydens, L.M.; Jong, S.D.; Lewi, P.J.; Smeyers-Verbeke, J. Handbook of Chemometrics and Qualimetrics: Part, A; Elsevier: Amsterdam, The Netherlands, 1997. [Google Scholar]
- Shaikshavali, P.; Madhusudana Reddy, T.; Venu Gopal, T.; Venkataprasad, G.; Kotakadi, V.S.; Palakollu, V.N.; Karpoormath, R. A simple sonochemical assisted synthesis of nanocomposite (ZnO/MWCNTs) for electrochemical sensing of Epinephrine in human serum and pharmaceutical formulation. Colloids Surf. A 2020, 584, 124038. [Google Scholar] [CrossRef]
- Crosnier de Lassichere, C.; Latapie, L.; Evrard, D.; Gros, P. New insight into the EC’ mechanism of uric acid regeneration in the presence of ascorbic acid on a poly(3,4-ethylenedioxithiophene) modified gold electrode. Electroanalysis 2018, 30, 1653–1658. [Google Scholar] [CrossRef]
- Chairam, S.; Sriraksa, W.; Amatatongchai, M.; Somsook, E. Electrocatalytic oxidation of ascorbic acid using a poly(aniline-co-m-ferrocenylaniline) modified glassy carbon electrode. Sensors 2011, 11, 10166–10179. [Google Scholar] [CrossRef] [PubMed]
- Kongkiatpaiboon, S.; Duangdee, N.; Chewchinda, S.; Poachanukoon, O.; Amnuaypattanapon, K. Development and validation of stability indicating HPLC method for determination of adrenaline tartrate. J. King Saud Univ. Sci. 2019, 31, 48–51. [Google Scholar] [CrossRef]
- Van de Velde, F.; Pirovani, M.E.; Cámara, M.S.; Güemes, D.R.; Bernardi, C.M.d.H. Optimization and validation of a UV–HPLC method for vitamin C determination in strawberries (Fragaria ananassa Duch.), using experimental designs. Food Anal. Methods 2012, 5, 1097–1104. [Google Scholar] [CrossRef]
- Wijemanne, N.; Soysa, P.; Wijesundara, S.; Perera, H. Development and validation of a simple highperformance liquid chromatography/UV Method for simultaneous determination of urinary uric acid, hypoxanthine, and creatinine in human urine. Int. J. Anal. Chem. 2018, 2018, 1647923. [Google Scholar] [CrossRef] [PubMed]
- Rozet, E.; Marini, R.D.; Ziemons, E.; Boulanger, B.; Hubert, P. Advances in validation, risk and uncertainty assessment of bioanalytical methods. J. Pharm. Biomed. Anal. 2011, 55, 848–858. [Google Scholar] [CrossRef]
- Desharnais, B.; Camirand-Lemyre, F.; Mireault, P.; Skinner, C.D. Procedure for the selection and validation of a calibration model I—Description and application. J. Anal. Toxicol. 2017, 41, 261–268. [Google Scholar] [CrossRef]
- Goldsmith, J.G. Modern Analytical Chemistry, 1st Edition (Harvey, David). J. Chem. Educ. 2000, 77, 705. [Google Scholar] [CrossRef]
- da Silva, C.P.; Emídio, E.S.; de Marchi, M.R.R. Method validation using weighted linear regression models for quantification of UV filters in water samples. Talanta 2015, 131, 221–227. [Google Scholar] [CrossRef]
- Food and Drug Administration (FDA). Bioanalytical Method Validation: Guidance for Industry; Food and Drug Administration: Hampton, VA, USA, 2018. [Google Scholar]
- Shi, K.; Shiu, K.-K. Determination of uric acid at electrochemically activated glassy carbon electrode. Electroanalysis 2001, 13, 1319–1325. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
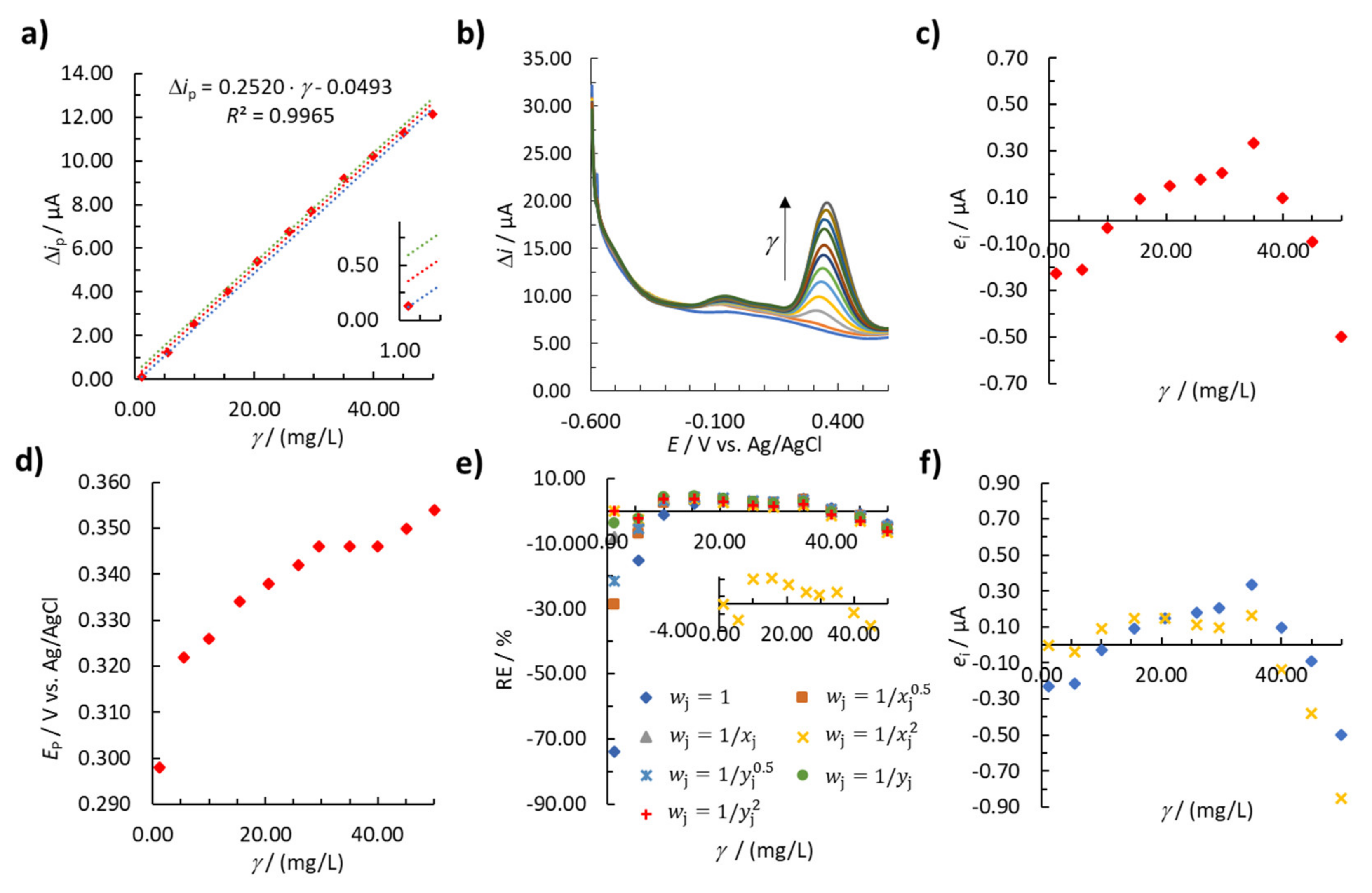{kind=link}
| LOD/(mg/L) | S/N | LOQ/(mg/L) | S/N | |
|---|---|---|---|---|
| EP (anodic sweep) | 0.62 | 4.78 | 1.10 | 11.48 |
| EP (cathodic sweep) | 0.25 | 3.99 | 0.49 | 10.23 |
| AA | 0.50 | 5.72 | 1.98 | 11.67 |
| UA | 0.62 | 3.56 | 1.22 | 10.85 |
| Analyte | EP (Anodic Sweep) | EP (Cathodic Sweep) | AA | UA |
|---|---|---|---|---|
| Linear concentration range/(mg/L) | 1.22–49.97 | 1.23–55.52 | 4.98–578.95 | 1.22–50.00 |
| b1 | 0.2259 | −0.5363 | 0.1307 | 0.4467 |
| b0 | 0.0348 | 0.1785 | 0.5533 | 0.5134 |
| QC/% | 1.78 | 0.40 | 1.39 | 1.70 |
| R2 | 0.9992 | 1.0000 | 0.9995 | 0.9994 |
| Normality | ||||
| Q-Q plot | passed | passed | passed | passed |
| K-S test | passed | passed | passed | passed |
| Homoscedasticity | ||||
| Residual plot | not random | random | not random | not random |
| Hartley’s test | not passed | not passed | not passed | not passed |
| Bartlett’s test | not passed | not passed | not passed | not passed |
| Cochran’s C test | passed | not passed | passed | passed |
| Model No. for (Anodic Sweep) | b1 | b0 | R2 | ∑|RE|/% | ||
|---|---|---|---|---|---|---|
| 1 (unweighted regression) | 1 | 0.2520 | 0.0493 | 0.9965 | 110.57 | |
| weighted regression models | 2 | 0.2570 | −0.0941 | 0.9977 | 60.99 | |
| 3 | 0.2601 | −0.1621 | 0.9984 | 38.63 | ||
| 4 | 0.2638 | −0.1919 | 0.9988 | 28.16 | ||
| 5 | 0.2573 | −0.1168 | 0.9979 | 54.34 | ||
| 6 | 0.2603 | −0.1761 | 0.9985 | 34.46 | ||
| 7 | 0.2633 | −0.1917 | 0.9988 | 28.62 |
| Analyte | Model No. | b1 | b0 | R2 | ∑|RE|/% | |
|---|---|---|---|---|---|---|
| EP (cathodic sweep) | 1 | 1 | −0.4797 | 0.1153 | 0.9994 | 12.18 |
| 7 | −0.4792 | 0.1084 | 0.9999 | 11.05 | ||
| AA | 1 | 1 | 0.1367 | 1.1455 | 0.9988 | 213.62 |
| 7 | 0.1425 | −0.1470 | 0.9993 | 25.03 | ||
| UA | 1 | 1 | 0.4373 | −0.3546 | 0.9999 | 32.90 |
| 4 | 0.4282 | −0.2078 | 0.9995 | 17.34 |
| γ /(mg/L) (Spiked) | γ /(mg/L) (Determined) | Recovery/% | RSD/% | |
|---|---|---|---|---|
| EP (anodic sweep) | 4.54 | 4.93 | 108.56 | 0.08 |
| 27.76 | 27.60 | 99.43 | 5.23 | |
| 47.59 | 45.99 | 96.64 | 13.64 | |
| EP (cathodic sweep) | 2.44 | 2.37 | 97.30 | 3.02 |
| 27.26 | 26.21 | 96.17 | 2.80 | |
| 52.91 | 50.56 | 95.55 | 0.57 | |
| AA | 24.39 | 21.26 | 87.16 | 3.15 |
| 298.25 | 290.48 | 97.39 | 1.27 | |
| 574.47 | 579.31 | 100.84 | 2.88 | |
| UA | 2.38 | 1.87 | 78.58 | 5.54 |
| 25.00 | 23.14 | 92.57 | 3.80 | |
| 47.96 | 41.09 | 85.68 | 6.55 |
| aγ/(mg/L) (Initially Found) | bγ/(mg/L) (Spiked) | cγ/(mg/L) (Found) | Recovery/% | RSD/% | Content of the Sample/(mg/L) | |
|---|---|---|---|---|---|---|
| EP (anodic) | 12.50 | 10.98 | 24.09 | 105.50 | 1.83 | 483.67 |
| EP (cathodic) | 6.68 | 11.59 | 17.72 | 95.21 | 0.85 | 452.08 |
| AA | 200.44 | 215.69 | 423.08 | 103.22 | 2.86 | 255.57 |
| UA | 3.46 | 25.00 | 31.77 | 113.23 | 7.98 | 345.41 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Majer, D.; Mastnak, T.; Finšgar, M. An Advanced Statistical Approach Using Weighted Linear Regression in Electroanalytical Method Development for Epinephrine, Uric Acid and Ascorbic Acid Determination. Sensors 2020, 20, 7056. https://doi.org/10.3390/s20247056
Majer D, Mastnak T, Finšgar M. An Advanced Statistical Approach Using Weighted Linear Regression in Electroanalytical Method Development for Epinephrine, Uric Acid and Ascorbic Acid Determination. Sensors. 2020; 20(24):7056. https://doi.org/10.3390/s20247056
Chicago/Turabian StyleMajer, David, Tinkara Mastnak, and Matjaž Finšgar. 2020. "An Advanced Statistical Approach Using Weighted Linear Regression in Electroanalytical Method Development for Epinephrine, Uric Acid and Ascorbic Acid Determination" Sensors 20, no. 24: 7056. https://doi.org/10.3390/s20247056
APA StyleMajer, D., Mastnak, T., & Finšgar, M. (2020). An Advanced Statistical Approach Using Weighted Linear Regression in Electroanalytical Method Development for Epinephrine, Uric Acid and Ascorbic Acid Determination. Sensors, 20(24), 7056. https://doi.org/10.3390/s20247056