Improving the Voltammetric Determination of Hg(II): A Comparison Between Ligand-Modified Glassy Carbon and Electrochemically Reduced Graphene Oxide Electrodes


 , ,
, ,  ,
, 
Abstract
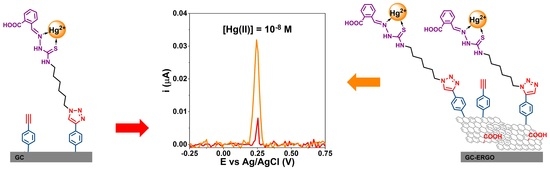
1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. Equipment
2.3. Procedures
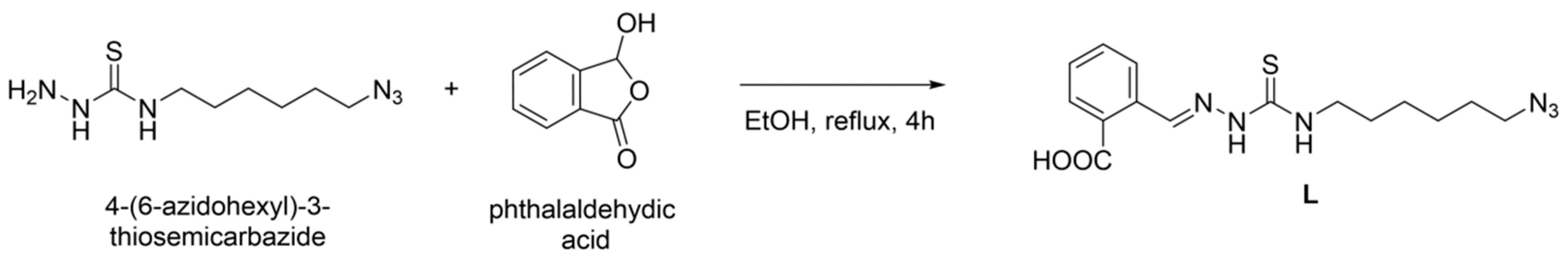
2.3.1. Synthesis of Ligand L
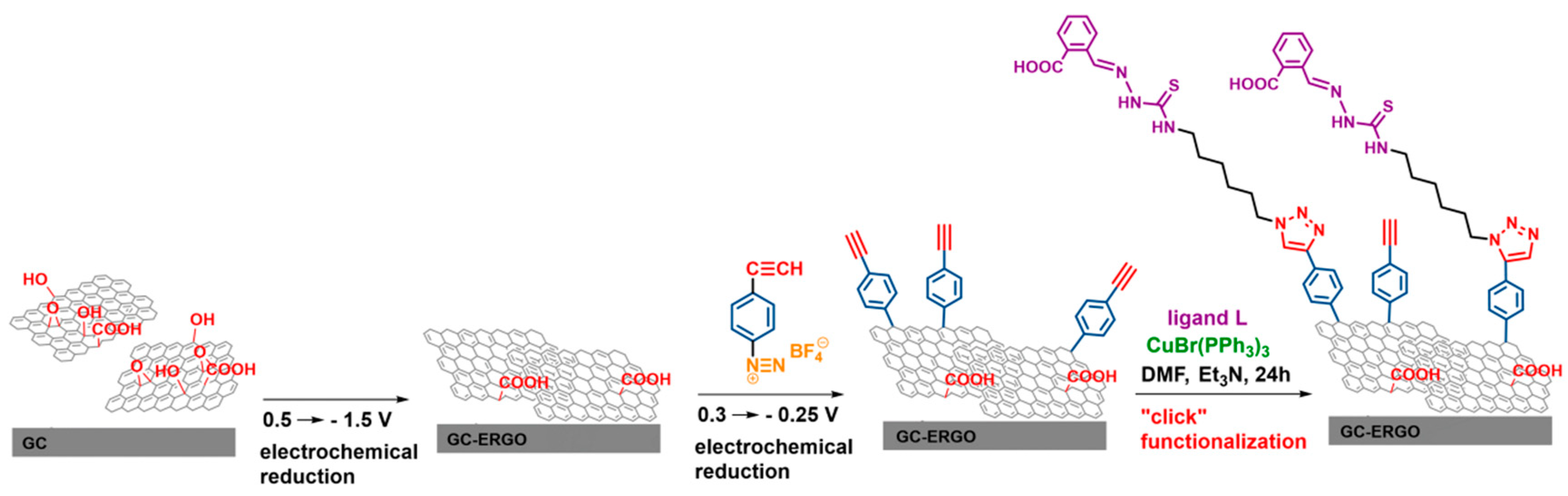
2.3.2. Electrode Preparation
3. Results and Discussion
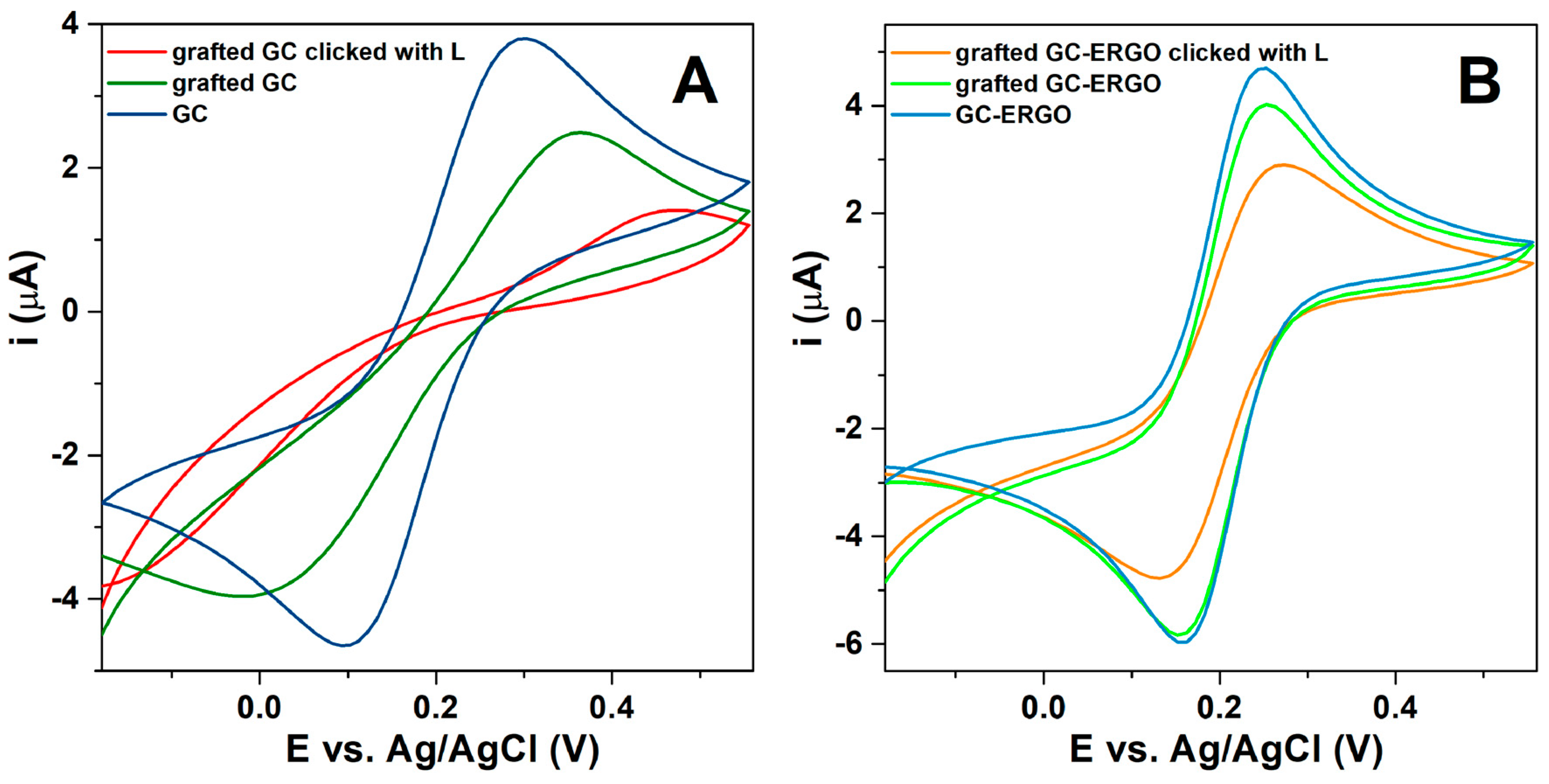
3.1. Preparation of Modified Electrodes
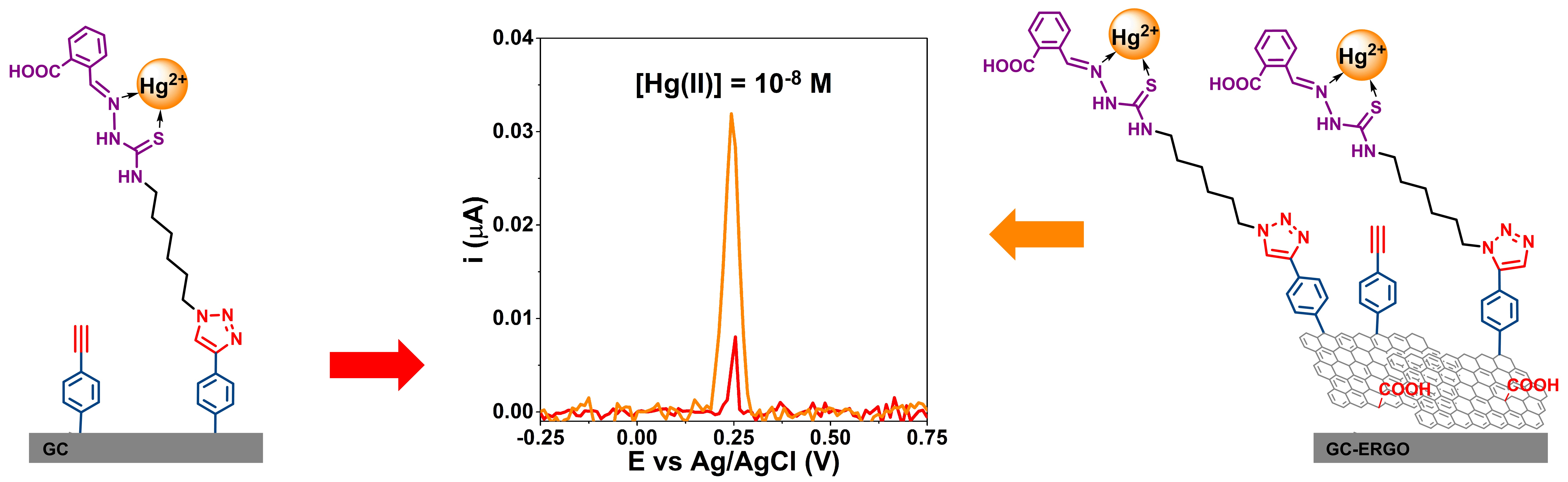
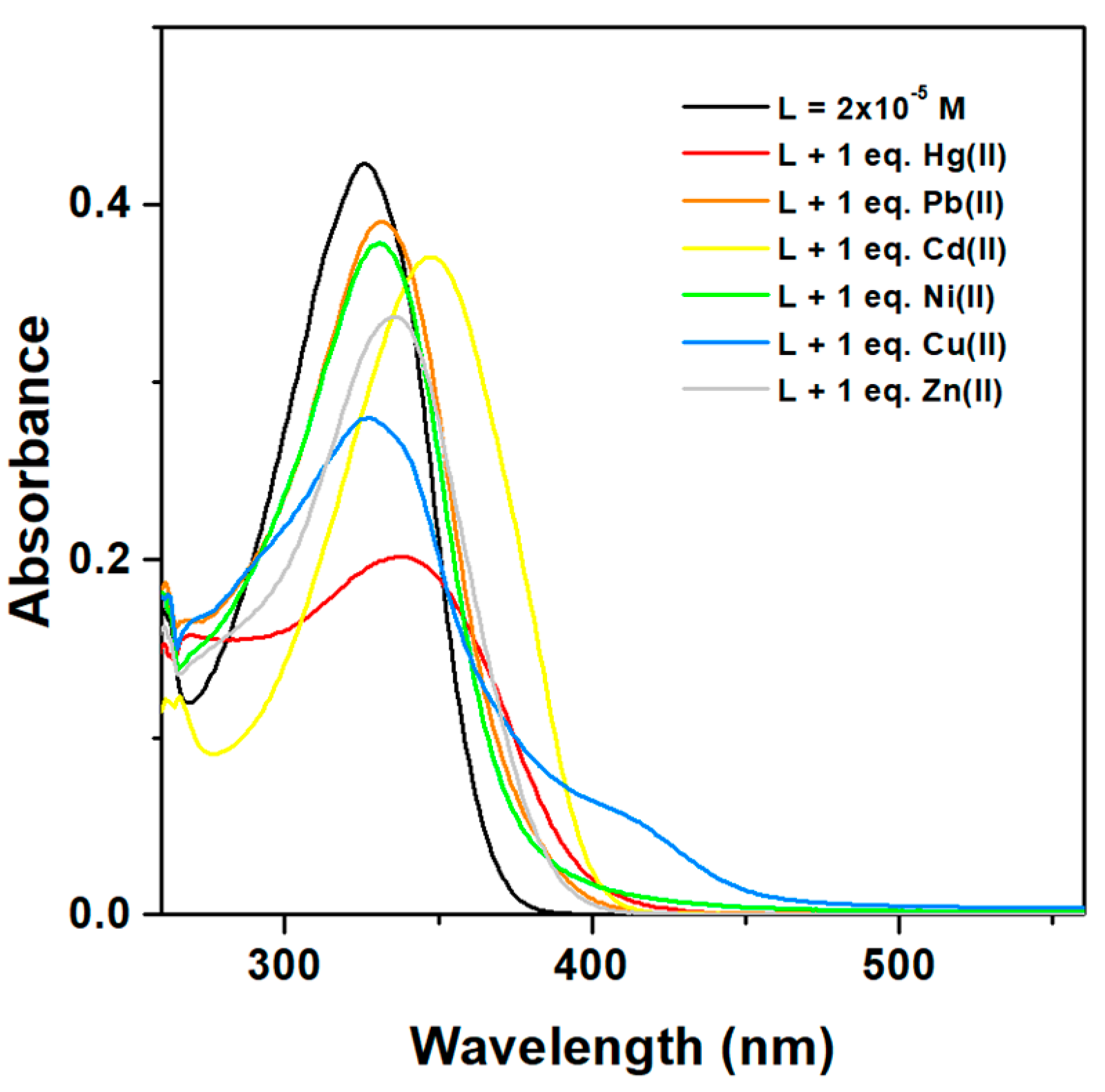
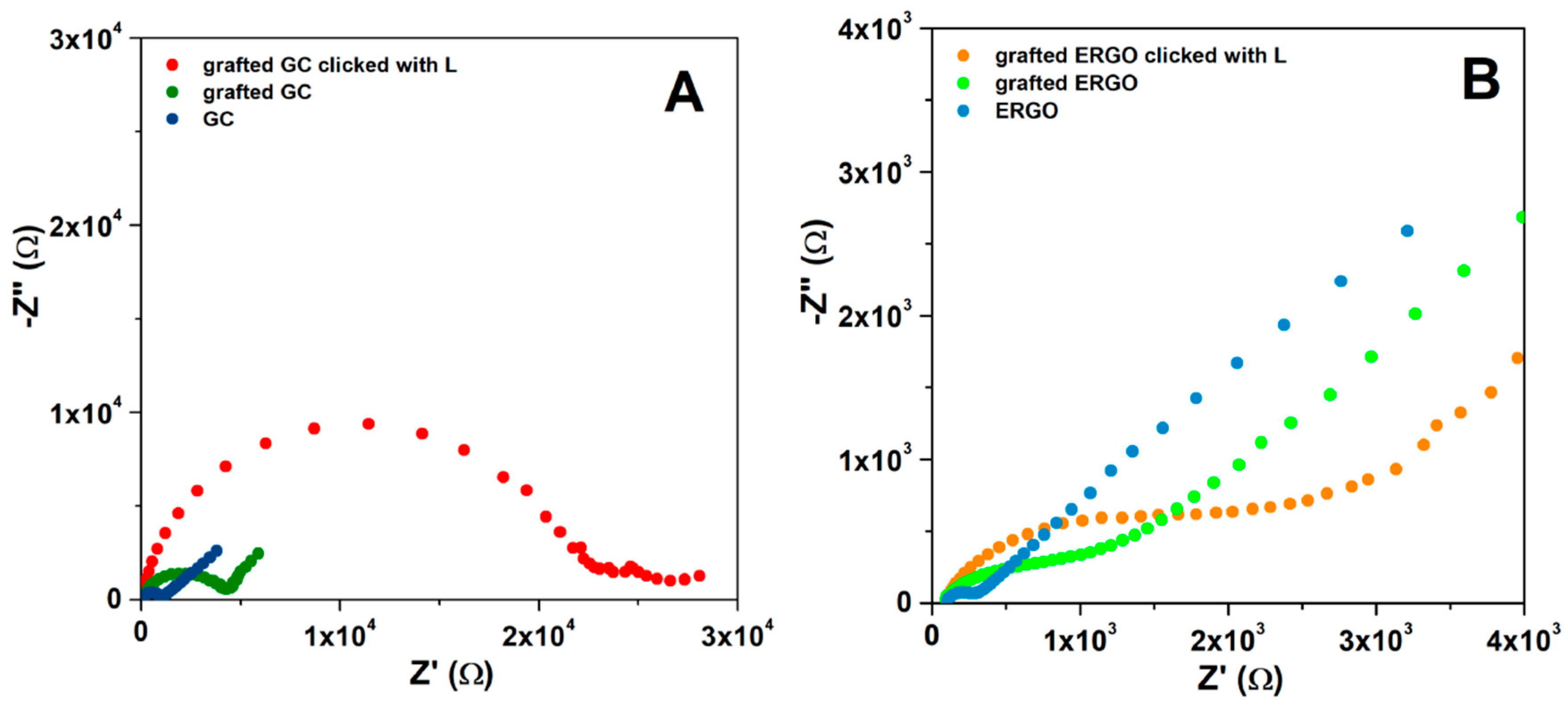
3.2. Characterization of Modified Electrodes
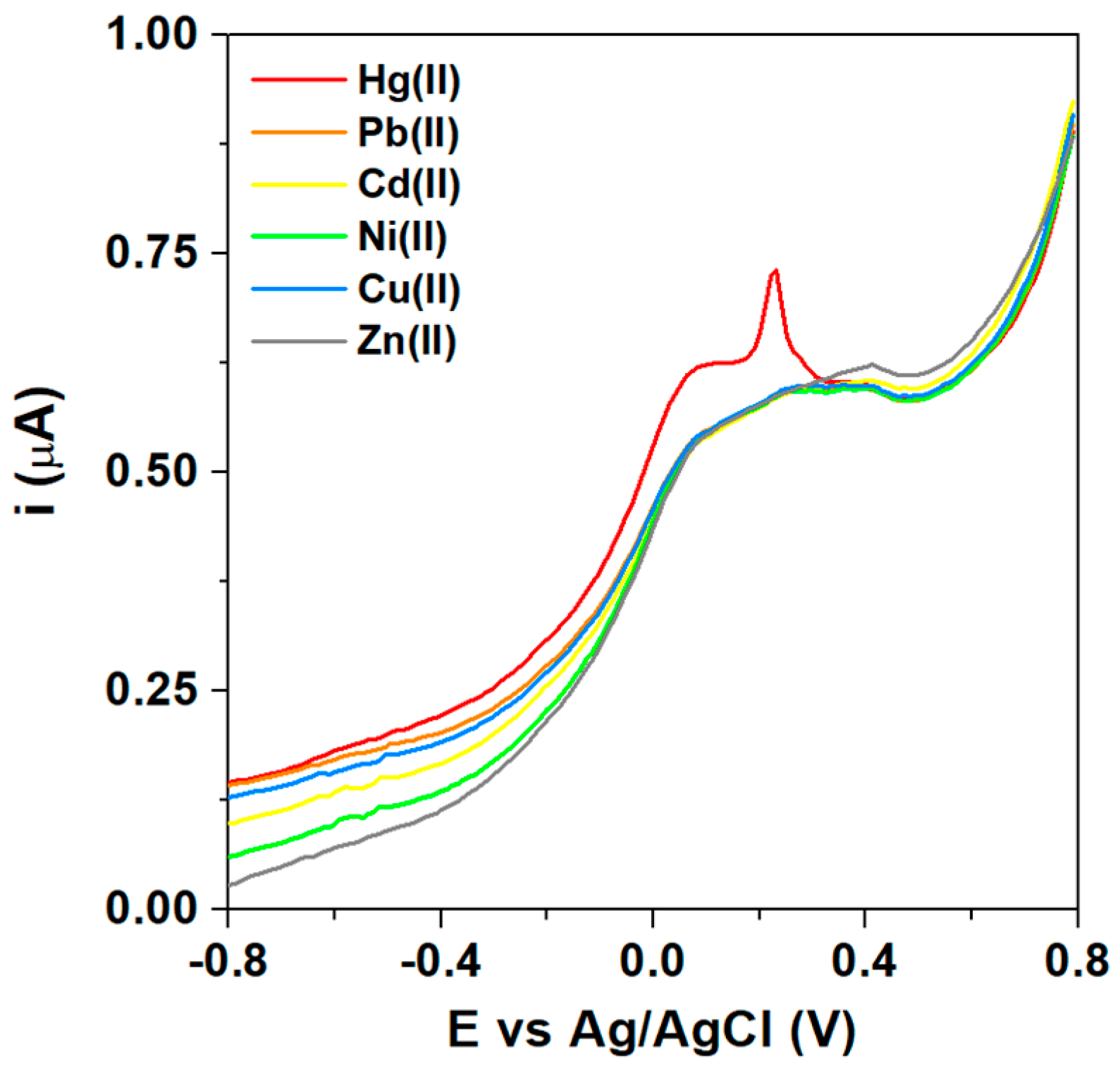
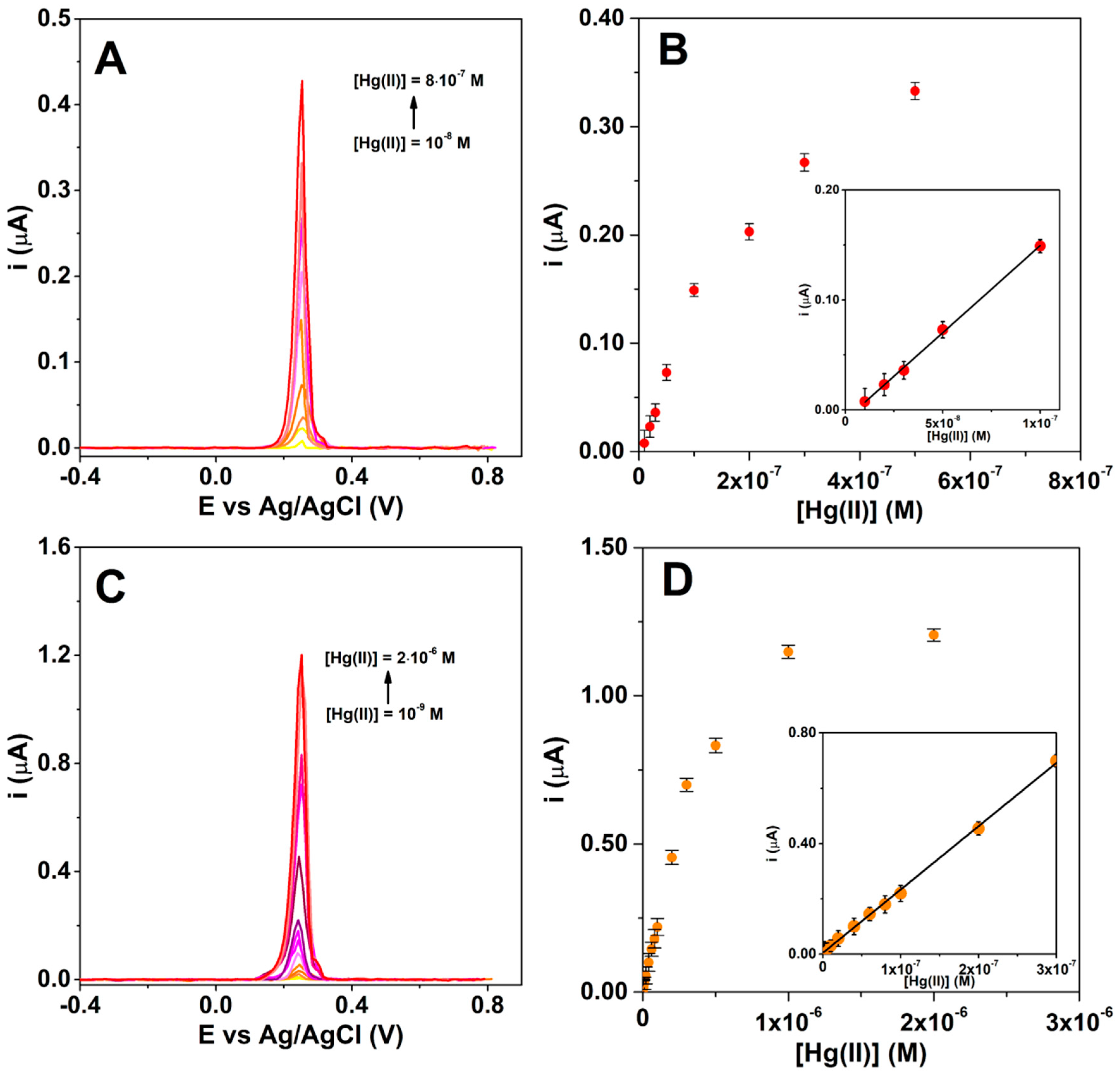
3.3. Voltammetric Detection of Hg(II)
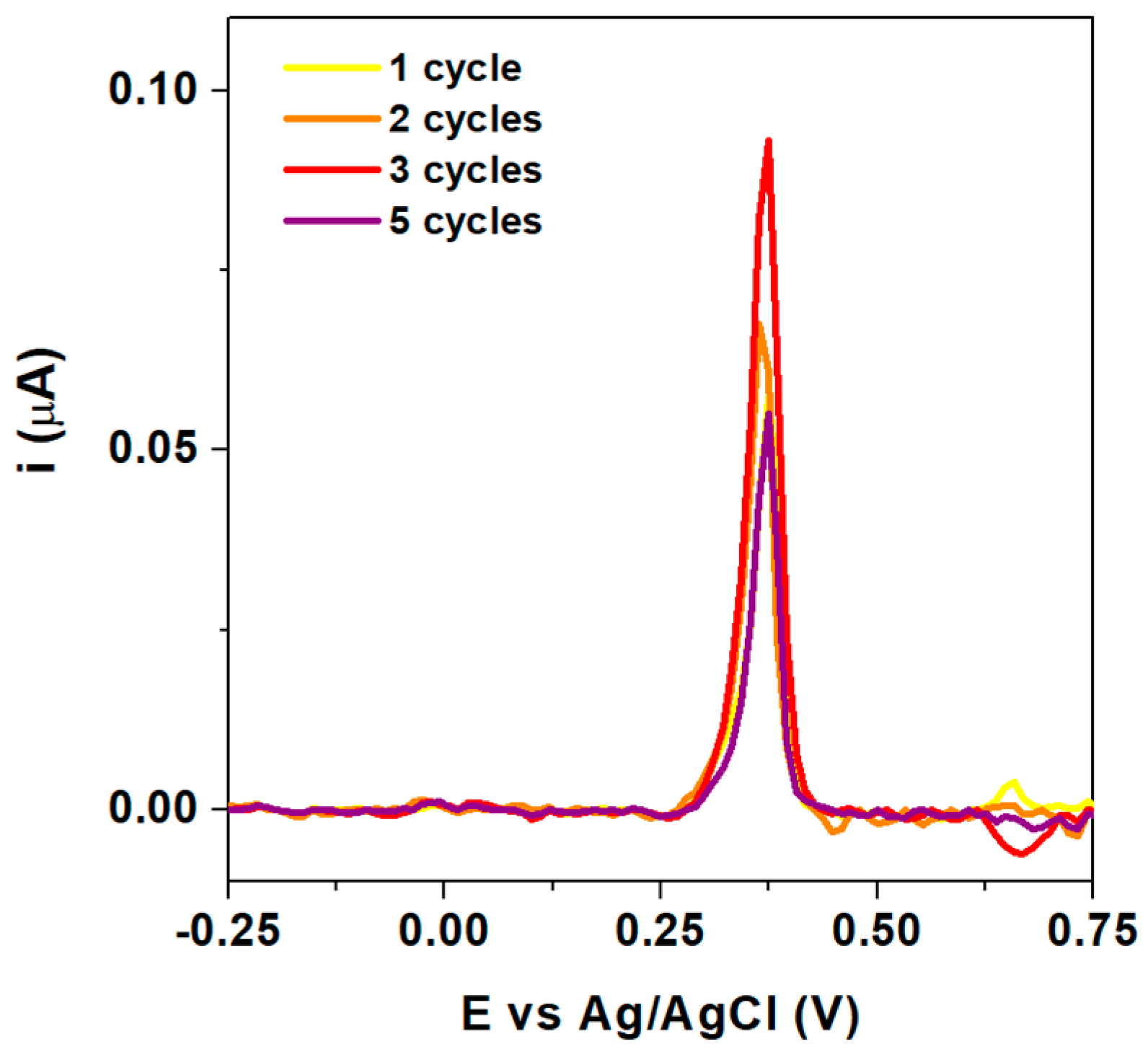
3.4. Reproducibility, Interference, and Real Sample Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Fu, Z.; Xi, S. The effects of heavy metals on human metabolism. Toxicol. Mech. Methods 2020, 30, 167–176. [Google Scholar] [CrossRef] [PubMed]
- Council Directive 98/83/EC of 3 November 1998 on the Quality of Water Intended for Human Consumption. Available online: http://data.europa.eu/eli/dir/1998/83/oj (accessed on 27 October 2015).
- Saleh, T.A.; Fadillah, G.; Ciptawati, E.; Khaled, M. Analytical methods for mercury speciation, detection, and measurement in water, oil, and gas. Trends Anal. Chem. 2020, 132, 116016. [Google Scholar] [CrossRef]
- Vasile, G.G.; Gheorghe, S.; Ene, C.; Serban, E.A.; Stoica, C. Total mercury in fish and benthic invertebrates collected from Olt River, Romania. Rev. Chim. 2019, 70, 263–268. [Google Scholar] [CrossRef]
- Wijngaard, R.R.; van der Perk, M.; van der Grift, B.; de Nijs, T.C.M.; Bierkens, M.F.P. The Impact of Climate Change on Metal Transport in a Lowland Catchment. Water Air Soil Pollut. 2017, 228, 107. [Google Scholar] [CrossRef] [PubMed]
- Shrestha, S.; Gunawardana, S.K.; Piman, T.; Babel, M.S. Assessment of the impact of climate change and mining activities on streamflow and selected metal’s loading in the Chindwin River, Myanmar. Environ. Res. 2020, 181, 108942. [Google Scholar] [CrossRef] [PubMed]
- Vasile, G.G.; Marin, N.M.; Cruceru, L.V.; Simion, M.; Galaon, T.; Lehr, C.B. Determination of ultra-trace mercury in water samples based on cold vapor atomic fluorescence spectrometry using a gold trap. Rev. Chim. 2016, 67, 1427–1432. [Google Scholar]
- Hu, P.Y.; Wang, X.; Yang, L.; Yang, H.Y.; Tang, Y.Y.; Luo, H.; Xiong, X.L.; Jiang, X.; Huang, K. Speciation of mercury by hydride generation ultraviolet atomization-atomic fluorescence spectrometry without chromatographic separation. Microchem. J. 2018, 143, 228–233. [Google Scholar] [CrossRef]
- Tassone, A.; Moretti, S.; Martino, M.; Pirrone, N.; Sprovieri, F.; Naccarato, A. Modification of the EPA method 1631E for the quantification of total mercury in natural waters. Methods X 2020, 7, 100987. [Google Scholar] [CrossRef]
- Janani, B.; Syed, A.; Raju, L.; Marraiki, N.; Elgorban, A.M.; Zaghloul, S.S.; Thomas, A.M.; Das, A.; Sudheer, K.S. Highly selective and effective environmental mercuric ion detection method based on starch modified Ag NPs in presence of glycine. Opt. Commun. 2020, 465, 125564. [Google Scholar] [CrossRef]
- Fernández, Z.H.; Rojas, L.A.V.; Álvarez, A.M.; Álvarez, J.R.E.; dos Santos Júnior, J.A.; González, I.P.; González, M.R.; Macias, N.A.; Sánchez, D.L.; Torres, D.H. Application of Cold Vapor-Atomic Absorption (CVAAS) Spectrophotometry and Inductively Coupled Plasma-Atomic Emission Spectrometry methods for cadmium, mercury and lead analyses of fish samples. Validation of the method of CVAAS. Food Control 2015, 48, 37–42. [Google Scholar] [CrossRef]
- Angyus, S.B.; Darvasi, E.; Ponta, M.; Petreus, D.; Etz, R.; Senila, M.; Frentiu, M.; Frentiu, T. Interference-free, green microanalytical method for total mercury and methylmercury determination in biological and environmental samples using small-sized electrothermal vaporization capacitively coupled plasma microtorch optical emission spectrometry. Talanta 2020, 217, 121067. [Google Scholar] [CrossRef] [PubMed]
- Mohibul Kabir, K.M.; Jampaiah, D.; Kandjani, A.E.; Mullett, M.; Tardio, J.; Sabri, Y.M.; Bhargava, S.K. Cold vapor integrated quartz crystal microbalance (CV-QCM) based detection of mercury ions with gold nanostructures. Sensor. Actuator. B Chem. 2019, 290, 453–458. [Google Scholar] [CrossRef]
- Samaz, M.; Akgul, B.; Yildrim, D.; Sazmaz, A. Mercury uptake and phytotoxicity in terrestrial plants grown naturally in the Gumuskoy (Kutahya) mining area, Turkey. Int. J. Phytoremediat. 2016, 18, 69–76. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; He, Q.; Wu, L.; Sun, J.; Zheng, F.; Li, L.; Liu, W.; Liu, J. Ultrasensitive speciation analysis of mercury in waters by headspace solid-phase microextraction coupled with gas chromatography-triple quadrupole mass spectrometry. Microchem. J. 2020, 153, 104459. [Google Scholar] [CrossRef]
- Jia, X.; Zhao, J.; Ren, H.; Wang, J.; Hong, Z.; Zhang, X. Zwitterion-functionalized polymer microspheres-based solid phase extraction method on-line combined with HPLC–ICP-MS for mercury speciation. Talanta 2019, 196, 592–599. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, A.G.-M.; Carrington, P.; Rowley-Neale, S.J.; Banks, C.E. Recent advances in portable heavy metal electrochemical sensing platforms. Environ. Sci.Water Res. Technol. 2020, 6, 2676–2690. [Google Scholar] [CrossRef]
- Gao, C.; Huang, X.-J. Voltammetric determination of mercury(II). Trends Analyt. Chem. 2013, 51, 1–12. [Google Scholar]
- Yáñez-Sedeño, P.; González-Cortés, A.; Campuzano, S.; Pingarrón, J.M. Copper(I)-Catalyzed Click Chemistry as a Tool for the Functionalization of Nanomaterials and the Preparation of Electrochemical (Bio)Sensors. Sensors 2019, 19, 2379. [Google Scholar] [CrossRef]
- Qiu, S.; Xie, L.; Gao, S.; Liu, Q.; Lin, Z.; Qiu, B.; Chena, G. Determination of copper(II) in the dairy product by an electrochemical sensor based on click chemistry. Anal. Chim. Acta 2011, 707, 57–61. [Google Scholar] [CrossRef]
- Tang, Y.; Zhang, J.; Tang, D.; Teng, L.; Lv, J.; Tang, D. Click-conjugation of nanogoldfunctionalized PAMAM dendrimer: Toward a novel electrochemical detection platform. Electroanalysis 2015, 27, 2280–2285. [Google Scholar] [CrossRef]
- Tang, D.; Zhang, J.; Tang, Y.; Teng, L.; Xia, B.; Tang, D. Hairpin DNA-dependent click conjugation of oligonucleotides for electrochemical monitoring of copper(II). Electroanalysis 2015, 27, 2513–2517. [Google Scholar] [CrossRef]
- Fomo, G.; Nwaji, N.; Nyokong, T. Low symmetric metallophthalocyanine modified electrode via click chemistry for simultaneous detection of heavy metals. J. Electroanal. Chem. 2018, 813, 58–66. [Google Scholar] [CrossRef]
- Ambrosi, A.; Chua, C.K.; Bonanni, A.; Pumera, M. Electrochemistry of Graphene and Related Materials. Chem. Rev. 2014, 114, 7150–7188. [Google Scholar] [CrossRef] [PubMed]
- Nag, A.; Mitra, A.; Mukhopadhyay, S.C. Graphene and its sensor-based applications: A review. Sens. Act. A Physical 2018, 270, 177–194. [Google Scholar] [CrossRef]
- Sang, M.; Shin, J.; Kim, K.; Yu, K.J. Electronic and Thermal Properties of Graphene and Recent Advances in Graphene Based Electronics Applications. Nanomaterials 2019, 9, 374. [Google Scholar] [CrossRef]
- Cernat, A.; Györfi, S.J.; Irimes, M.-B.; Tertiș, M.; Bodoki, A.; Pralea, I.-E.; Suciu, M.; Cristea, C. Click chemistry on azide-functionalized graphene oxide. Electrochem. Commun. 2019, 98, 23–27. [Google Scholar] [CrossRef]
- Rowley-Neale, S.J.; Randviir, E.P.; Abo Dena, A.S.; Banks, C.E. An overview of recent applications of reduced graphene oxide as a basis of electroanalytical sensing platforms. Appl. Mater. Today 2018, 10, 218–226. [Google Scholar] [CrossRef]
- Raicopol, M.D.; Chira, N.A.; Pandele, A.M.; Hanganu, A.; Ivanov, A.A.; Tecuceanu, V.; Bugean, I.G.; Buica, G.-O. Electrodes modified with clickable thiosemicarbazone ligands for sensitive voltammetric detection of Hg(II) ions. Sens. Actuators B. Chem. 2020, 313, 128030. [Google Scholar] [CrossRef]
- Chen, L.; Tang, Y.; Wang, K.; Liu, C.; Luo, S. Direct electrodeposition of reduced graphene oxide on glassy carbon electrode and its electrochemical application. Electrochem. Commun. 2011, 13, 133–137. [Google Scholar] [CrossRef]
- Buica, G.-O.; Ivanov, A.A.; Lazar, I.-G.; Tatu (Arnold), G.-L.; Omocea, C.; Birzan, L.; Ungureanu, E.-M. Colorimetric and voltammetric sensing of mercury ions using 2,2′-(ethane-1,2-diylbis((2-(azulen-2-ylamino)-2-oxoethyl)azanediyl)) diacetic acid. J. Electroanal. Chem. 2019, 849, 113351. [Google Scholar] [CrossRef]
- Buica, G.-O.; Lazar, I.-G.; Saint-Aman, E.; Tecuceanu, V.; Dumitriu, C.; Ivanov, A.A.; Stoian, A.B.; Ungureanu, E.-M. Ultrasensitive modified electrode based on poly(1H-pyrrole-1-hexanoic acid) for Pb(II) detection. Sens. Actuators B 2017, 246, 434–443. [Google Scholar] [CrossRef]
- Water Quality–Determination of Mercury–Method Using Atomic Absorption Spectrometry (AAS) with and without Enrichment. Available online: https://www.iso.org/standard/51964.html (accessed on 14 April 2012).
- Water quality—Determination of Mercury—Method Using Atomic Fluorescence Spectrometry. Available online: https://www.iso.org/standard/38502.html. (accessed on 13 June 2006).
- Salinas, F.; Espinosa-Mansilla, A.; López-Martínez, L.; López-de-Alba, P.L. Selective extraction-spectrophotometric determination of microamounts of palladium in catalysts. Chem. Anal. (Warsaw) 2001, 46, 239–248. [Google Scholar]
- Lobana, T.S.; Sharma, R.; Bawa, G.; Khanna, S. Bonding and structure trends of thiosemicarbazone derivatives of metals—An overview. Coord. Chem. Rev. 2009, 253, 977–1055. [Google Scholar] [CrossRef]
- Padhyé, S.; Kauffman, G.B. Transition metal complexes of semicarbazones and thiosemicarbazones. Coord. Chem. Rev. 1985, 63, 127–160. [Google Scholar] [CrossRef]
- Allongue, P.; Delamar, M.; Desbat, B.; Fagebaume, O.; Hitmi, R.; Pinson, J.; Savéant, J.-M. Covalent Modification of Carbon Surfaces by Aryl Radicals Generated from the Electrochemical Reduction of Diazonium Salts. J. Am. Chem. Soc. 1997, 119, 201–207. [Google Scholar] [CrossRef]
- McCreery, R.L. Advanced Carbon Electrode Materials for Molecular Electrochemistry. Chem. Rev. 2008, 108, 2646–2687. [Google Scholar] [CrossRef]
- Velický, M.; Toth, P.S.; Woods, C.R.; Novoselov, K.S.; Dryfe, R.A.W. Electrochemistry of the Basal Plane versus Edge Plane of Graphite Revisited. J. Phys. Chem. C 2019, 123, 11677–11685. [Google Scholar] [CrossRef]
- Ray, K.; McCreery, R.L. Spatially Resolved Raman Spectroscopy of Carbon Electrode Surfaces: Observations of Structural and Chemical Heterogeneity. Anal. Chem. 1997, 69, 4680–4687. [Google Scholar] [CrossRef]
- Kariuki, J.K.; McDermott, M.T. Nucleation and Growth of Functionalized Aryl Films on Graphite Electrodes. Langmuir 1999, 15, 6534–6540. [Google Scholar] [CrossRef]
- Koehler, F.M.; Jacobsen, A.; Ensslin, K.; Stampfer, C.; Stark, W.J. Selective Chemical Modification of Graphene Surfaces: Distinction Between Single- and Bilayer Graphene. Small 2010, 6, 1125–1130. [Google Scholar] [CrossRef]
- Paulus, G.L.C.; Wang, Q.H.; Strano, M.S. Covalent Electron Transfer Chemistry of Graphene with Diazonium Salts. Acc. Chem. Res. 2013, 46, 160–170. [Google Scholar] [CrossRef] [PubMed]
- Hossain, M.Z.; Walsh, M.A.; Hersam, M.C. Scanning Tunneling Microscopy, Spectroscopy, and Nanolithography of Epitaxial Graphene Chemically Modified with Aryl Moieties. J. Am. Chem. Soc. 2010, 132, 15399–15403. [Google Scholar] [CrossRef] [PubMed]
- Rice, R.J.; Pontikos, N.M.; McCreery, R.L. Quantitative correlations of heterogeneous electron-transfer kinetics with surface properties of glassy carbon electrodes. J. Am. Chem. Soc. 1990, 112, 4617–4622. [Google Scholar] [CrossRef]
- Kariuki, J.K.; McDermott, M.T. Formation of Multilayers on Glassy Carbon Electrodes via the Reduction of Diazonium Salts. Langmuir 2001, 17, 5947–5951. [Google Scholar] [CrossRef]
- Raicopol, M.D.; Andronescu, C.; Atasiei, R.; Hanganu, A.; Vasile, E.; Brezoiu, A.M.; Pilan, L. Organic layers via aryl diazonium electrochemistry: Towards modifying platinum electrodes for interference free glucose biosensors. Electrochim. Acta 2016, 206, 226–237. [Google Scholar] [CrossRef]
- Bard, A.J.; Faulkner, L.R. Electrochemical Methods: Fundamentals and Applications, 2nd ed.; John Wiley & Sons: New York, NY, USA, 2001; p. 237. [Google Scholar]
- Matsuda, H.; Ayabe, Y.Z. Zur Theorie der Randles-Sevčikschen Kathodenstrahl-Polarographie. Elektrochem 1955, 59, 494–503. [Google Scholar]
- Nicholson, R.S. Theory and Application of Cyclic Voltammetry for Measurement of Electrode Reaction Kinetics. Anal. Chem. 1965, 37, 1351–1355. [Google Scholar] [CrossRef]
- Lavagnini, I.; Antiochia, R.; Magno, F. An Extended Method for the Practical Evaluation of the Standard Rate Constant from Cyclic Voltammetric Data. Electroanalysis 2004, 16, 505–506. [Google Scholar] [CrossRef]
- Cumba, L.R.; Foster, C.W.; Brownson, D.A.C.; Smith, J.P.; Iniesta, J.; Thakur, B.; do Carmo, D.R.; Banks, C.E. Can the mechanical activation (polishing) of screen-printed electrodes enhance their electroanalytical response? Analyst 2016, 141, 2791–2799. [Google Scholar] [CrossRef]
- Bishop, G.W.; Ahiadu, B.K.; Smith, J.L.; Patterson, J.D. Use of Redox Probes for Characterization of Layer-by-Layer Gold Nanoparticle-Modified Screen-Printed Carbon Electrodes. J. Electrochem. Soc. 2017, 164, B23–B28. [Google Scholar] [CrossRef]
- Chen, P.; McCreery, R.L. Control of Electron Transfer Kinetics at Glassy Carbon Electrodes by Specific Surface Modification. Anal. Chem. 1996, 68, 3958–3965. [Google Scholar] [CrossRef]
- Saby, C.; Ortiz, B.; Champagne, G.Y.; Bélanger, D. Electrochemical Modification of Glassy Carbon Electrode Using Aromatic Diazonium Salts. 1. Blocking Effect of 4-Nitrophenyl and 4-Carboxyphenyl Groups. Langmuir 1997, 13, 6805–6813. [Google Scholar] [CrossRef]
- Greenwood, J.; Phan, T.H.; Fujita, Y.; Li, Z.; Ivasenko, O.; Vanderlinden, W.; Van Gorp, H.; Frederickx, W.; Lu, G.; Tahara, K.; et al. Covalent Modification of Graphene and Graphite Using Diazonium Chemistry: Tunable Grafting and Nanomanipulation. ACS Nano. 2015, 9, 5520–5535. [Google Scholar] [CrossRef]
- Zhang, Y.; Hao, H.; Wang, L. Effect of morphology and defect density on electron transfer of electrochemically reduced graphene oxide. Appl. Surf. Sci. 2016, 390, 385–392. [Google Scholar] [CrossRef]
- Barrow, N.J.; Cox, V.C. The effects of pH and chloride concentration on mercury sorption. I. By goethite. J. Soil. Sci. 1992, 43, 295–304. [Google Scholar] [CrossRef]
- Xu, L.; Yuan, R.; Chai, Y.Q. Mercury(II) ion potentiometric sensor based on a sulfur schiff’s base 1-(2-hydroxy-1,2-diphenylethylidene) thiosemicarbazide as ionophore. Chem. Lett. 2005, 34, 440–441. [Google Scholar]
- Ghazy, S.E.-S.; El-Reash, G.M.A.; Al-Gammal, O.A.; Yousef, T. Flotation-separation of toxic metal ions from aqueous solutions using thiosemicarbazide derivatives as chelating agents and oleic acid as a surfactant. Eur. J. Chem. 2010, 1, 76–82. [Google Scholar] [CrossRef]
- Wang, J. Voltammetry following nonelectrolytic preconcentration. In Electroanalytical Chemistry: A Series of Advances; Bard, A.J., Rubinstein, I., Eds.; Marcel Dekker: New York, NY, USA, 1989; Volume 16, pp. 1–89. [Google Scholar]
- Taverniers, I.; De Loose, M.; Van Bockstaele, E. Trends in Quality in the Analytical Laboratory. II. Analytical method validation and quality assurance. Trends Anal. Chem. 2004, 23, 535–552. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Electrode | Redox Probe | (V) | (V) | (V) | (A) | (A) | (cm s−1) | ||
|---|---|---|---|---|---|---|---|---|---|
| GC | FcMeOH | 0.20 | 0.12 | 0.08 | 3.4 × 10−6 | 3.4 × 10−6 | 1.3 | 0.96 | 5.6 × 10−3 |
| GC-ERGO | 0.21 | 0.14 | 0.07 | 5.2 × 10−6 | 5.2 × 10−6 | 2.5 | 0.98 | 1.1 × 10−2 | |
| GC | Fe(CN)6 3− | 0.30 | 0.10 | 0.20 | 5.4 × 10−6 | 5.5 × 10−6 | 0.087 | - | 3.6 × 10−4 |
| GC grafted a) | 0.36 | 0.02 | 0.34 | 3.7 × 10−6 | 4.1 × 10−6 | 0.031 | - | 1.3 × 10−4 | |
| GC|click|L | 0.46 | −0.17 | 0.63 | - | - | 0.002 | - | 8.3 × 10−6 | |
| GC-ERGO | 0.24 | 0.15 | 0.09 | 6.8 × 10−6 | 6.8 × 10−6 | 0.83 | - | 3.4 × 10−3 | |
| GC-ERGO grafted a) | 0.25 | 0.15 | 0.10 | 6.3 × 10−6 | 6.3 × 10−6 | 0.59 | - | 2.4 × 10−3 | |
| GC-ERGO|click|L | 0.27 | 0.13 | 0.14 | 5.1 × 10−6 | 5.2 × 10−6 | 0.24 | - | 9.9 × 10−4 |
| Electrode | Atomic Concentration (%) | Atomic Ratios | ||||||
|---|---|---|---|---|---|---|---|---|
| C 1s | O 1s | N 1s | Hg 4f | C/O | C/N | C/Hg | N/Hg | |
| GC|click|L | 83.0 | 15.9 | 1.1 | - | 5.6 | 64.2 | - | - |
| GC|click|L + Hg(II) a) | 82.9 | 15.3 | 1.1 | 0.7 | 5.6 | 60.8 | 106.9 | 1.2 |
| GC-ERGO | 90.6 | 8.2 | 1.2 | - | 10.4 | 68.3 | - | - |
| GC-ERGO grafted b) | 86.9 | 11.0 | 2.1 | - | 7.3 | 34.0 | - | - |
| GC-ERGO|click|L | 85.2 | 12.3 | 2.5 | - | 6.8 | 37.2 | - | - |
| GC-ERGO|click|L + Hg(II) a) | 85.1 | 11.6 | 2.0 | 1.3 | 7.1 | 34.5 | 42.6 | 1.2 |
| Electrode | Linearity Domain (M) | Linear Regression Equation | Limit of Detection (M) |
|---|---|---|---|
| GC|click|L | 10−8 ÷ 10−7 | i (A) = 8 × 10−9 + 1.58C (M); R2 = 0.998 | 7 × 10−9 |
| GC-ERGO|click|L | 10−9 ÷ 3 × 10−7 | i (A) = 4.5 × 10−9 + 2.3C (M); R2 = 0.9996 | 8 × 10−10 |
| Sample | Added Hg(II) (M) | Method | |||
|---|---|---|---|---|---|
| GC|Click|L | GC-ERGO|Click|L | VG-AAS | CV-AFS | ||
| Tap water | - | ND a | ND | ND | ND |
| Spiked tap water I | 2.5 × 10−8 | 2.47 (±0.46) × 10−8 | 2.48 (±0.52) × 10−8 | 2.61 (±0.31) × 10−8 | 2.54 (±0.25) × 10−8 |
| Spiked tap water II | 1 × 10−7 | 0.97 (±0.18) × 10−7 | 0.95 (±0.20) × 10−7 | 0.93 (±0.11) × 10−7 | 1.02 (±0.10) × 10−7 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Raicopol, M.D.; Pandele, A.M.; Dascălu, C.; Vasile, E.; Hanganu, A.; Vasile, G.-G.; Bugean, I.G.; Pirvu, C.; Stanciu, G.; Buica, G.-O. Improving the Voltammetric Determination of Hg(II): A Comparison Between Ligand-Modified Glassy Carbon and Electrochemically Reduced Graphene Oxide Electrodes. Sensors 2020, 20, 6799. https://doi.org/10.3390/s20236799
Raicopol MD, Pandele AM, Dascălu C, Vasile E, Hanganu A, Vasile G-G, Bugean IG, Pirvu C, Stanciu G, Buica G-O. Improving the Voltammetric Determination of Hg(II): A Comparison Between Ligand-Modified Glassy Carbon and Electrochemically Reduced Graphene Oxide Electrodes. Sensors. 2020; 20(23):6799. https://doi.org/10.3390/s20236799
Chicago/Turabian StyleRaicopol, Matei D., Andreea M. Pandele, Constanţa Dascălu, Eugeniu Vasile, Anamaria Hanganu, Gabriela-Geanina Vasile, Ioana Georgiana Bugean, Cristian Pirvu, Gabriela Stanciu, and George-Octavian Buica. 2020. "Improving the Voltammetric Determination of Hg(II): A Comparison Between Ligand-Modified Glassy Carbon and Electrochemically Reduced Graphene Oxide Electrodes" Sensors 20, no. 23: 6799. https://doi.org/10.3390/s20236799
APA StyleRaicopol, M. D., Pandele, A. M., Dascălu, C., Vasile, E., Hanganu, A., Vasile, G.-G., Bugean, I. G., Pirvu, C., Stanciu, G., & Buica, G.-O. (2020). Improving the Voltammetric Determination of Hg(II): A Comparison Between Ligand-Modified Glassy Carbon and Electrochemically Reduced Graphene Oxide Electrodes. Sensors, 20(23), 6799. https://doi.org/10.3390/s20236799