Broadband Time-Resolved Absorption and Dispersion Spectroscopy of Methane and Ethane in a Plasma Using a Mid-Infrared Dual-Comb Spectrometer

 , , and
, , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Absorption and Dispersion Spectroscopy
- (i)
- The intensity of dispersion lines is always proportional to the amount of gas species, whereas absorption lines can saturate at strong absorptions and high concentrations. Therefore, phase spectroscopy provides a larger dynamic range to measure highly absorbing samples.
- (ii)
- Dispersion spectra can be immune to intensity fluctuations, as the phase depends only on the refractive index and not on the laser intensity [49].
2.2. Experimental Setup
2.2.1. Mid-Infrared Dual-Comb Source
2.2.2. Dual-Comb Spectrometer Design for Absorption/Dispersion Spectroscopy
2.2.3. Discharge System and Gas Mixtures
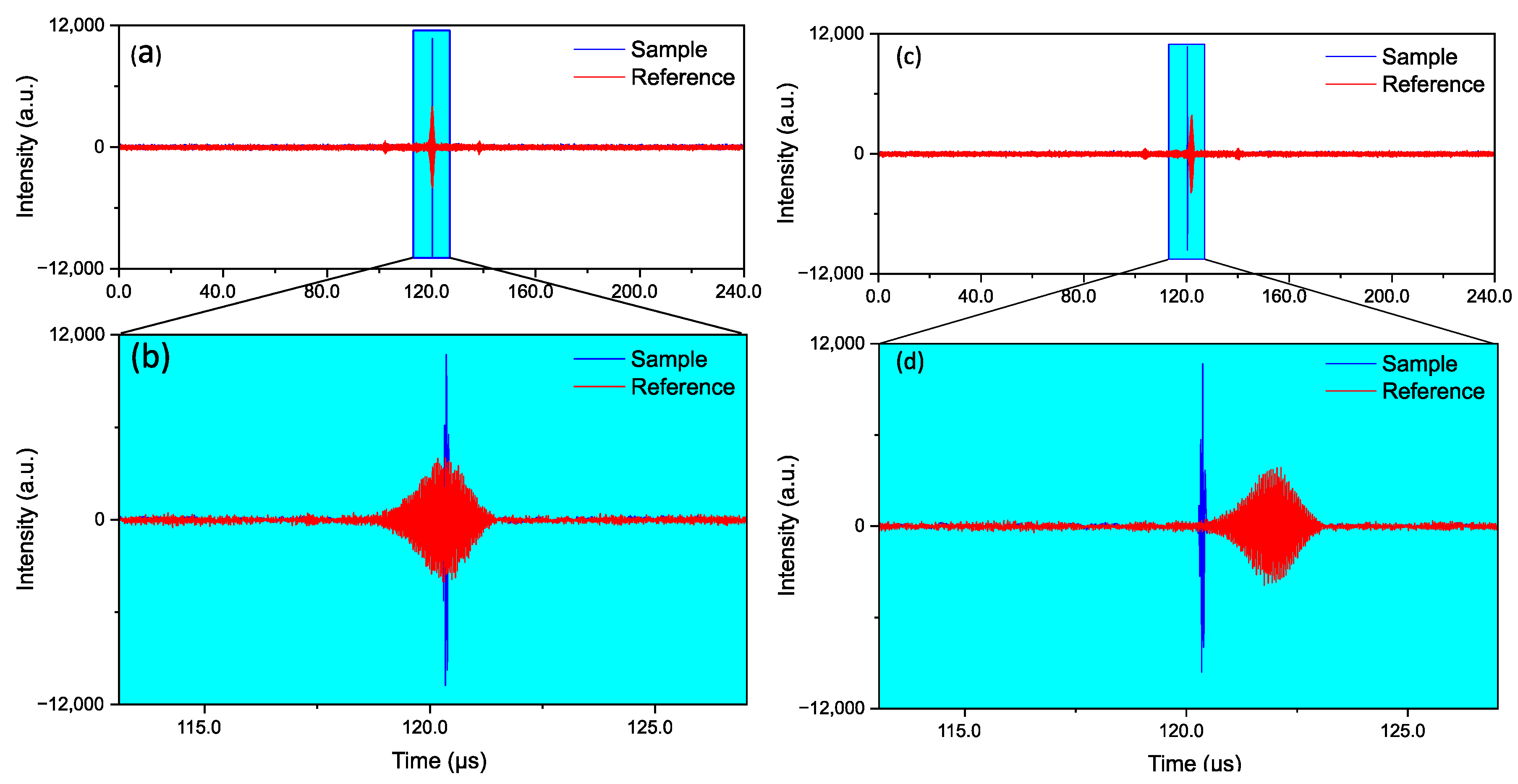
2.2.4. Data Acquisition and Synchronization
2.2.5. Data Processing
3. Results and Discussion
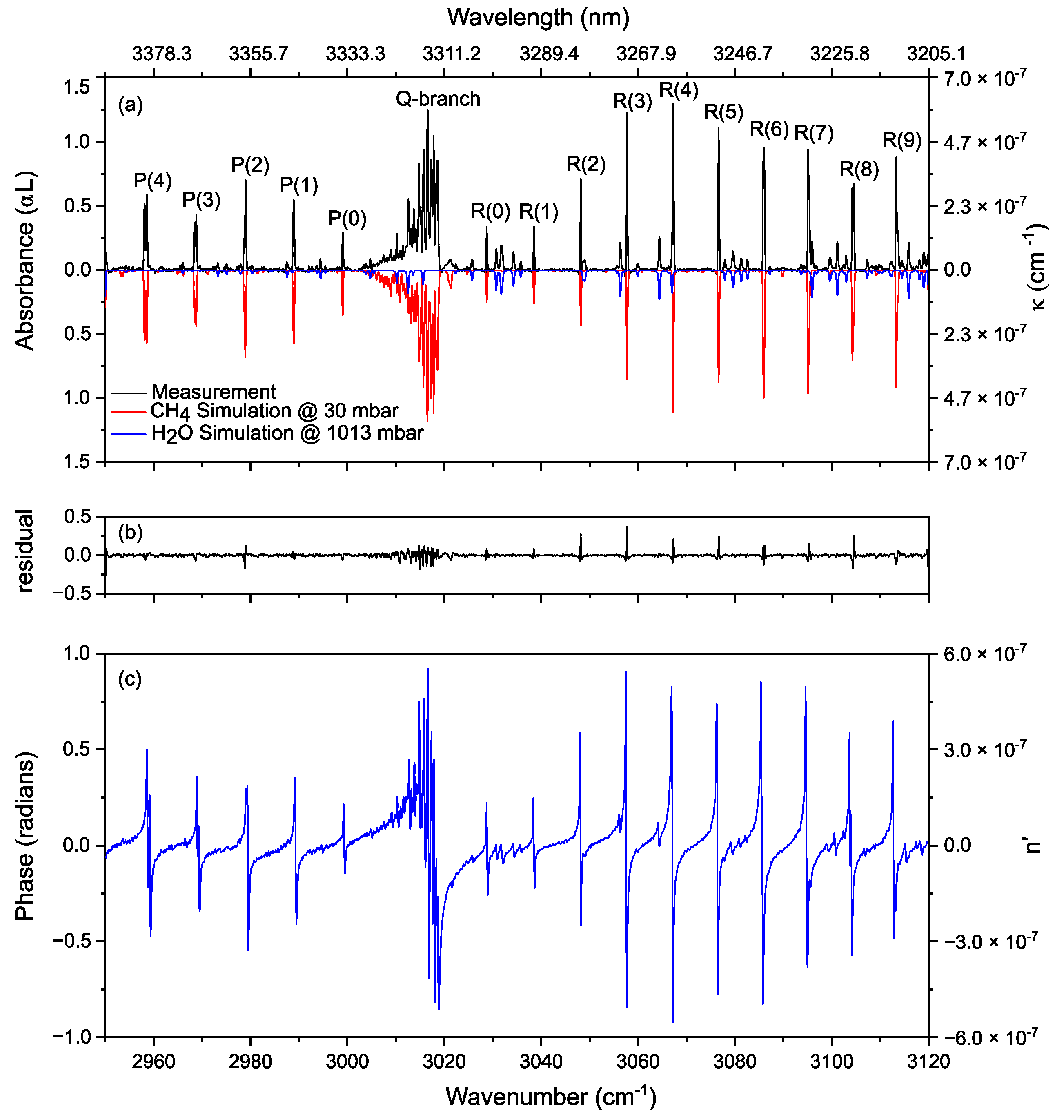
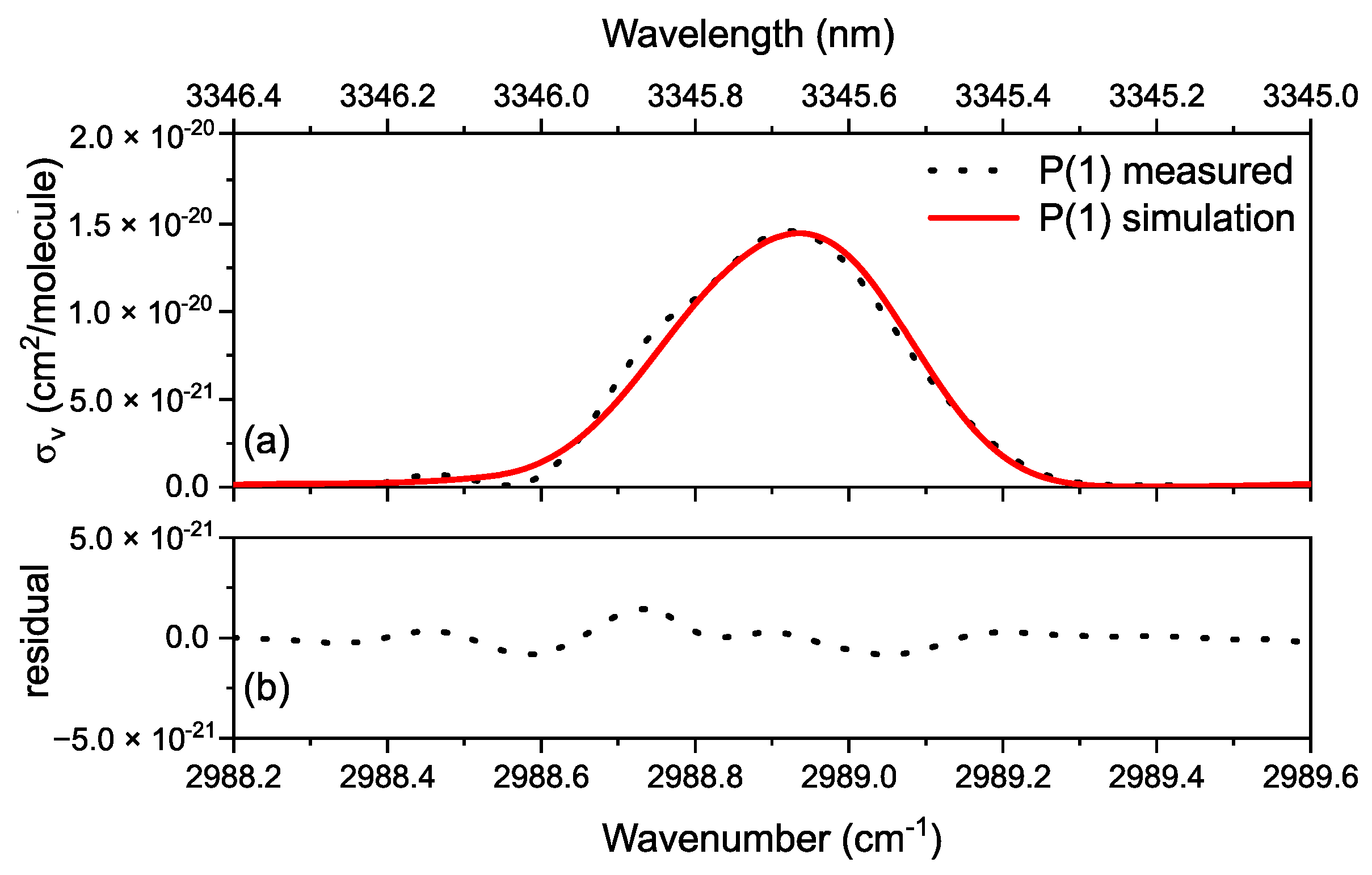
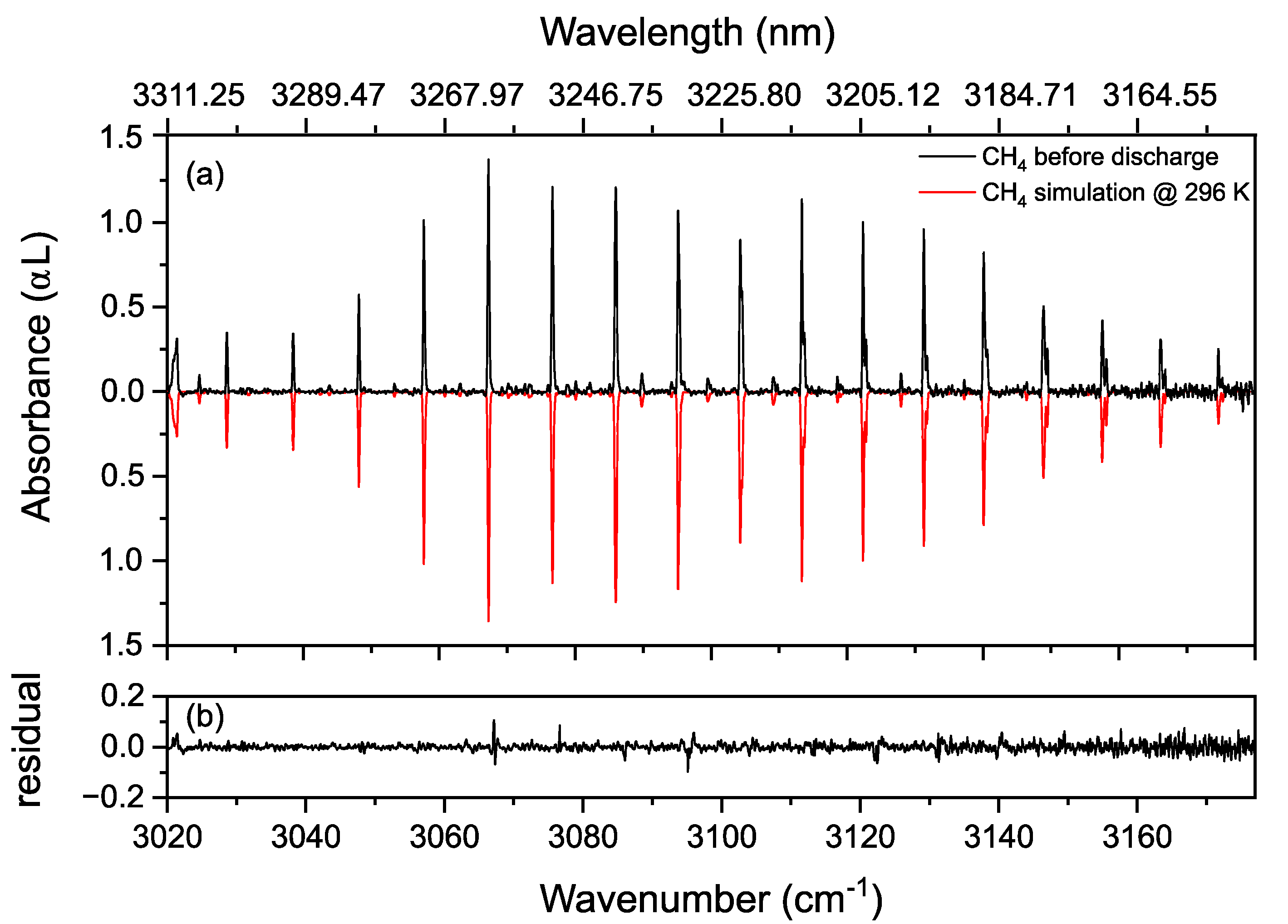
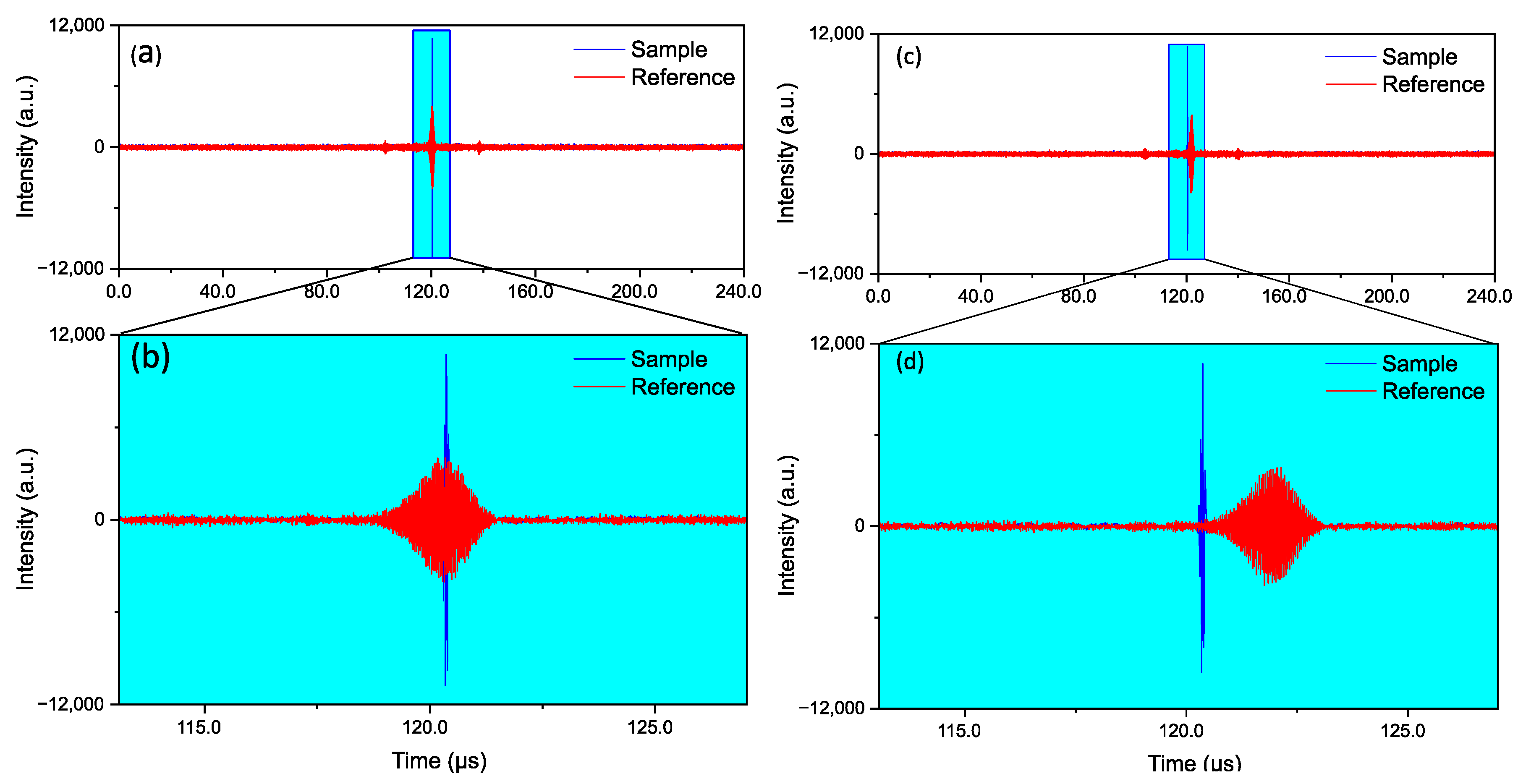
3.1. Proof of Principle
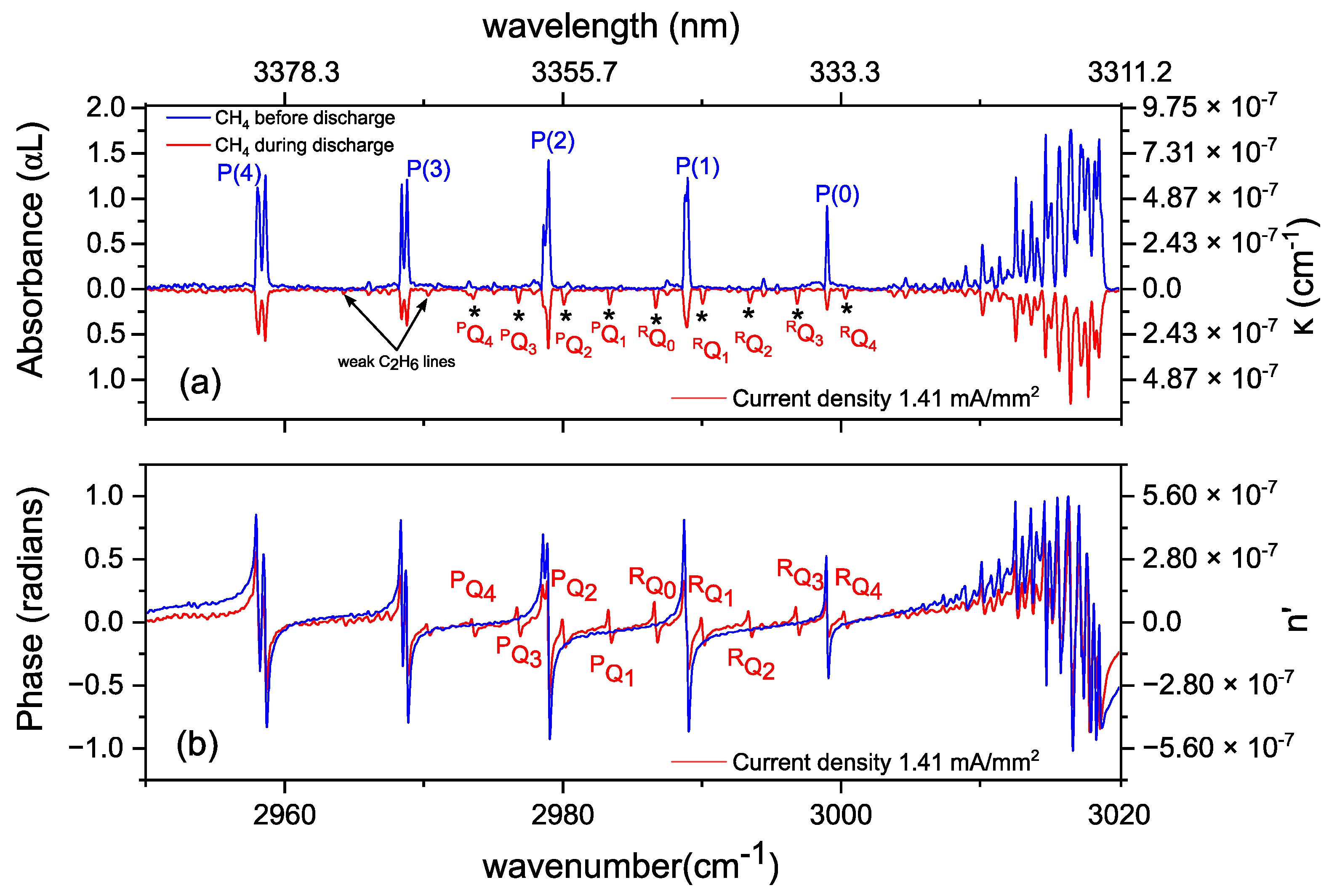
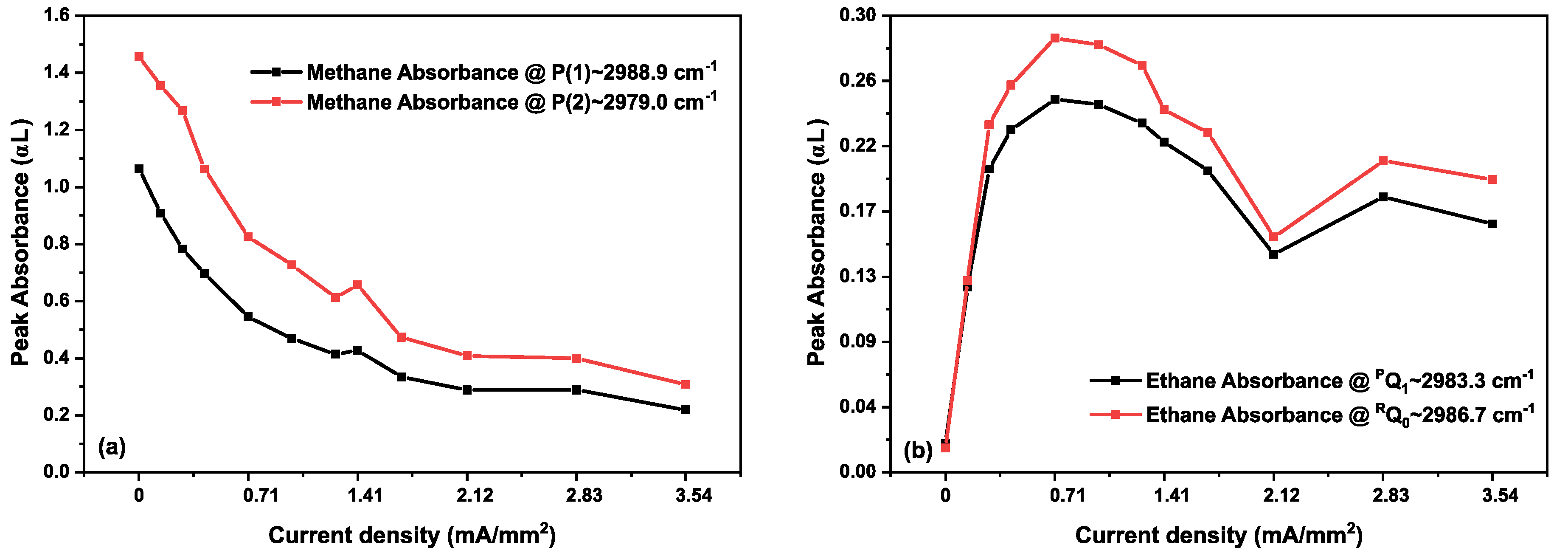
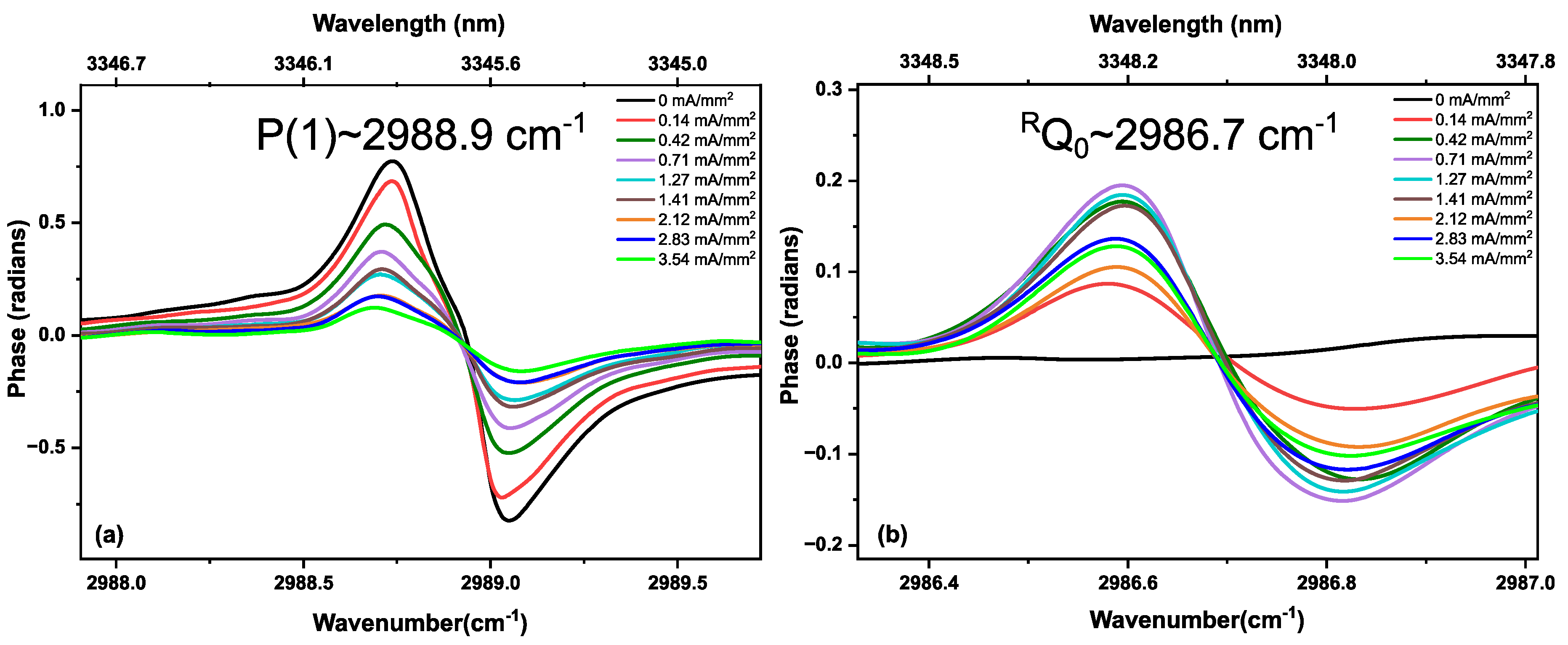
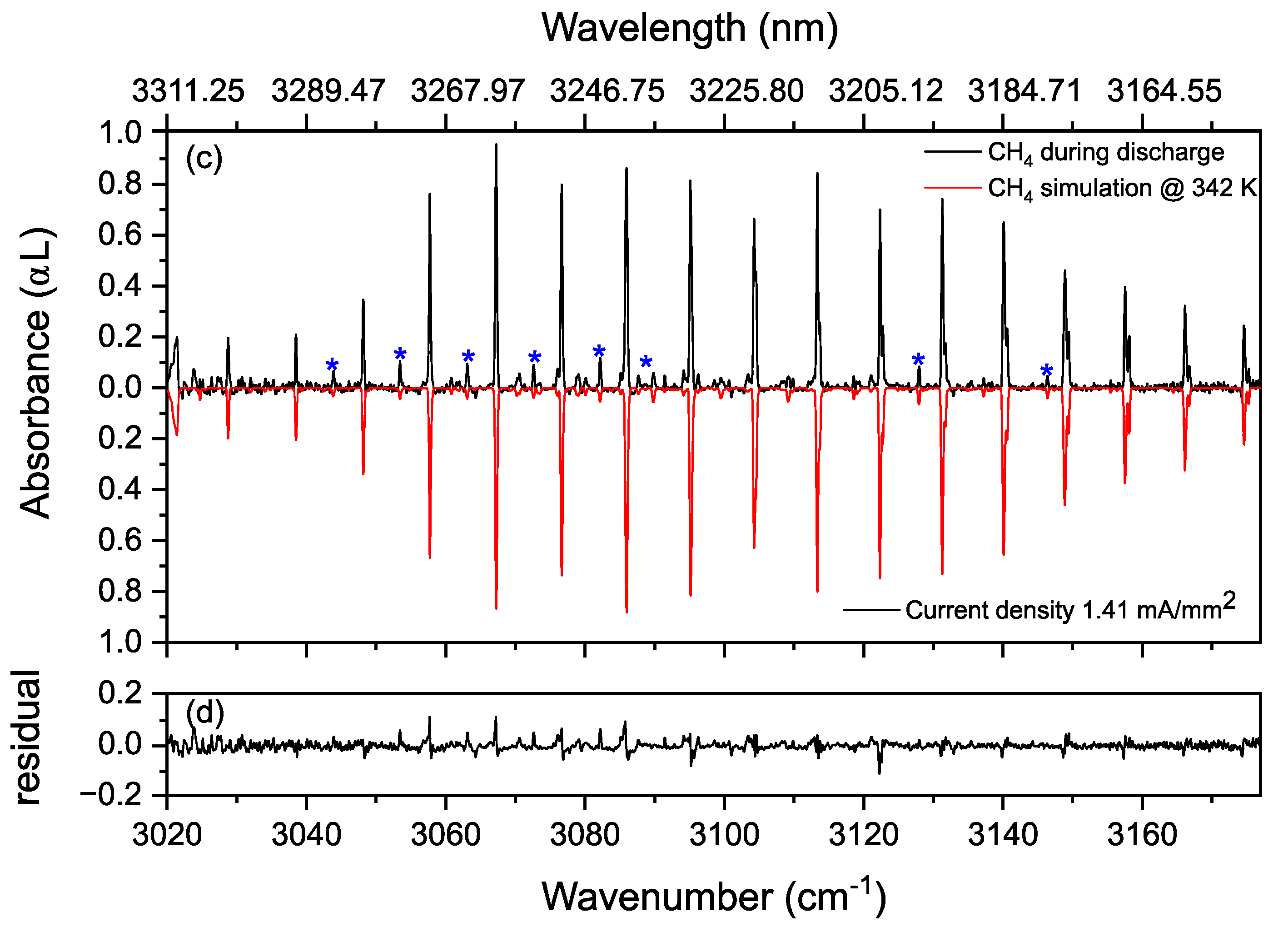
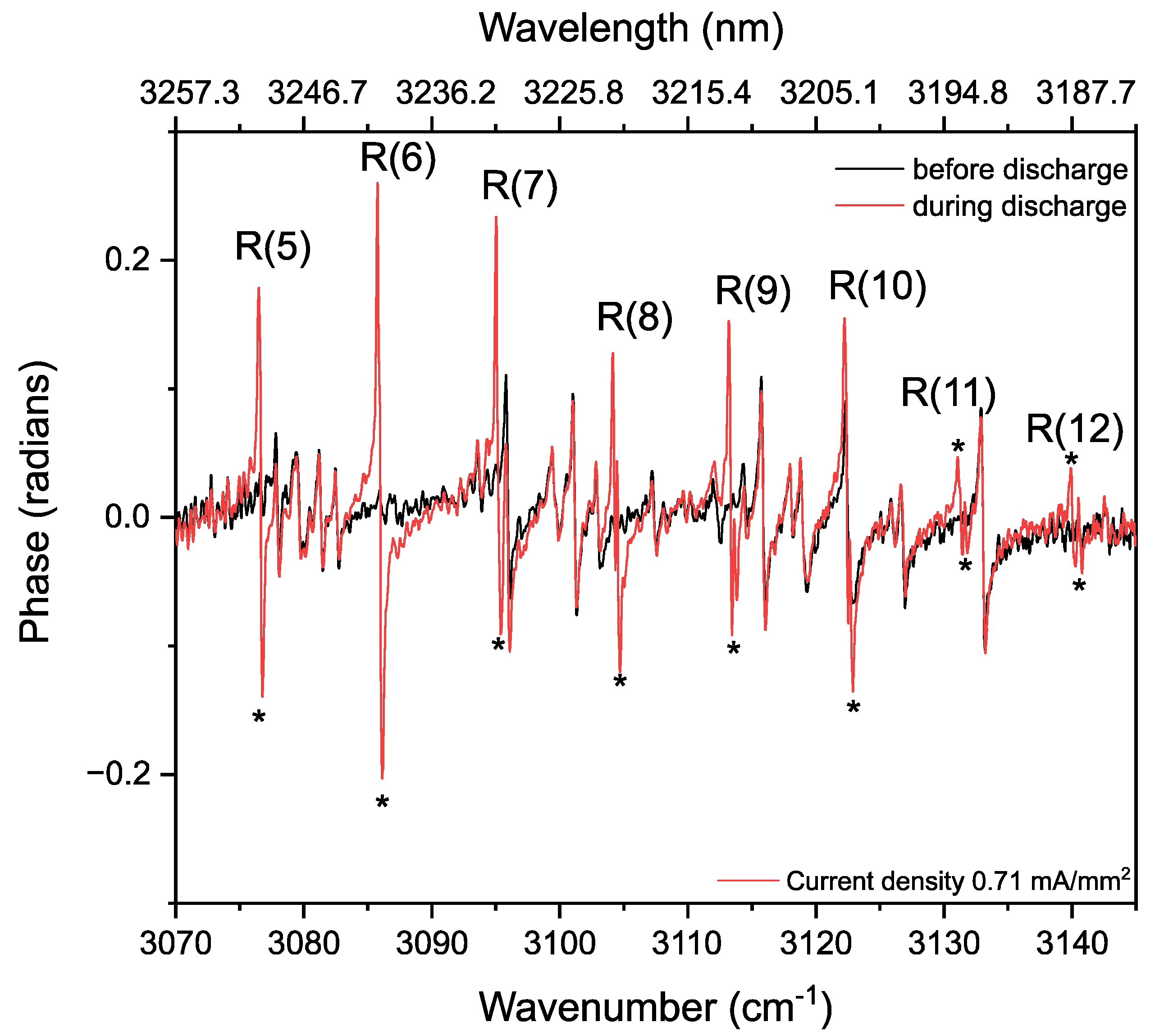
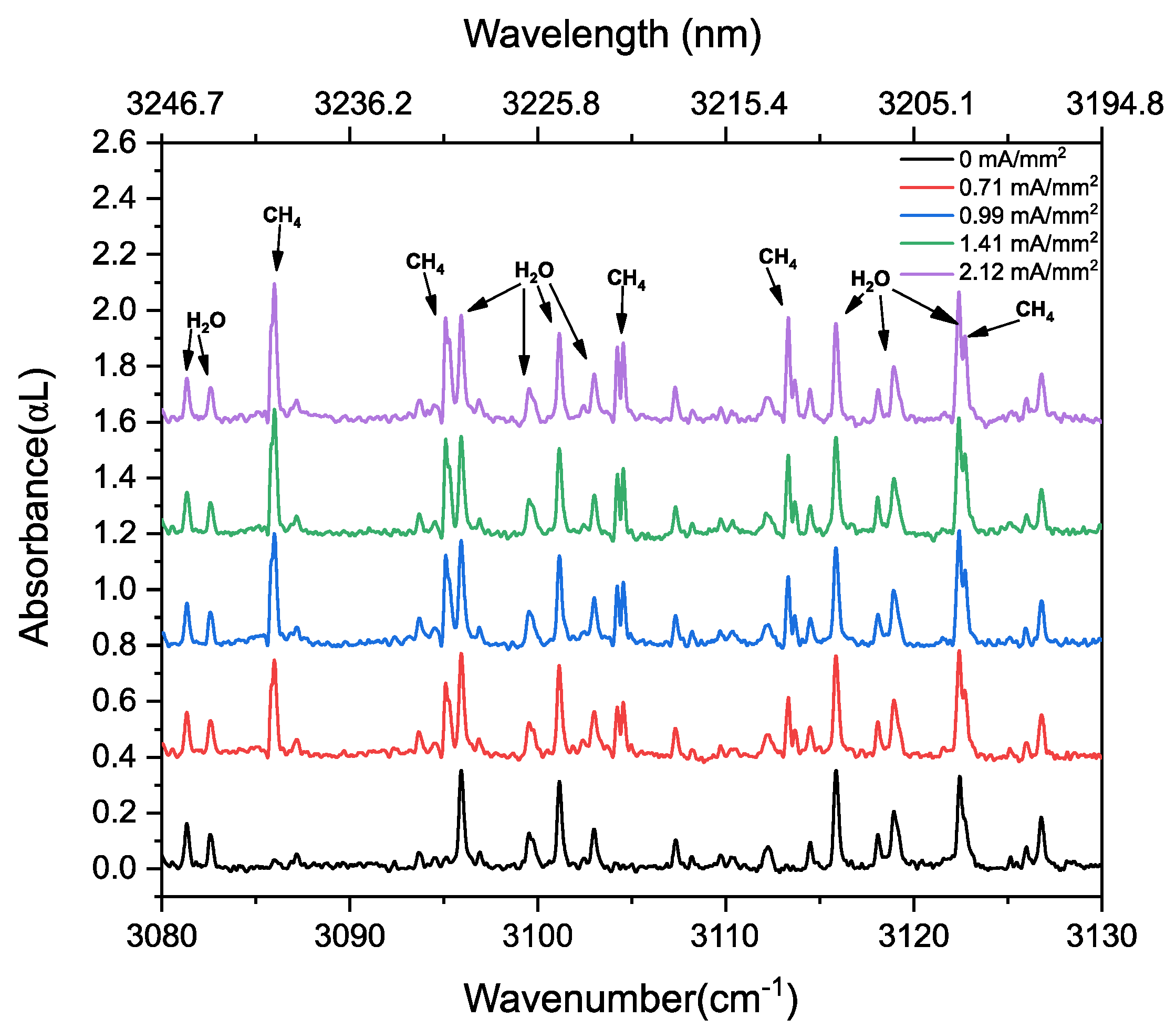
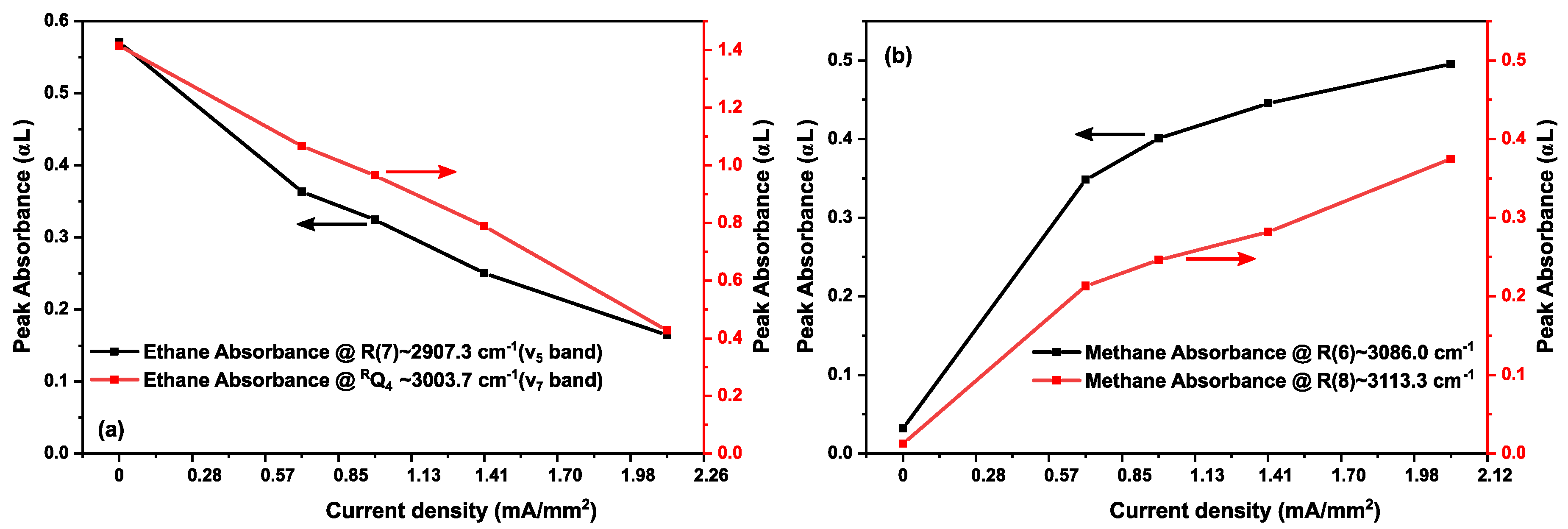
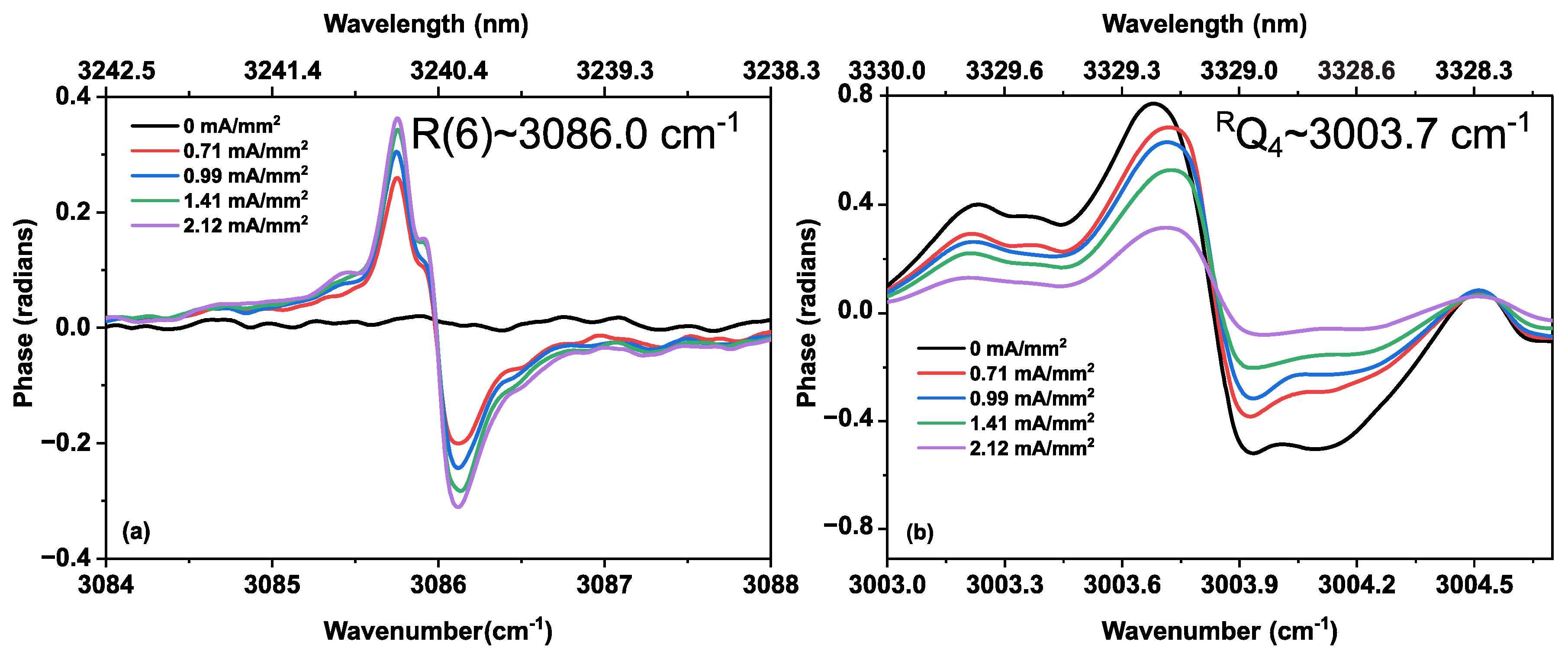
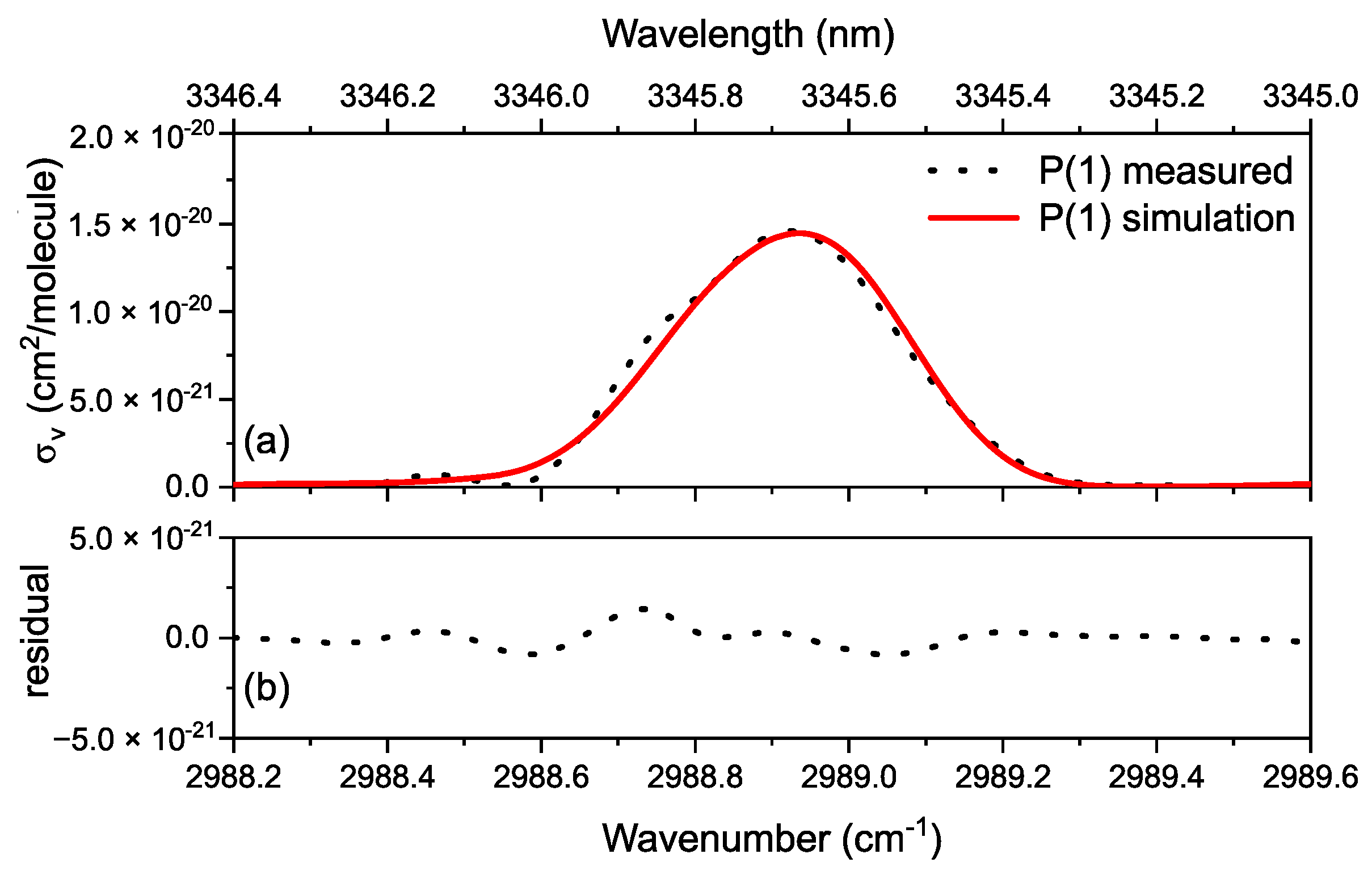
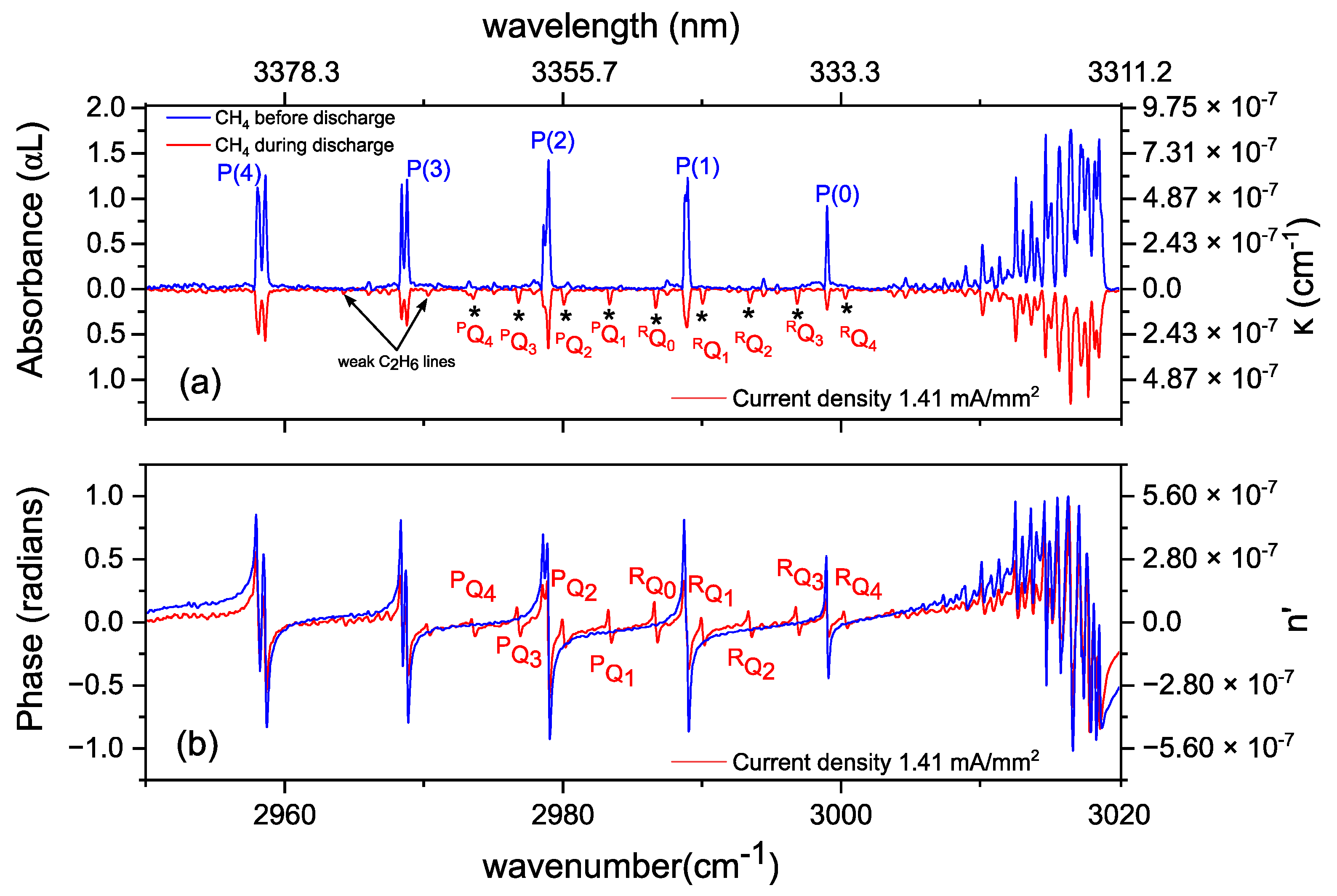
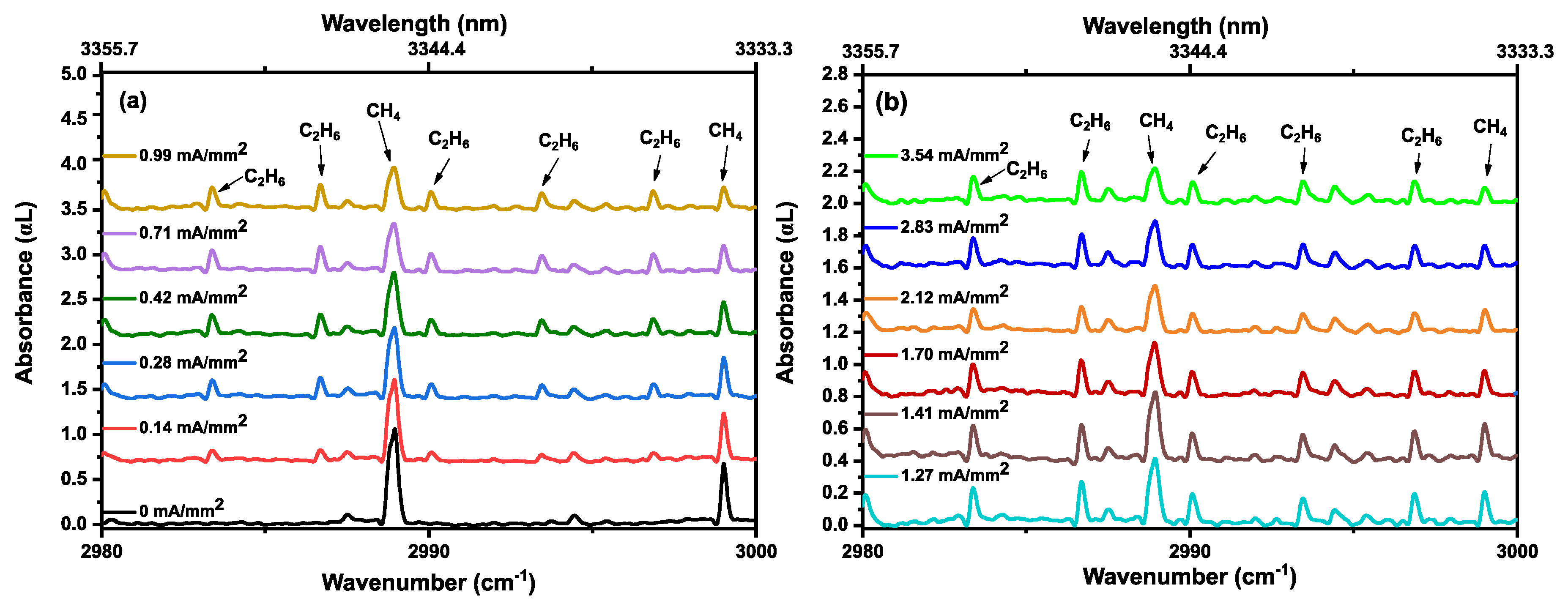
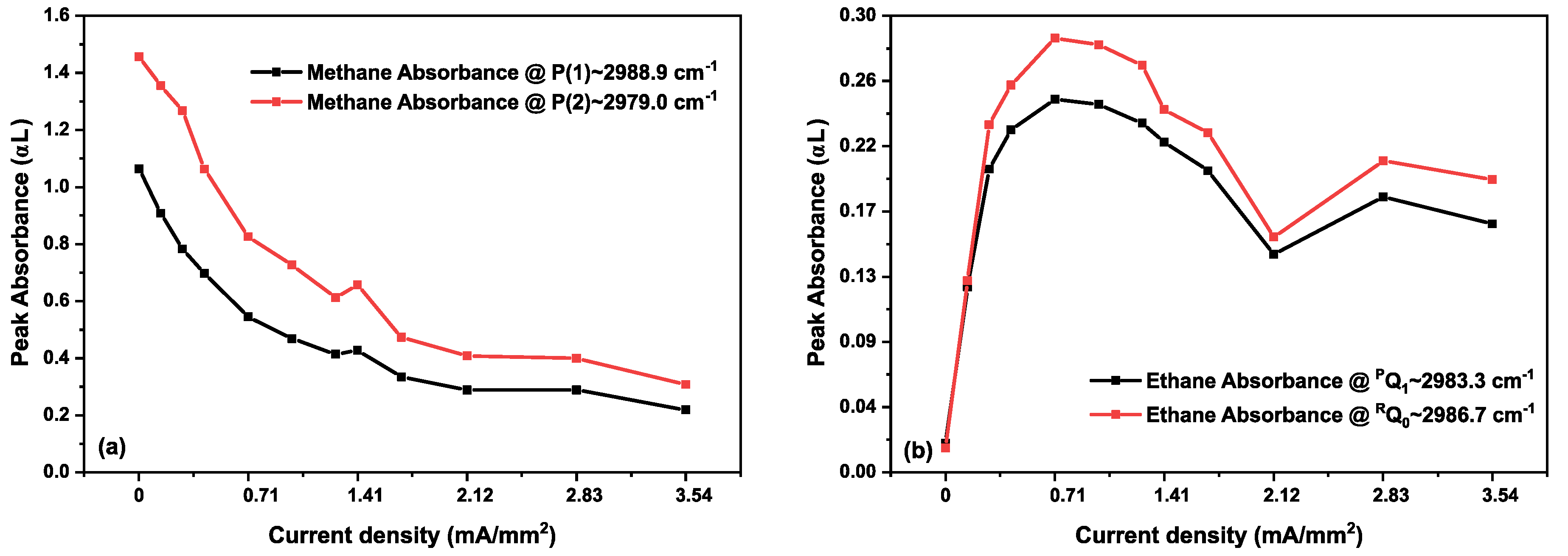
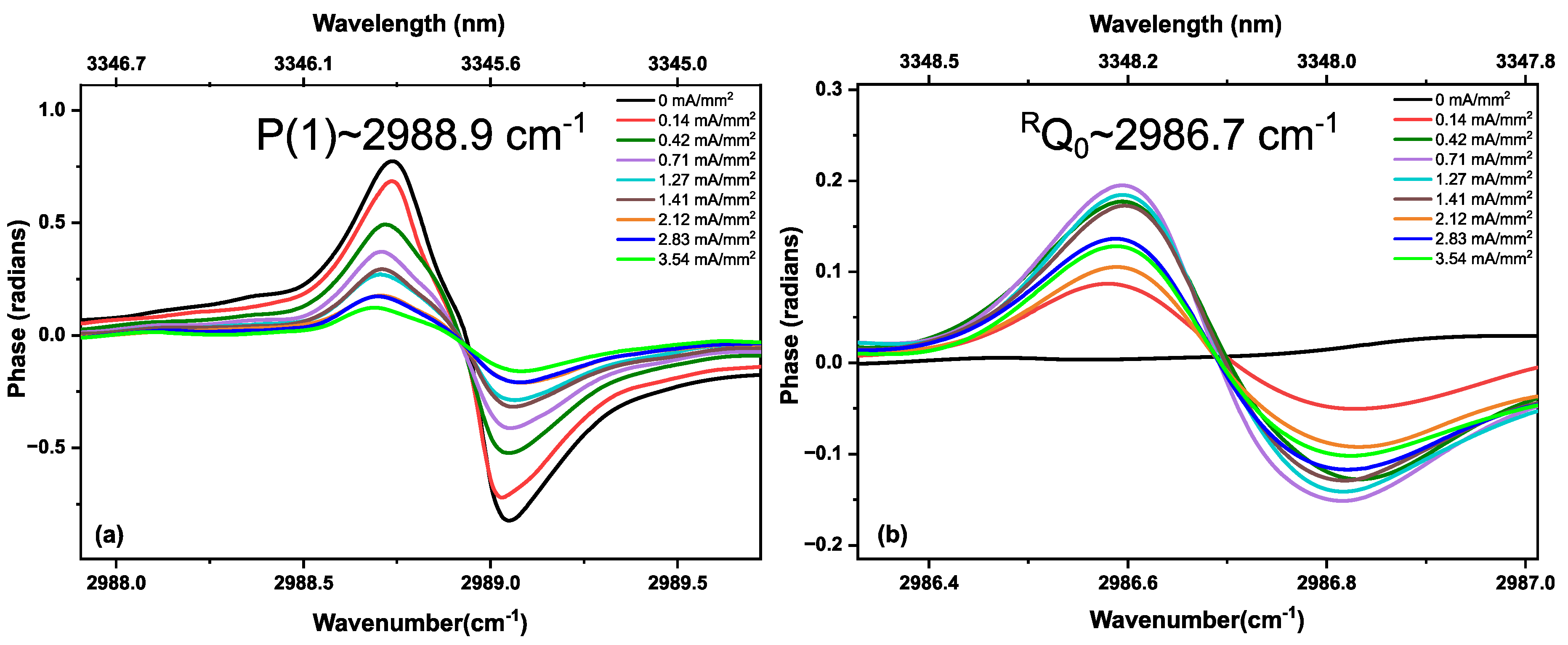
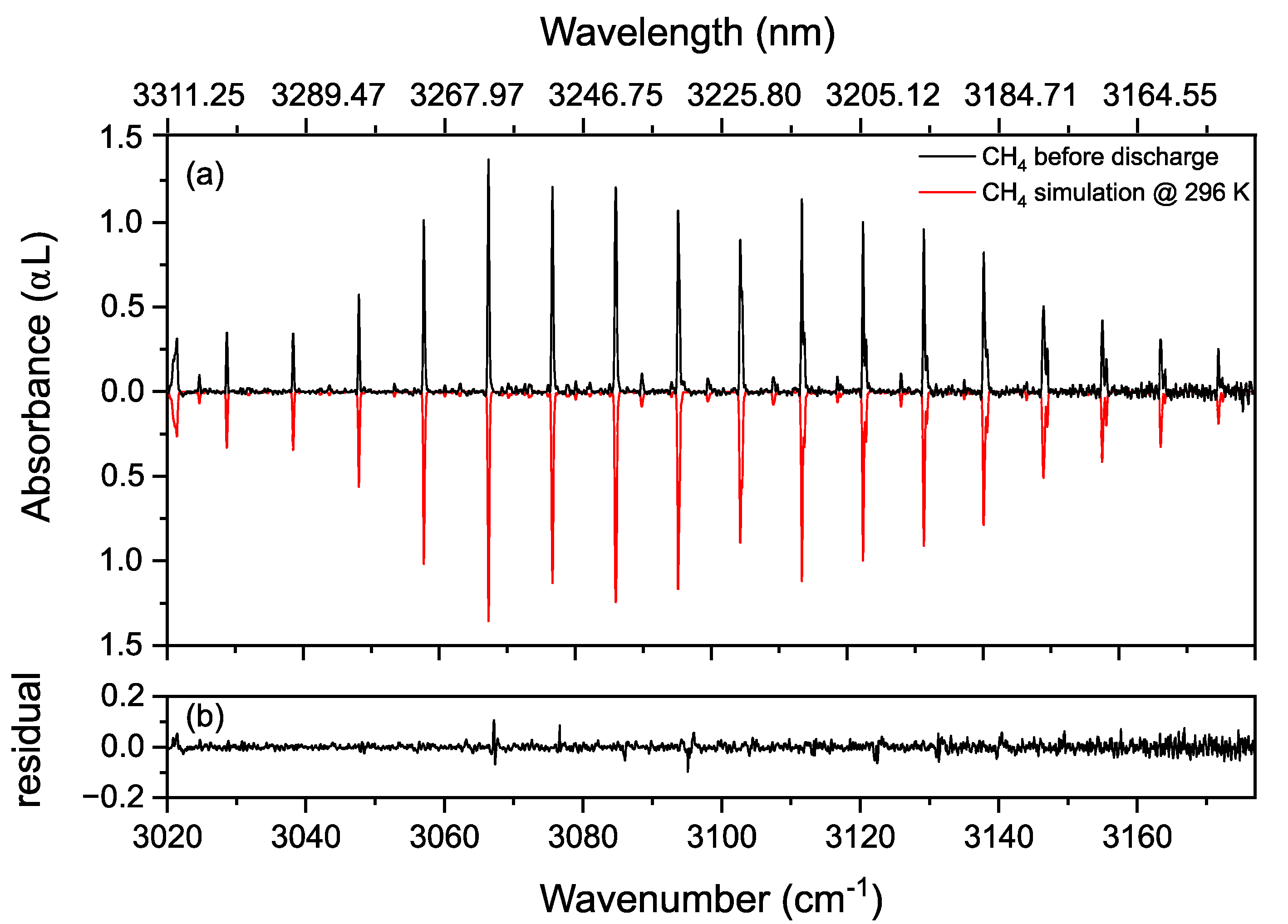
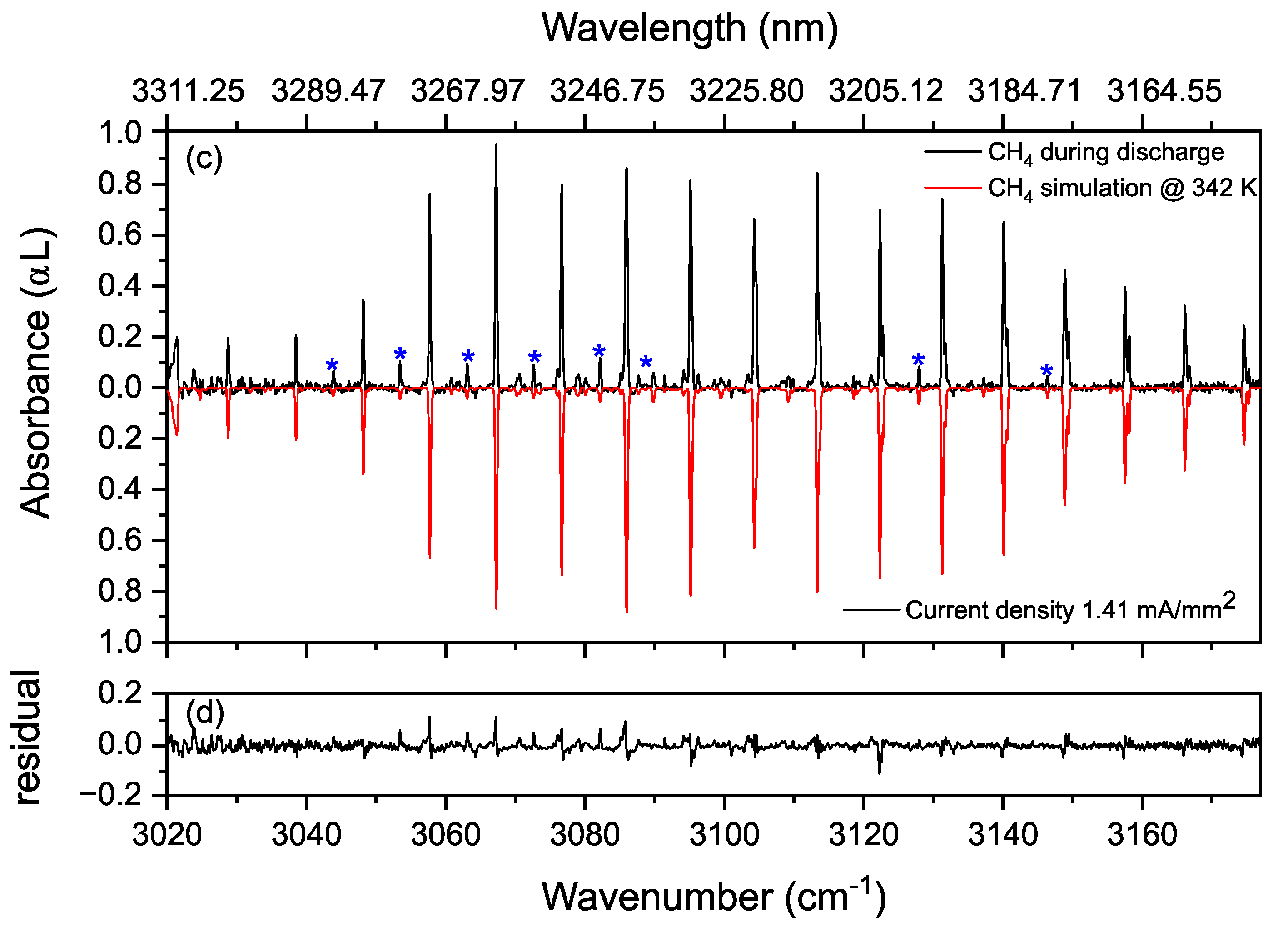
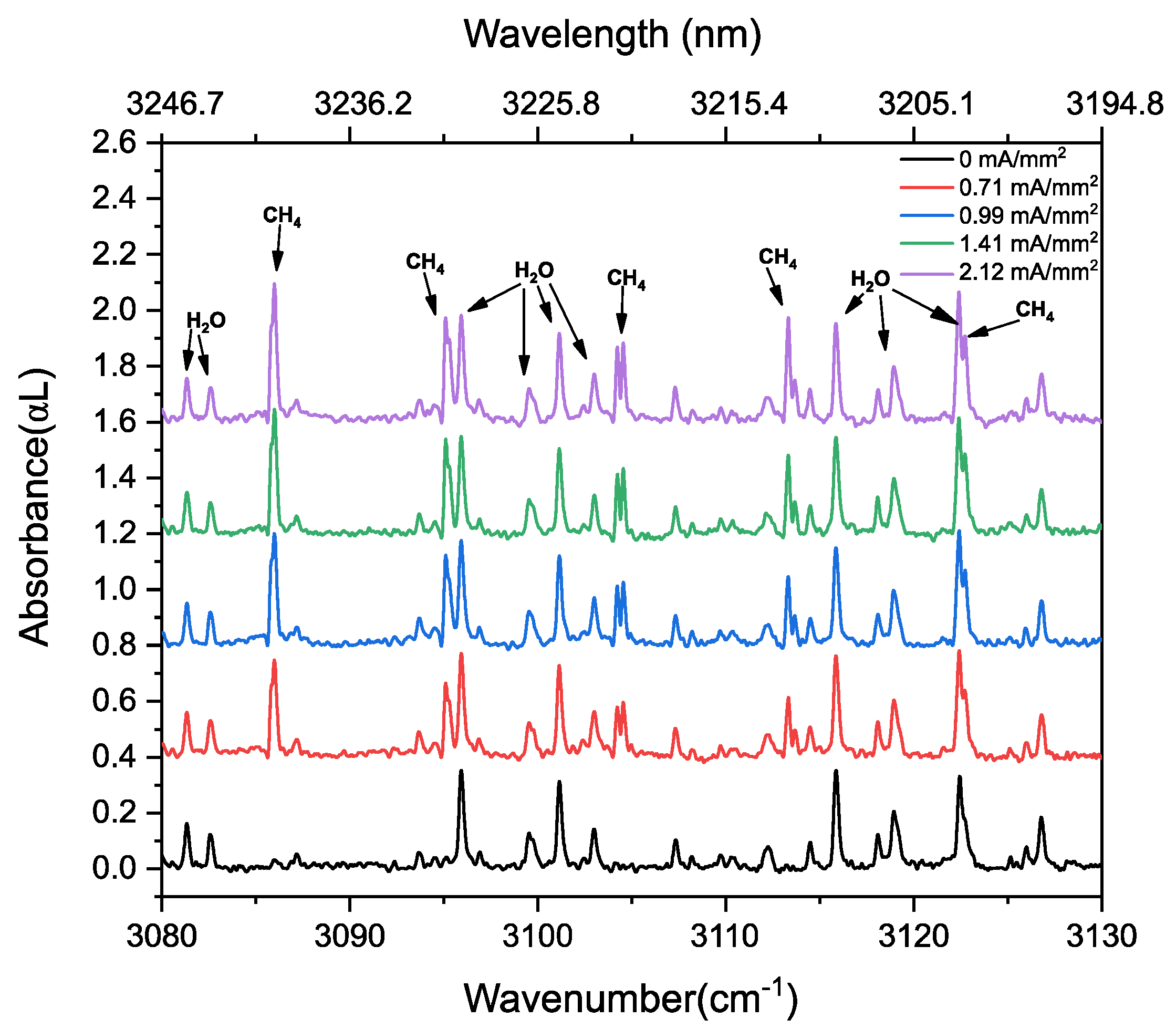
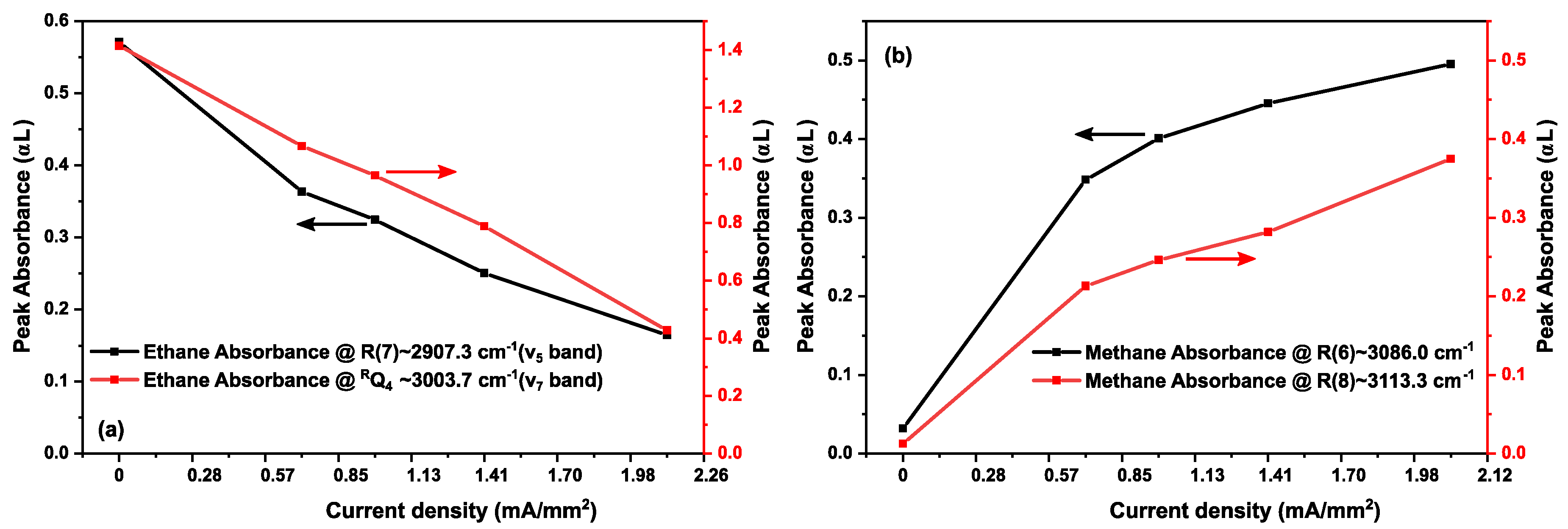
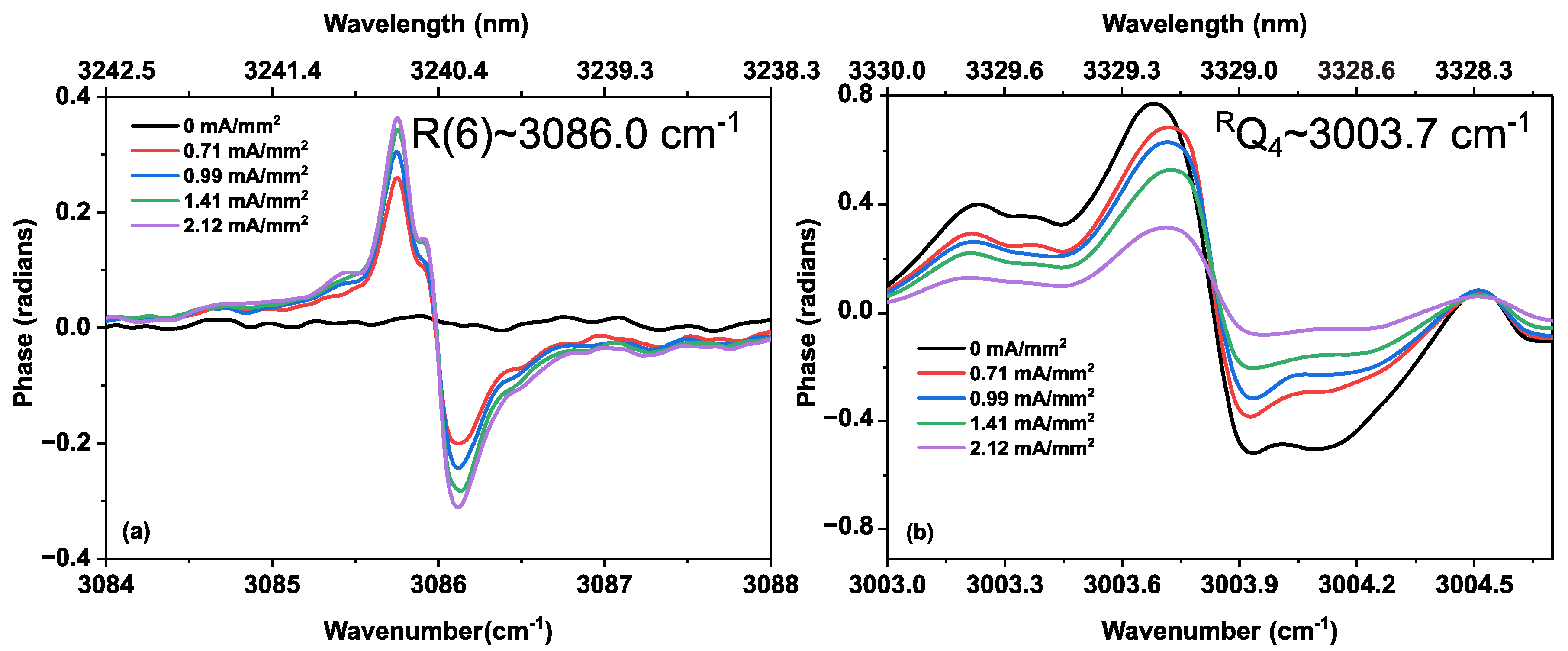
3.2. Methane in a Static (DC) Discharge
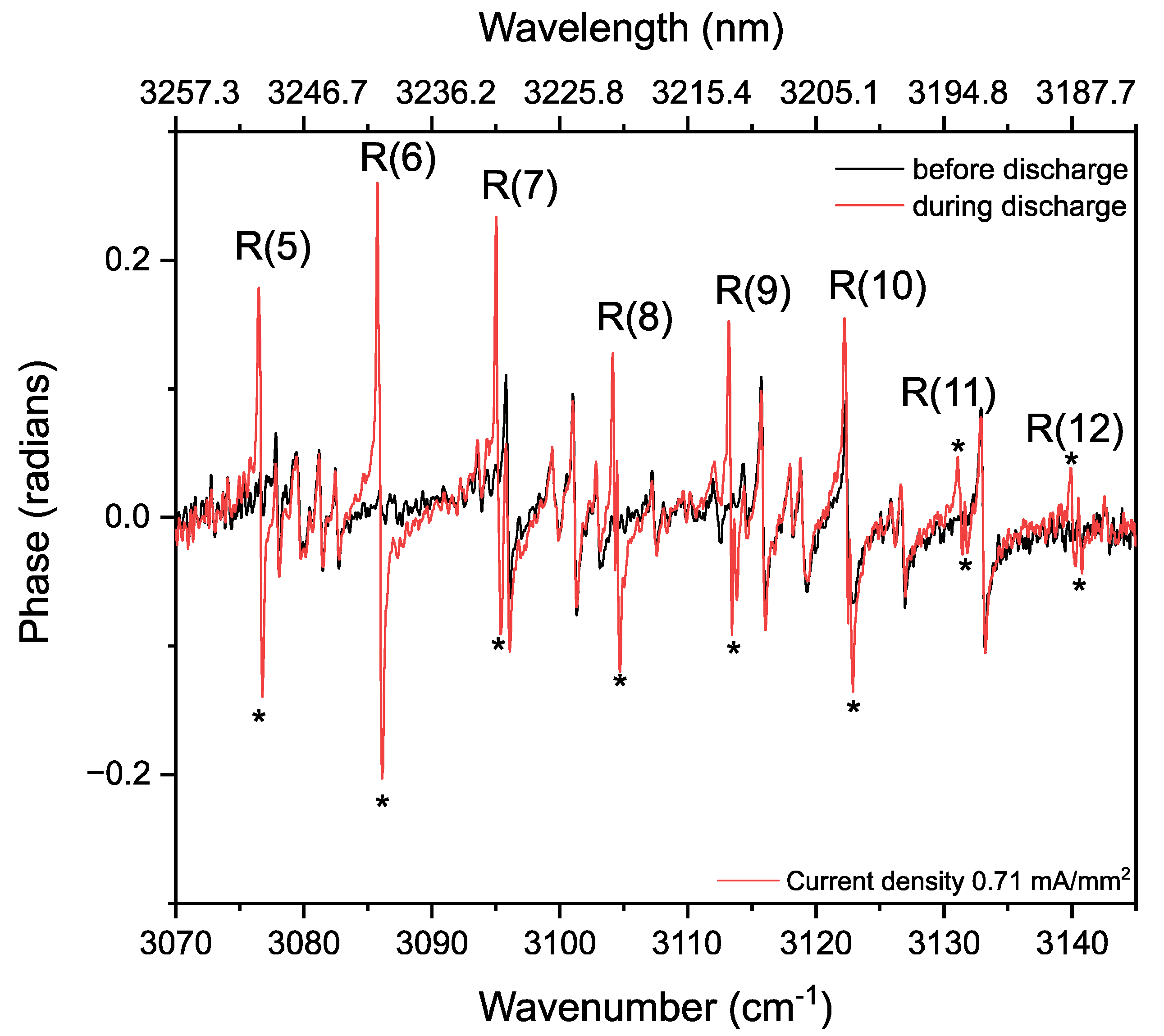
Rotational Temperature of Methane in a Static Discharge Plasma
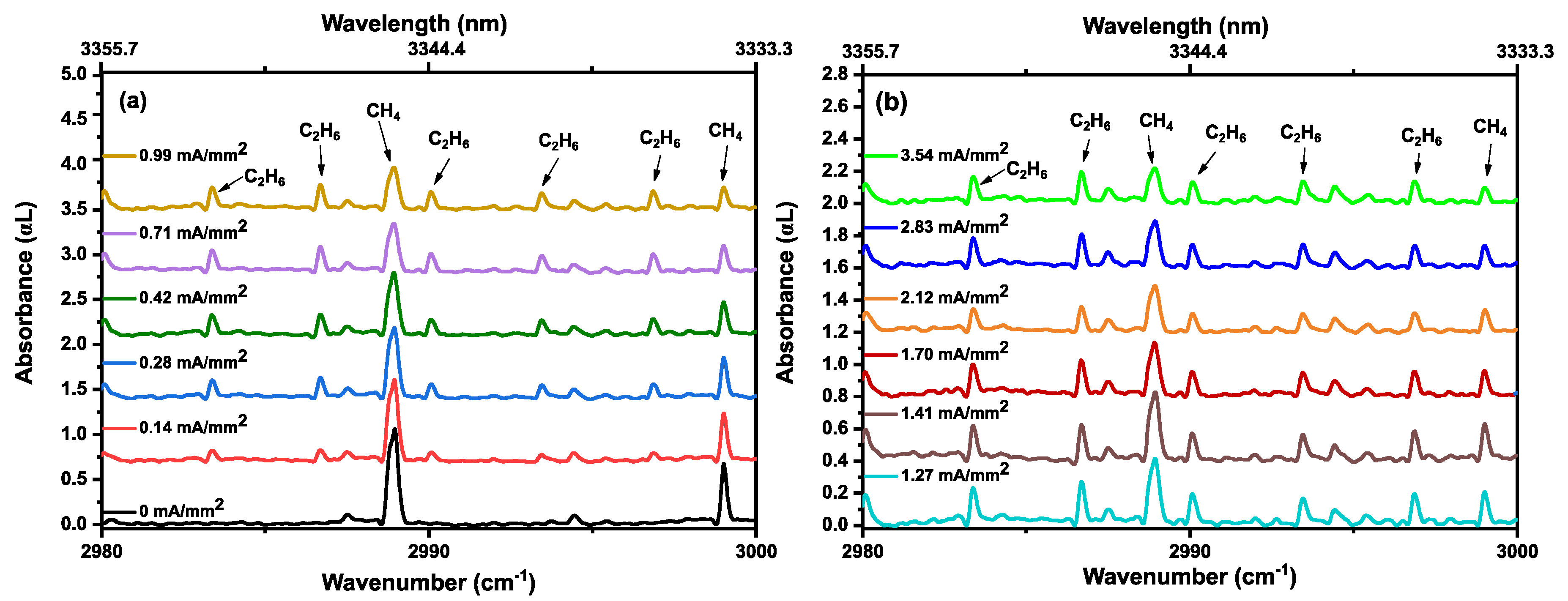
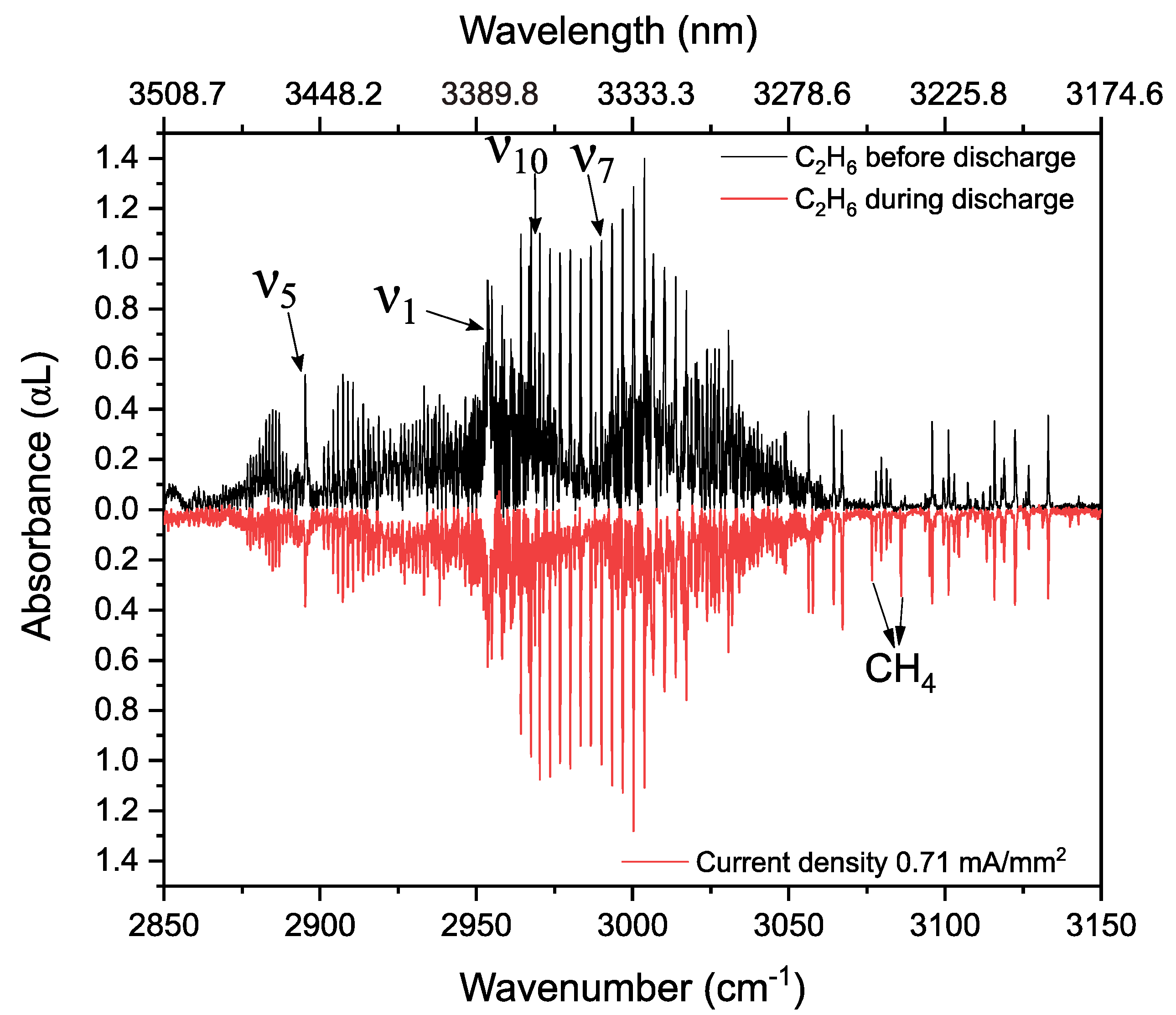
3.3. Ethane in a Static Discharge
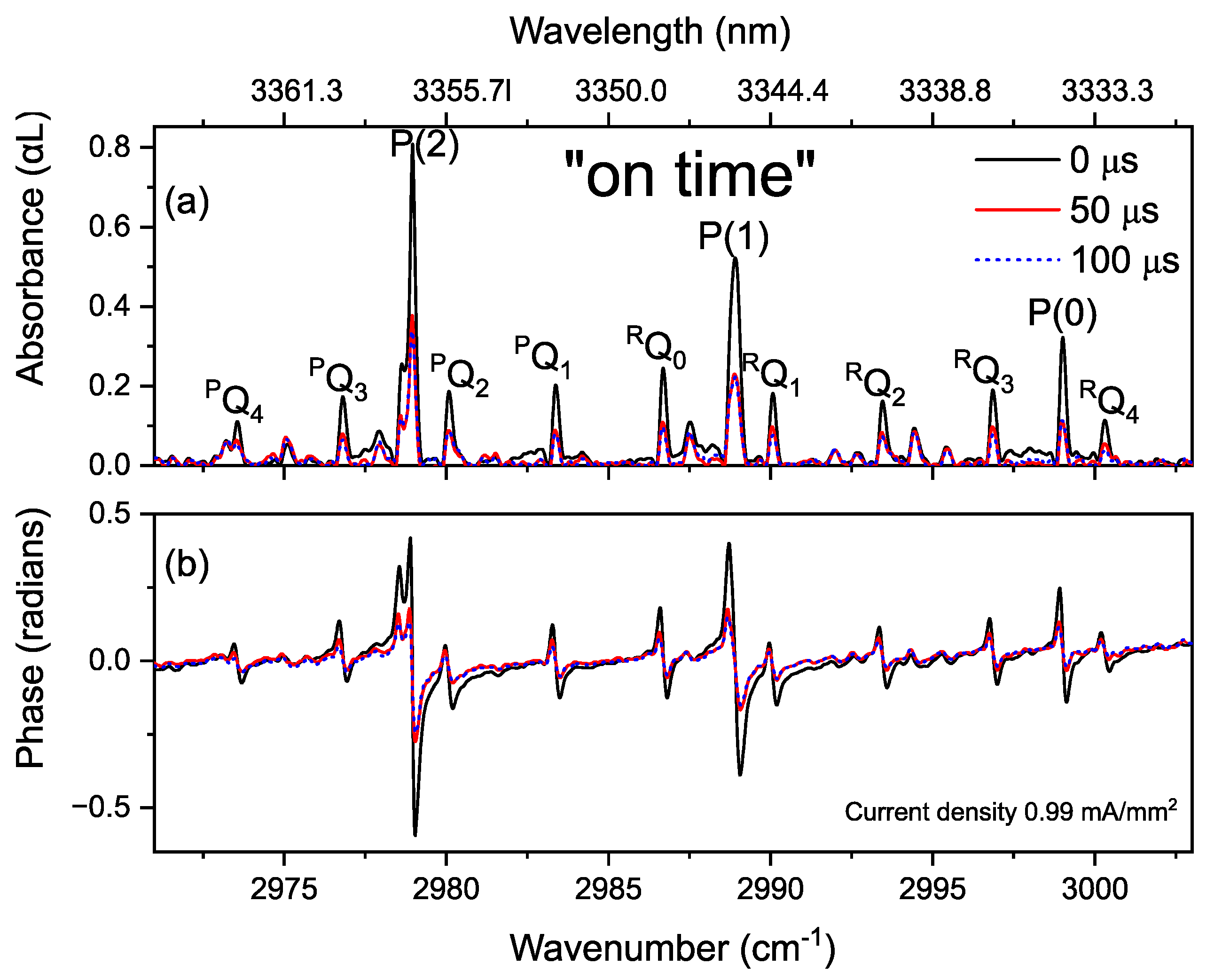
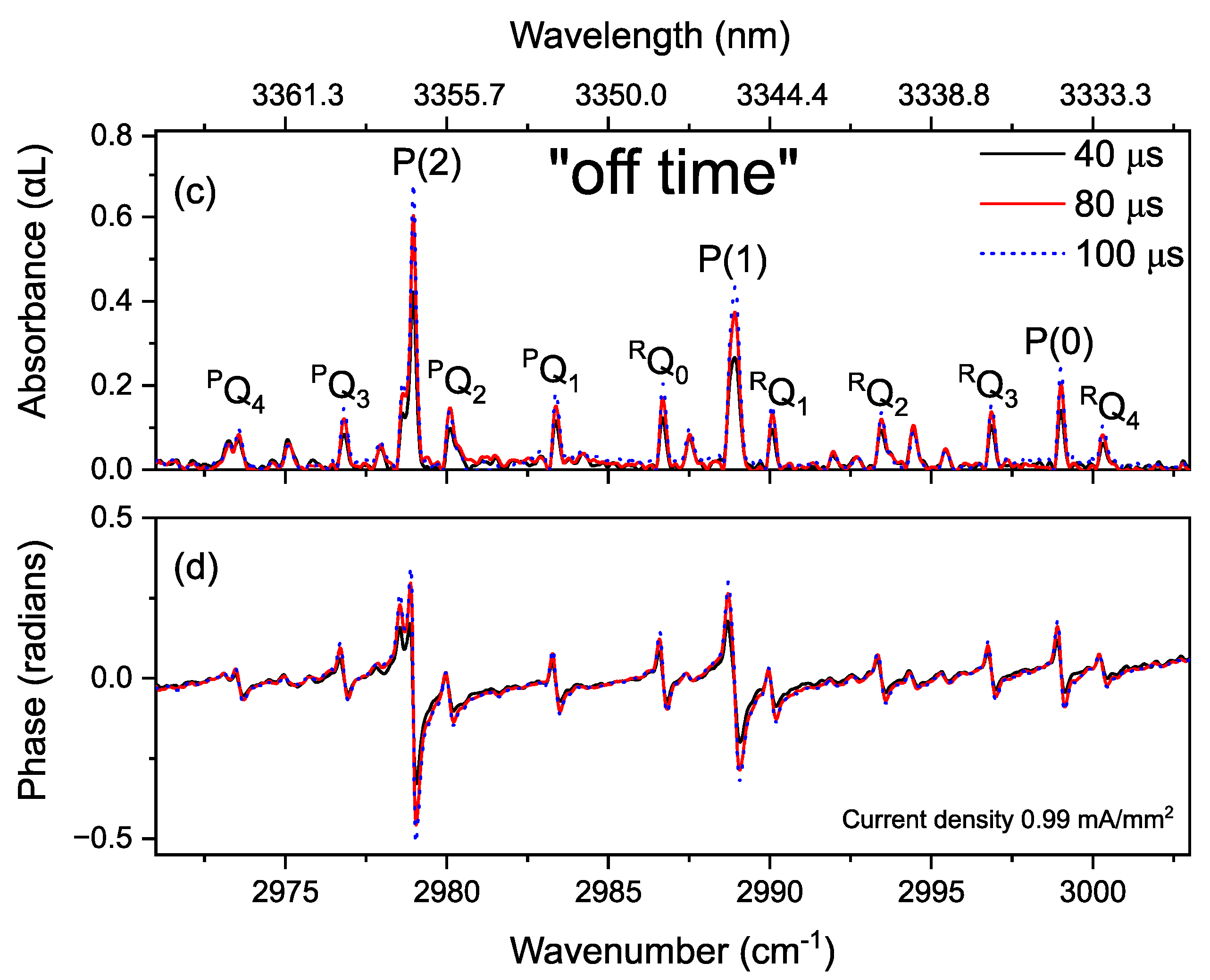
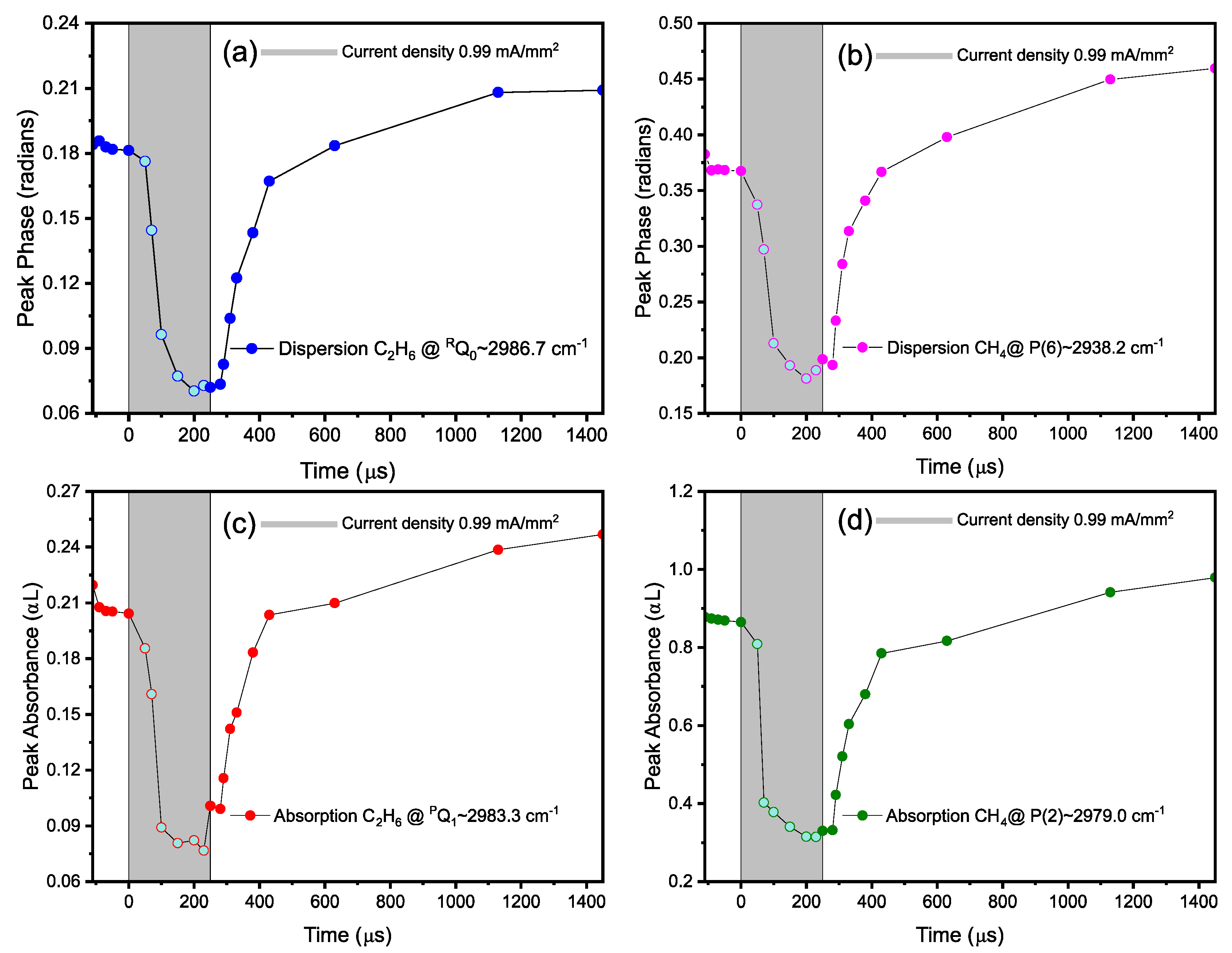
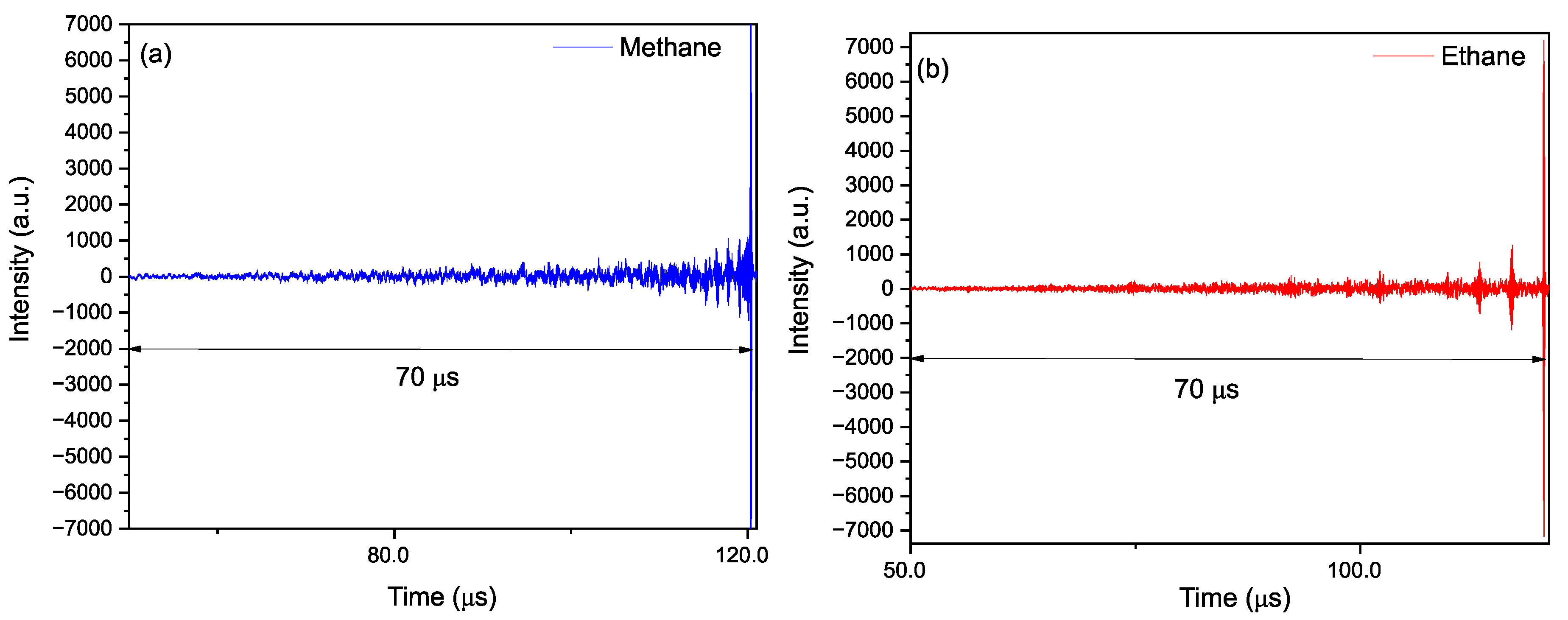
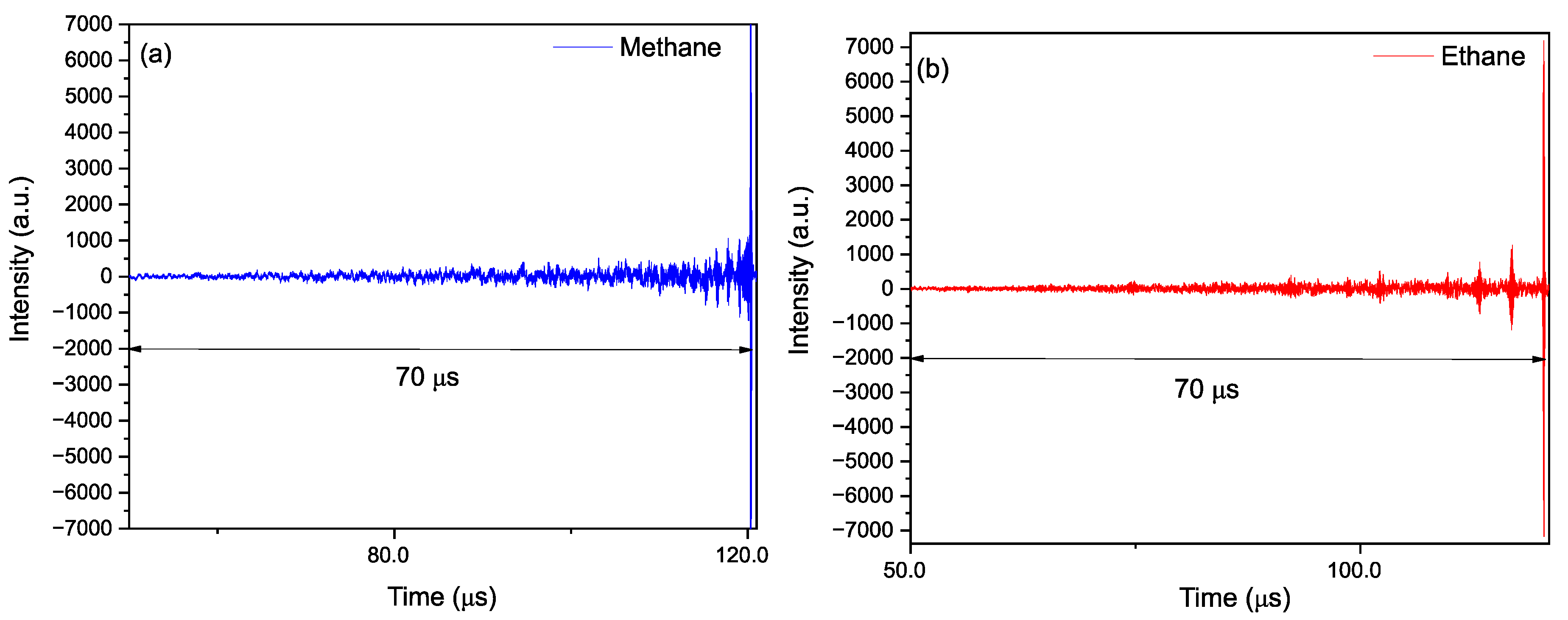
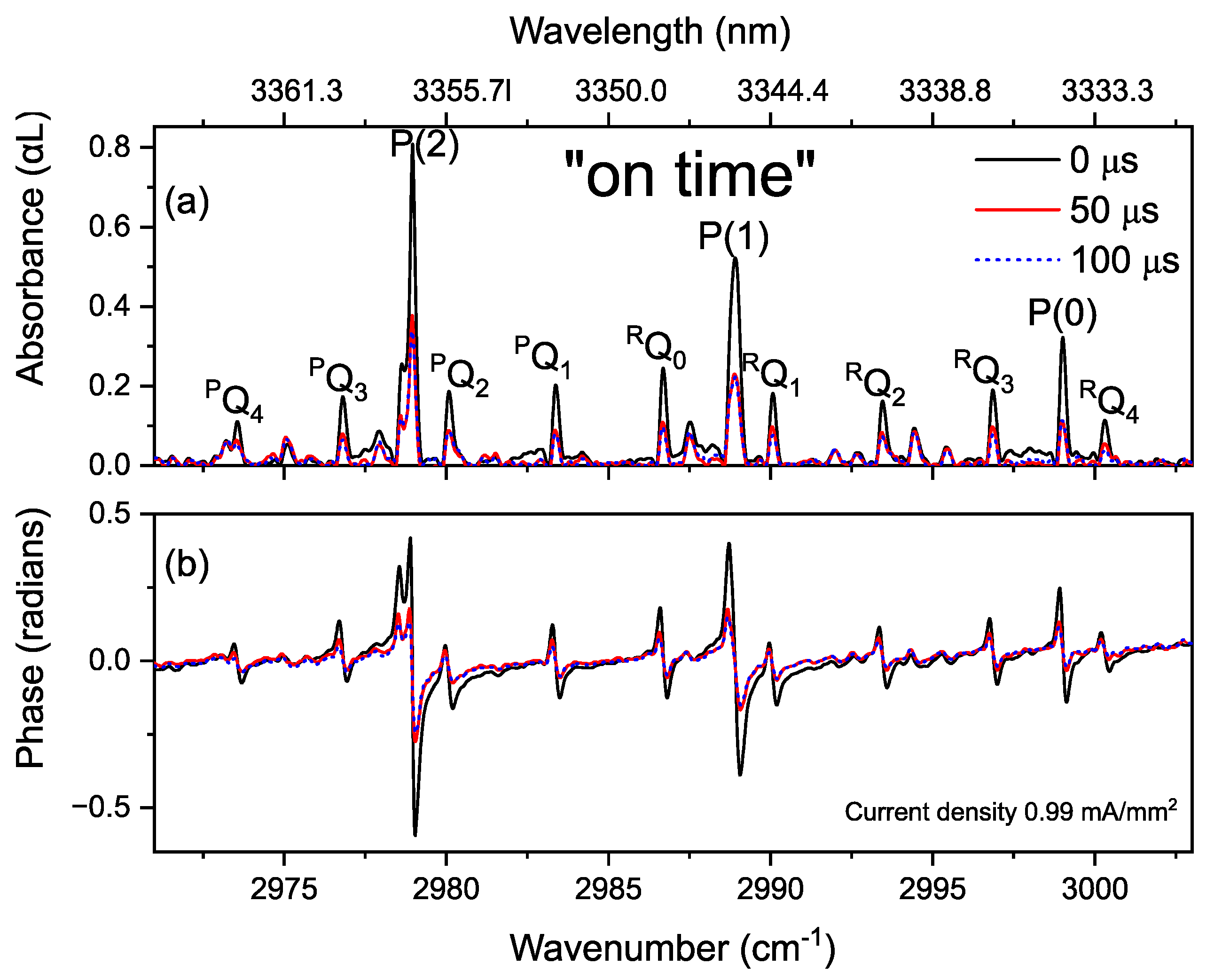
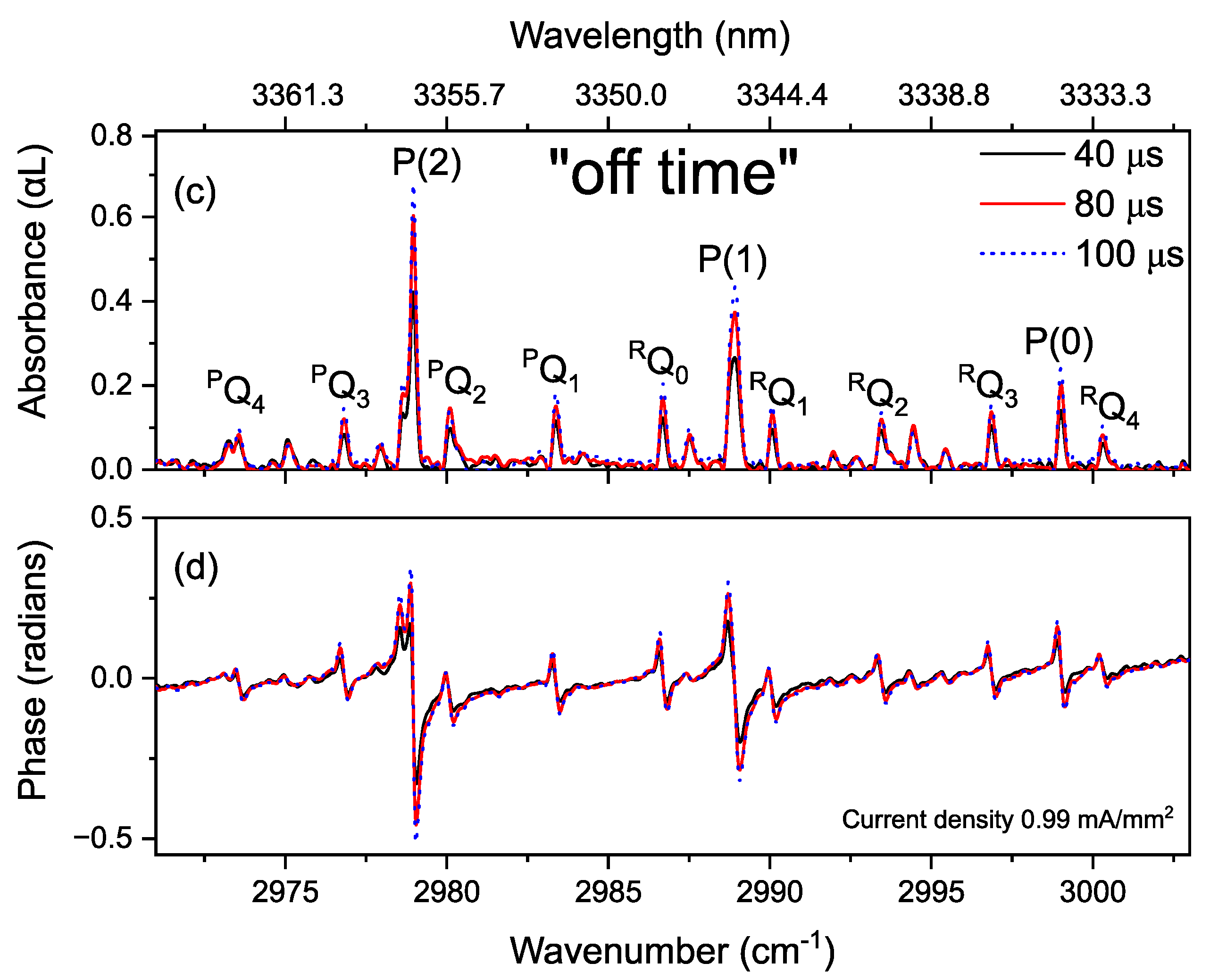
3.4. Time-Resolved Dynamics at Microseconds Timescale
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A
References
- Snoeckx, R.; Setareh, M.; Aerts, R.; Simon, P.; Maghari, A.; Bogaerts, A. Influence of N2 concentration in a CH4/N2 dielectric barrier discharge used for CH4 conversion into H2. Int. J. Hydrog. Energy 2013, 38, 16098–16120. [Google Scholar] [CrossRef]
- Xu, C.; Tu, X. Plasma-assisted methane conversion in an atmospheric pressure dielectric barrier discharge reactor. J. Energy Chem. 2013, 22, 420–425. [Google Scholar] [CrossRef]
- Vastola, F.J.; Wightman, J.P. The rearrangement of acetylene, benzene, ethane, ethylene, methane and naphthalene in a microwave discharge. J. Appl. Chem. 1964, 14, 69–73. [Google Scholar] [CrossRef]
- Li, X.-S.; Zhu, A.-M.; Wang, K.-J.; Xu, Y.; Song, Z.-M. Methane conversion to C2 hydrocarbons and hydrogen in atmospheric non-thermal plasmas generated by different electric discharge techniques. Catal. Today 2004, 98, 617–624. [Google Scholar] [CrossRef]
- Gautier, T.; Carrasco, N.; Stefanovic, I.; Sikimic, B.; Cernogora, G.; Winter, J. Methane Conversion in a N2-CH4 Radiofrequency Discharge. Plasma Process. Polym. 2014, 11, 472–481. [Google Scholar] [CrossRef]
- Majumdar, A.; Behnke, J.F.; Hippler, R.; Matyash, K.; Schneider, R. Chemical Reaction Studies in CH4/Ar and CH4/N2 Gas Mixtures of a Dielectric Barrier Discharge. J. Phys. Chem. A 2005, 109, 9371–9377. [Google Scholar] [CrossRef] [PubMed]
- Cheesman, A.; Smith, J.A.; Ashfold, M.N.R.; Langford, N.; Wright, S.; Duxbury, G. Application of a Quantum Cascade Laser for Time-Resolved, in Situ Probing of CH4/H2 and C2H2/H2 Gas Mixtures during Microwave Plasma Enhanced Chemical Vapor Deposition of Diamond. J. Phys. Chem. A 2006, 110, 2821–2828. [Google Scholar] [CrossRef] [PubMed]
- Zhao, G.-B.; John, S.; Zhang, J.-J.; Wang, L.; Muknahallipatna, S.; Hamann, J.C.; Ackerman, J.F.; Argyle, M.D.; Plumb, O.A. Methane conversion in pulsed corona discharge reactors. Chem. Eng. J. 2006, 125, 67–79. [Google Scholar] [CrossRef] [Green Version]
- Scapinello, M.; Delikonstantis, E.; Stefanidis, G.D. The panorama of plasma-assisted non-oxidative methane reforming. Chem. Eng. Process. Process Intensif. 2017, 117, 120–140. [Google Scholar] [CrossRef]
- Fujii, T.; Syouji, K. Mass spectrometric studies of the neutral and ionic products in a methane/oxygen microwave discharge plasma. J. Phys. Chem. 1993, 97, 11380–11384. [Google Scholar] [CrossRef]
- Burlacov, I.; Hamann, S.; Spies, H.J.; Dalke, A.; Röpcke, J.; Biermann, H. A Novel Approach of Plasma Nitrocarburizing Using a Solid Carbon Active Screen—a Proof of Concept. Htm J. Heat Treat. Mater. 2017, 72, 254–259. [Google Scholar] [CrossRef]
- Hamann, S.; Burlacov, I.; Spies, H.J.; Biermann, H.; Röpcke, J. Spectroscopic investigations of plasma nitriding processes: A comparative study using steel and carbon as active screen materials. J. Appl. Phys. 2017, 121, 153301. [Google Scholar] [CrossRef]
- Cunge, G.; Vempaire, D.; Touzeau, M.; Sadeghi, N. Broadband and time-resolved absorption spectroscopy with light emitting diodes: Application to etching plasma monitoring. Appl. Phys. Lett. 2007, 91, 231503. [Google Scholar] [CrossRef]
- Bergevin, J.; Wu, T.-H.; Yeak, J.; Brumfield, B.E.; Harilal, S.S.; Phillips, M.C.; Jones, R.J. Dual-comb spectroscopy of laser-induced plasmas. Nat. Commun. 2018, 9, 1273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cunge, G.; Fouchier, M.; Brihoum, M.; Bodart, P.; Touzeau, M.; Sadeghi, N. Vacuum UV broad-band absorption spectroscopy: A powerful diagnostic tool for reactive plasma monitoring. J. Phys. D Appl. Phys. 2011, 44, 122001. [Google Scholar] [CrossRef]
- Röpcke, J.; Lombardi, G.; Rousseau, A.; Davies, P. Application of mid-infrared tuneable diode laser absorption spectroscopy to plasma diagnostics: A review. Plasma Sources Sci. Technol. 2006, 15, S148. [Google Scholar] [CrossRef]
- Allah, Z.A.; Sawtell, D.; Kasyutich, V.L.; Martin, P.A. FTIR and QCL diagnostics of the decomposition of volatile organic compounds in an atmospheric pressure dielectric packed bed plasma reactor. J. Phys. Conf. Ser. 2009, 157, 012001. [Google Scholar] [CrossRef]
- Hempel, F.; Davies, P.B.; Loffhagen, D.; Mechold, L.; Röpcke, J. Diagnostic studies of H2–Ar–N2 microwave plasmas containing methane or methanol using tunable infrared diode laser absorption spectroscopy. Plasma Sources Sci. Technol. 2003, 12, S98–S110. [Google Scholar] [CrossRef]
- Röpcke, J.; Revalde, G.; Osiac, M.; Li, K.; Meichsner, J. Tunable Diode Laser Absorption Studies of Hydrocarbons in RF Plasmas Containing Hexamethyldisiloxane. Plasma Chem. Plasma Process. 2002, 22, 137–157. [Google Scholar] [CrossRef]
- Welzel, S.; Hempel, F.; Hübner, M.; Lang, N.; Davies, P.B.; Röpcke, J. Quantum Cascade Laser Absorption Spectroscopy as a Plasma Diagnostic Tool: An Overview. Sensors 2010, 10, 6861–6900. [Google Scholar] [CrossRef] [Green Version]
- Röpcke, J.; Davies, P.B.; Hamann, S.; Hannemann, M.; Lang, N.; Van Helden, J.-P.H. Applying Quantum Cascade Laser Spectroscopy in Plasma Diagnostics. Photonics 2016, 3, 45. [Google Scholar] [CrossRef]
- Keilmann, F.; Gohle, C.; Holzwarth, R. Time-domain mid-infrared frequency-comb spectrometer. Opt. Lett. 2004, 29, 1542–1544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coddington, I.; Newbury, N.; Swann, W. Dual-comb spectroscopy. Optica 2016, 3, 414–426. [Google Scholar] [CrossRef] [Green Version]
- Schiller, S. Spectrometry with frequency combs. Opt. Lett. 2002, 27, 766–768. [Google Scholar] [CrossRef] [Green Version]
- Weichman, M.L.; Changala, P.B.; Ye, J.; Chen, Z.; Yan, M.; Picqué, N. Broadband molecular spectroscopy with optical frequency combs. J. Mol. Spectrosc. 2019, 355, 66–78. [Google Scholar] [CrossRef]
- Villares, G.; Hugi, A.; Blaser, S.; Faist, J. Dual-comb spectroscopy based on quantum-cascade-laser frequency combs. Nat. Commun. 2014, 5, 5192. [Google Scholar] [CrossRef]
- Schliesser, A.; Picqué, N.; Hänsch, T.W. Mid-infrared frequency combs. Nat. Photonics 2012, 6, 440–449. [Google Scholar] [CrossRef] [Green Version]
- Ycas, G.; Giorgetta, F.R.; Friedlein, J.T.; Herman, D.; Cossel, K.C.; Baumann, E.; Newbury, N.R.; Coddington, I. Compact mid-infrared dual-comb spectrometer for outdoor spectroscopy. Opt. Express 2020, 28, 14740–14752. [Google Scholar] [CrossRef]
- Chen, Z.; Hänsch, T.W.; Picqué, N. Mid-infrared feed-forward dual-comb spectroscopy. Proc. Natl. Acad. Sci. USA 2019, 116, 3454. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Gardiner, T.; Reid, D.T. Mid-infrared dual-comb spectroscopy with an optical parametric oscillator. Opt. Lett. 2013, 38, 3148–3150. [Google Scholar] [CrossRef] [Green Version]
- Jin, Y.; Cristescu, S.M.; Harren, F.J.M.; Mandon, J. Two-crystal mid-infrared optical parametric oscillator for absorption and dispersion dual-comb spectroscopy. Opt. Lett. 2014, 39, 3270–3273. [Google Scholar] [CrossRef]
- Muraviev, A.V.; Smolski, V.O.; Loparo, Z.E.; Vodopyanov, K.L. Massively parallel sensing of trace molecules and their isotopologues with broadband subharmonic mid-infrared frequency combs. Nat. Photonics 2018, 12, 209–214. [Google Scholar] [CrossRef]
- Ycas, G.; Giorgetta, F.R.; Baumann, E.; Coddington, I.; Herman, D.; Diddams, S.A.; Newbury, N.R. High-coherence mid-infrared dual-comb spectroscopy spanning 2.6 to 5.2 μm. Nat. Photonics 2018, 12, 202–208. [Google Scholar] [CrossRef] [Green Version]
- Gianella, M.; Nataraj, A.; Tuzson, B.; Jouy, P.; Kapsalidis, F.; Beck, M.; Mangold, M.; Hugi, A.; Faist, J.; Emmenegger, L. High-resolution and gapless dual comb spectroscopy with current-tuned quantum cascade lasers. Opt. Express 2020, 28, 6197–6208. [Google Scholar] [CrossRef] [PubMed]
- Pinkowski, N.H.; Ding, Y.; Strand, C.L.; Hanson, R.K.; Horvath, R.; Geiser, M. Dual-comb spectroscopy for high-temperature reaction kinetics. Meas. Sci. Technol. 2020, 31, 055501. [Google Scholar] [CrossRef] [Green Version]
- Zhang, G.; Horvath, R.; Liu, D.; Geiser, M.; Farooq, A. QCL-Based Dual-Comb Spectrometer for Multi-Species Measurements at High Temperatures and High Pressures. Sensors 2020, 20, 3602. [Google Scholar] [CrossRef] [PubMed]
- Hoghooghi, N.; Cole, R.K.; Rieker, G.B. 11-{\mu}s Time-resolved, Continuous Dual-Comb Spectroscopy with Spectrally Filtered Mode-locked Frequency Combs. arXiv 2020, arXiv:2005.13050. [Google Scholar]
- Klocke, J.L.; Mangold, M.; Allmendinger, P.; Hugi, A.; Geiser, M.; Jouy, P.; Faist, J.; Kottke, T. Single-Shot Sub-microsecond Mid-infrared Spectroscopy on Protein Reactions with Quantum Cascade Laser Frequency Combs. Anal. Chem. 2018, 90, 10494–10500. [Google Scholar] [CrossRef] [Green Version]
- Luo, P.-L.; Horng, E.-C. Simultaneous determination of transient free radicals and reaction kinetics by high-resolution time-resolved dual-comb spectroscopy. Commun. Chem. 2020, 3, 95. [Google Scholar] [CrossRef]
- Abbas, M.A.; Pan, Q.; Mandon, J.; Cristescu, S.M.; Harren, F.J.M.; Khodabakhsh, A. Time-resolved mid-infrared dual-comb spectroscopy. Sci. Rep. 2019, 9, 17247. [Google Scholar] [CrossRef] [Green Version]
- Birch, J.R. Dispersive Fourier transform spectroscopy. Microchim. Acta 1987, 93, 105–122. [Google Scholar] [CrossRef]
- Bell, E.E. The use of asymmetric interferograms in transmittance measurements. J. Phys. Colloq. 1967, 28, C2–C18. [Google Scholar] [CrossRef]
- Giorgetta, F.R.; Rieker, G.B.; Baumann, E.; Swann, W.C.; Sinclair, L.C.; Kofler, J.; Coddington, I.; Newbury, N.R. Broadband Phase Spectroscopy over Turbulent Air Paths. Phys. Rev. Lett. 2015, 115, 103901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coddington, I.; Swann, W.C.; Newbury, N.R. Coherent Multiheterodyne Spectroscopy Using Stabilized Optical Frequency Combs. Phys. Rev. Lett. 2008, 100, 013902. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rutkowski, L.; Johansson, A.C.; Zhao, G.; Hausmaninger, T.; Khodabakhsh, A.; Axner, O.; Foltynowicz, A. Sensitive and broadband measurement of dispersion in a cavity using a Fourier transform spectrometer with kHz resolution. Opt. Express 2017, 25, 21711–21718. [Google Scholar] [CrossRef]
- Johansson, A.C.; Rutkowski, L.; Filipsson, A.; Hausmaninger, T.; Zhao, G.; Axner, O.; Foltynowicz, A. Broadband calibration-free cavity-enhanced complex refractive index spectroscopy using a frequency comb. Opt. Express 2018, 26, 20633–20648. [Google Scholar] [CrossRef] [Green Version]
- Weeks, R.R.D.; Zhang, Y.; Lecaplain, C.; Yeak, J.; Harilal, S.S.; Phillips, M.C.; Jones, R.J. In Proceedings of the Time-Resolved Dual Frequency Comb Phase Spectroscopy of Laser-Induced Plasmas, Conference on Lasers and Electro-Optics, San Jose, CA, USA, 5 May 2019; Optical Society of America: San Jose, CA, USA, 2019.
- Cygan, A.; Wcislo, P.; Wojtewicz, S.; Maslowski, P.; Hodges, J.T.; Ciurylo, R.; Lisak, D. One-dimensional frequency-based spectroscopy. Opt. Express 2015, 23, 14472–14486. [Google Scholar] [CrossRef]
- Nikodem, M.; Wysocki, G. Molecular dispersion spectroscopy—new capabilities in laser chemical sensing. Ann. N. Y. Acad. Sci. 2012, 1260, 101–111. [Google Scholar] [CrossRef] [Green Version]
- Bjorklund, G.C. Frequency-modulation spectroscopy: A new method for measuring weak absorptions and dispersions. Opt. Lett. 1980, 5, 15–17. [Google Scholar] [CrossRef]
- Ye, J.; Ma, L.-S.; Hall, J.L. Ultrasensitive detections in atomic and molecular physics: Demonstration in molecular overtone spectroscopy. J. Opt. Soc. Am. B 1998, 15, 6–15. [Google Scholar] [CrossRef] [Green Version]
- Foltynowicz, A.; Schmidt, F.M.; Ma, W.; Axner, O. Noise-immune cavity-enhanced optical heterodyne molecular spectroscopy: Current status and future potential. Appl. Phys. B 2008, 92, 313. [Google Scholar] [CrossRef]
- Martín-Mateos, P.; Hayden, J.; Acedo, P.; Lendl, B. Heterodyne Phase-Sensitive Dispersion Spectroscopy in the Mid-Infrared with a Quantum Cascade Laser. Anal. Chem. 2017, 89, 5916–5922. [Google Scholar] [CrossRef] [PubMed]
- Wysocki, G.; Weidmann, D. Molecular dispersion spectroscopy for chemical sensing using chirped mid-infrared quantum cascade laser. Opt. Express 2010, 18, 26123–26140. [Google Scholar] [CrossRef] [PubMed]
- Lewicki, R.; Doty, J.H.; Curl, R.F.; Tittel, F.K.; Wysocki, G. Ultrasensitive detection of nitric oxide at 5.33 μm by using external cavity quantum cascade laser-based Faraday rotation spectroscopy. Proc. Natl. Acad. Sci. USA 2009, 106, 12587–12592. [Google Scholar] [CrossRef] [Green Version]
- Mandon, J.; Guelachvili, G.; Picqué, N. Fourier transform spectroscopy with a laser frequency comb. Nat. Photonics 2009, 3, 99–102. [Google Scholar] [CrossRef]
- Gohle, C.; Stein, B.; Schliesser, A.; Udem, T.; Hänsch, T.W. Frequency Comb Vernier Spectroscopy for Broadband, High-Resolution, High-Sensitivity Absorption and Dispersion Spectra. Phys. Rev. Lett. 2007, 99, 263902. [Google Scholar] [CrossRef] [Green Version]
- Abbas, M.A.; Khodabakhsh, A.; Pan, Q.; Mandon, J.; Cristescu, S.M.; Harren, F.J.M. Mid-infrared dual-comb spectroscopy with absolute frequency calibration using a passive optical reference. Opt. Express 2019, 27, 19282–19291. [Google Scholar] [CrossRef] [Green Version]
- Gordon, I.E.; Rothman, L.S.; Hill, C.; Kochanov, R.V.; Tan, Y.; Bernath, P.F.; Birk, M.; Boudon, V.; Campargue, A.; Chance, K.V.; et al. The HITRAN2016 molecular spectroscopic database. J. Quant. Spectrosc. Radiat. Transf. 2017, 203, 3–69. [Google Scholar] [CrossRef]
- Adler, F.; Maslowski, P.; Foltynowicz, A.; Cossel, K.C.; Briles, T.C.; Hartl, I.; Ye, J. Mid-infrared Fourier transform spectroscopy with a broadband frequency comb. Opt. Express 2010, 18, 21861–21872. [Google Scholar] [CrossRef]
- Pine, A.S.; Lafferty, W.J. Torsional splittings and assignments of the doppler-limited spectrum of ethane in the c-h stretching region. J. Res. Natl. Bur. Stand. 1982, 87, 237–256. [Google Scholar] [CrossRef]
- Alrefae, M.; Es-sebbar, E.-t.; Farooq, A. Absorption cross-section measurements of methane, ethane, ethylene and methanol at high temperatures. J. Mol. Spectrosc. 2014, 303, 8–14. [Google Scholar] [CrossRef] [Green Version]
- Sahlberg, A.-L.; Zhou, J.; Aldén, M.; Li, Z. Investigation of ro-vibrational spectra of small hydrocarbons at elevated temperatures using infrared degenerate four-wave mixing. J. Raman Spectrosc. 2016, 47, 1130–1139. [Google Scholar] [CrossRef] [Green Version]
- NIST Chemical Kinetics Database. Available online: https://kinetics.nist.gov/kinetics/index.jsp (accessed on 28 October 2020).
- Savinov, S.Y.; Lee, H.; Song, H.K.; Na, B.-K. A kinetic study on the conversion of methane to higher hydrocarbons in a radio-frequency discharge. Korean J. Chem. Eng. 2004, 21, 601–610. [Google Scholar] [CrossRef]
- Goldenstein, C.S.; Spearrin, R.M.; Jeffries, J.B.; Hanson, R.K. Infrared laser-absorption sensing for combustion gases. Prog. Energy Combust. Sci. 2017, 60, 132–176. [Google Scholar] [CrossRef] [Green Version]
- Sanchez-Gonzalez, R.; Kim, Y.; Rosocha, L.A.; Abbate, S. Methane and Ethane Decomposition in an Atmospheric-Pressure Plasma Jet. IEEE Trans. Plasma Sci. 2007, 35, 1669–1676. [Google Scholar] [CrossRef]
- Heintze, M.; Magureanu, M. Methane conversion into acetylene in a microwave plasma: Optimization of the operating parameters. J. Appl. Phys. 2002, 92, 2276–2283. [Google Scholar] [CrossRef]
- Gao, Y.; Zhang, S.; Sun, H.; Wang, R.; Tu, X.; Shao, T. Highly efficient conversion of methane using microsecond and nanosecond pulsed spark discharges. Appl. Energy 2018, 226, 534–545. [Google Scholar] [CrossRef]
- Kado, S.; Sekine, Y.; Nozaki, T.; Okazaki, K. Diagnosis of atmospheric pressure low temperature plasma and application to high efficient methane conversion. Catal. Today 2004, 89, 47–55. [Google Scholar] [CrossRef]
- Coddington, I.; Swann, W.C.; Newbury, N.R. Time-domain spectroscopy of molecular free-induction decay in the infrared. Opt. Lett. 2010, 35, 1395–1397. [Google Scholar] [CrossRef]
- Roy, J.; Deschênes, J.-D.; Potvin, S.; Genest, J. Continuous real-time correction and averaging for frequency comb interferometry. Opt. Express 2012, 20, 21932–21939. [Google Scholar] [CrossRef]
- Sterczewski, L.A.; Westberg, J.; Wysocki, G. Computational coherent averaging for free-running dual-comb spectroscopy. Opt. Express 2019, 27, 23875–23893. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, Z.; Ni, K.; Zhou, Q.; Wu, G. Digital correction method for realizing a phase-stable dual-comb interferometer. Opt. Express 2018, 26, 16813–16823. [Google Scholar] [CrossRef] [PubMed]
- Fendt, P.; Retzer, U.; Ulrich, H.; Will, S.; Zigan, L. Stability Analysis of the Fluorescent Tracer 1-Methylnaphthalene for IC Engine Applications by Supercontinuum Laser Absorption Spectroscopy. Sensors 2020, 20, 2871. [Google Scholar] [CrossRef] [PubMed]
- Makowiecki, A.S.; Herman, D.I.; Hoghooghi, N.; Strong, E.F.; Cole, R.K.; Ycas, G.; Giorgetta, F.R.; Lapointe, C.B.; Glusman, J.F.; Daily, J.W.; et al. Mid-infrared dual frequency comb spectroscopy for combustion analysis from 2.8 to 5 µm. Proc. Combust. Inst. 2020. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abbas, M.A.; Dijk, L.v.; Jahromi, K.E.; Nematollahi, M.; Harren, F.J.M.; Khodabakhsh, A. Broadband Time-Resolved Absorption and Dispersion Spectroscopy of Methane and Ethane in a Plasma Using a Mid-Infrared Dual-Comb Spectrometer. Sensors 2020, 20, 6831. https://doi.org/10.3390/s20236831
Abbas MA, Dijk Lv, Jahromi KE, Nematollahi M, Harren FJM, Khodabakhsh A. Broadband Time-Resolved Absorption and Dispersion Spectroscopy of Methane and Ethane in a Plasma Using a Mid-Infrared Dual-Comb Spectrometer. Sensors. 2020; 20(23):6831. https://doi.org/10.3390/s20236831
Chicago/Turabian StyleAbbas, Muhammad Ali, Luuk van Dijk, Khalil Eslami Jahromi, Mohammadreza Nematollahi, Frans J. M. Harren, and Amir Khodabakhsh. 2020. "Broadband Time-Resolved Absorption and Dispersion Spectroscopy of Methane and Ethane in a Plasma Using a Mid-Infrared Dual-Comb Spectrometer" Sensors 20, no. 23: 6831. https://doi.org/10.3390/s20236831
APA StyleAbbas, M. A., Dijk, L. v., Jahromi, K. E., Nematollahi, M., Harren, F. J. M., & Khodabakhsh, A. (2020). Broadband Time-Resolved Absorption and Dispersion Spectroscopy of Methane and Ethane in a Plasma Using a Mid-Infrared Dual-Comb Spectrometer. Sensors, 20(23), 6831. https://doi.org/10.3390/s20236831