LSPR Biosensing Approach for the Detection of Microtubule Nucleation

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
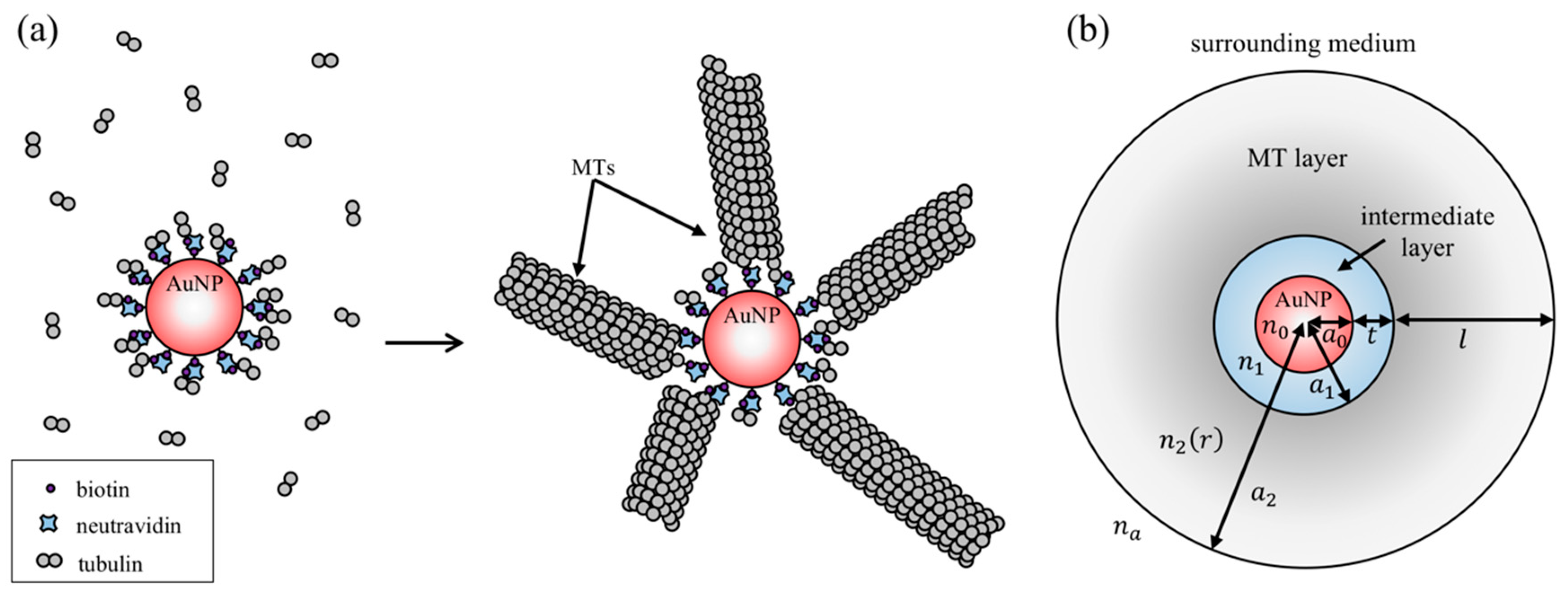
2.1. Theoretical Modeling
2.2. Sample Preparation
2.3. Fluorescence Microscopy
2.4. Ultraviolet-Visible Spectroscopy and Data Analysis
3. Results and Discussion
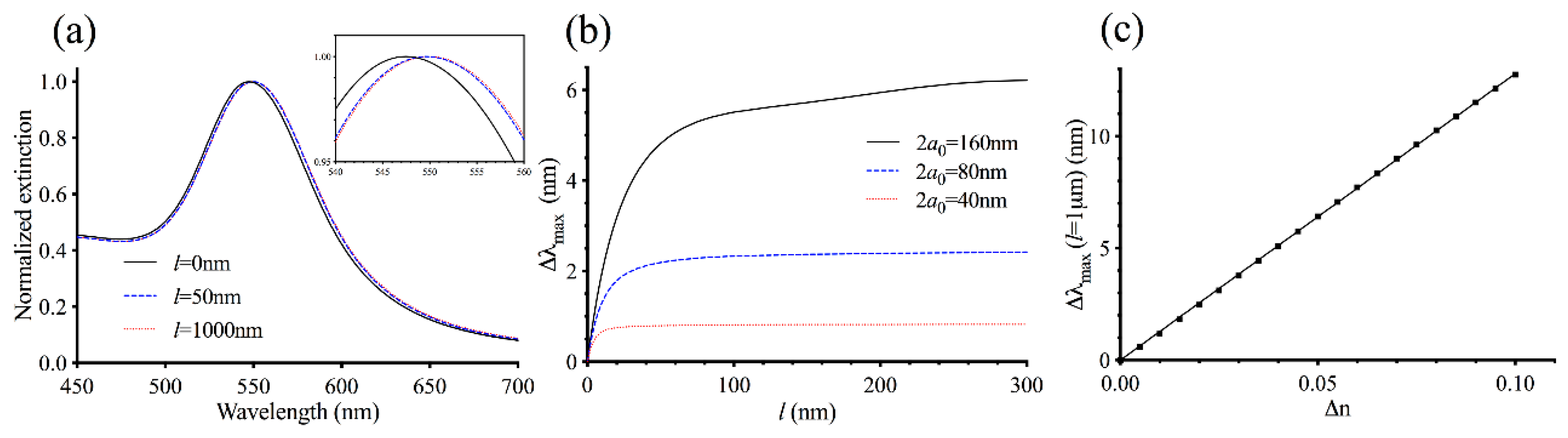
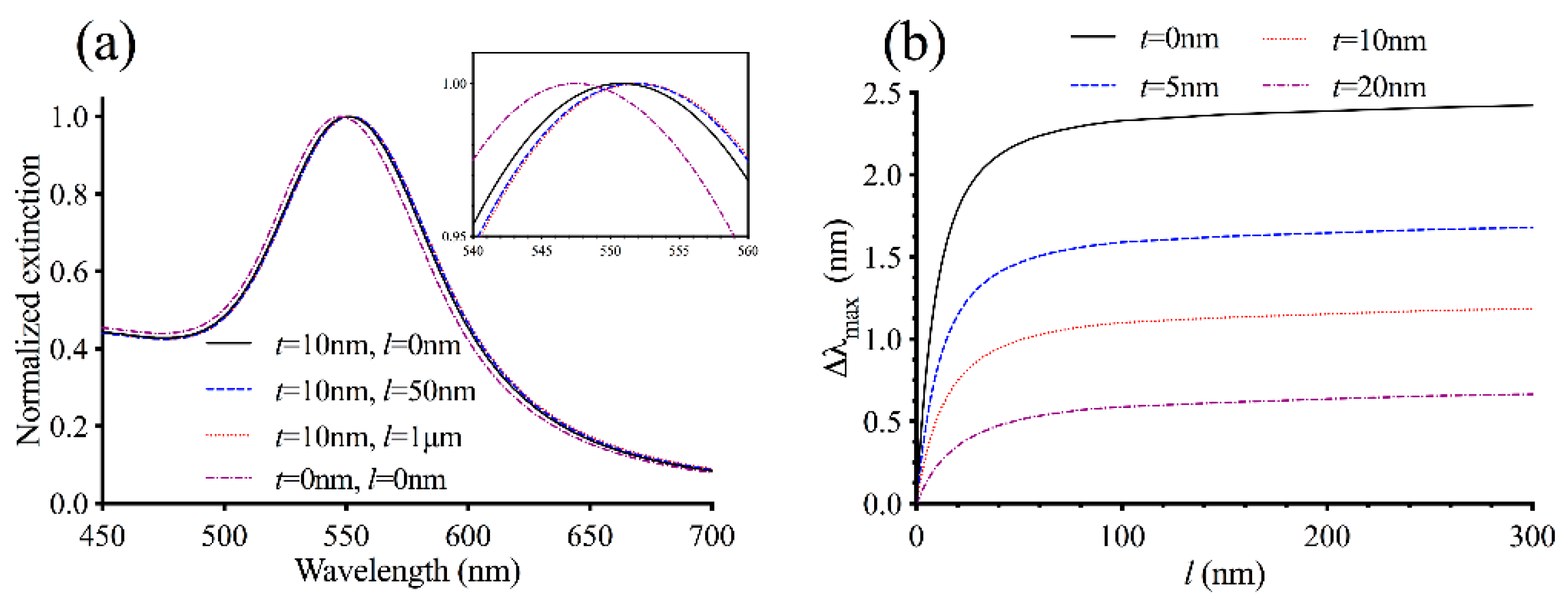
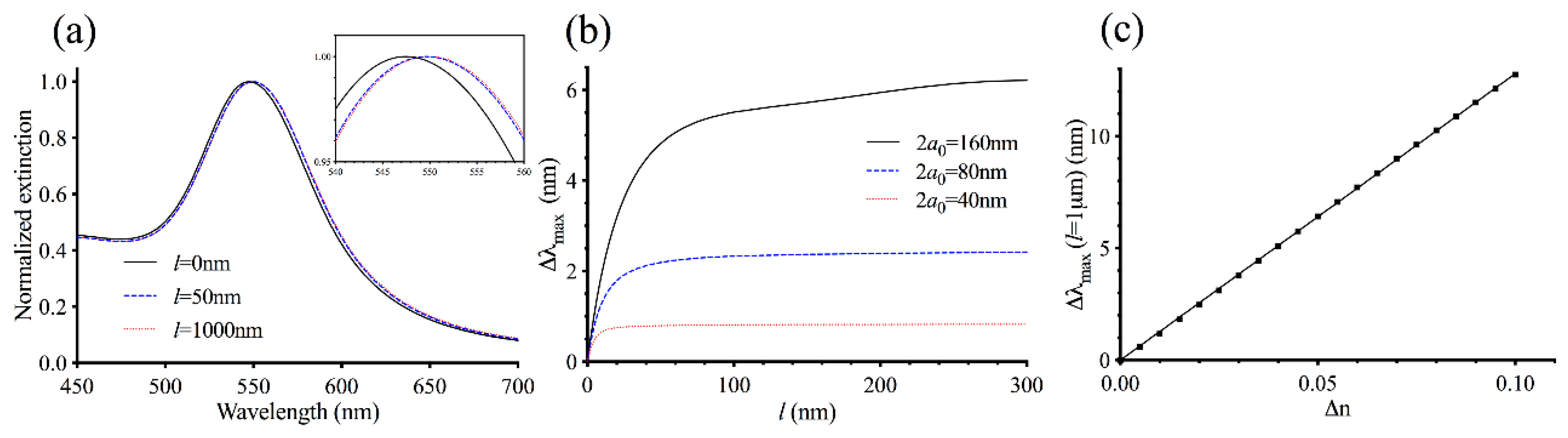
3.1. Theoretical Modeling
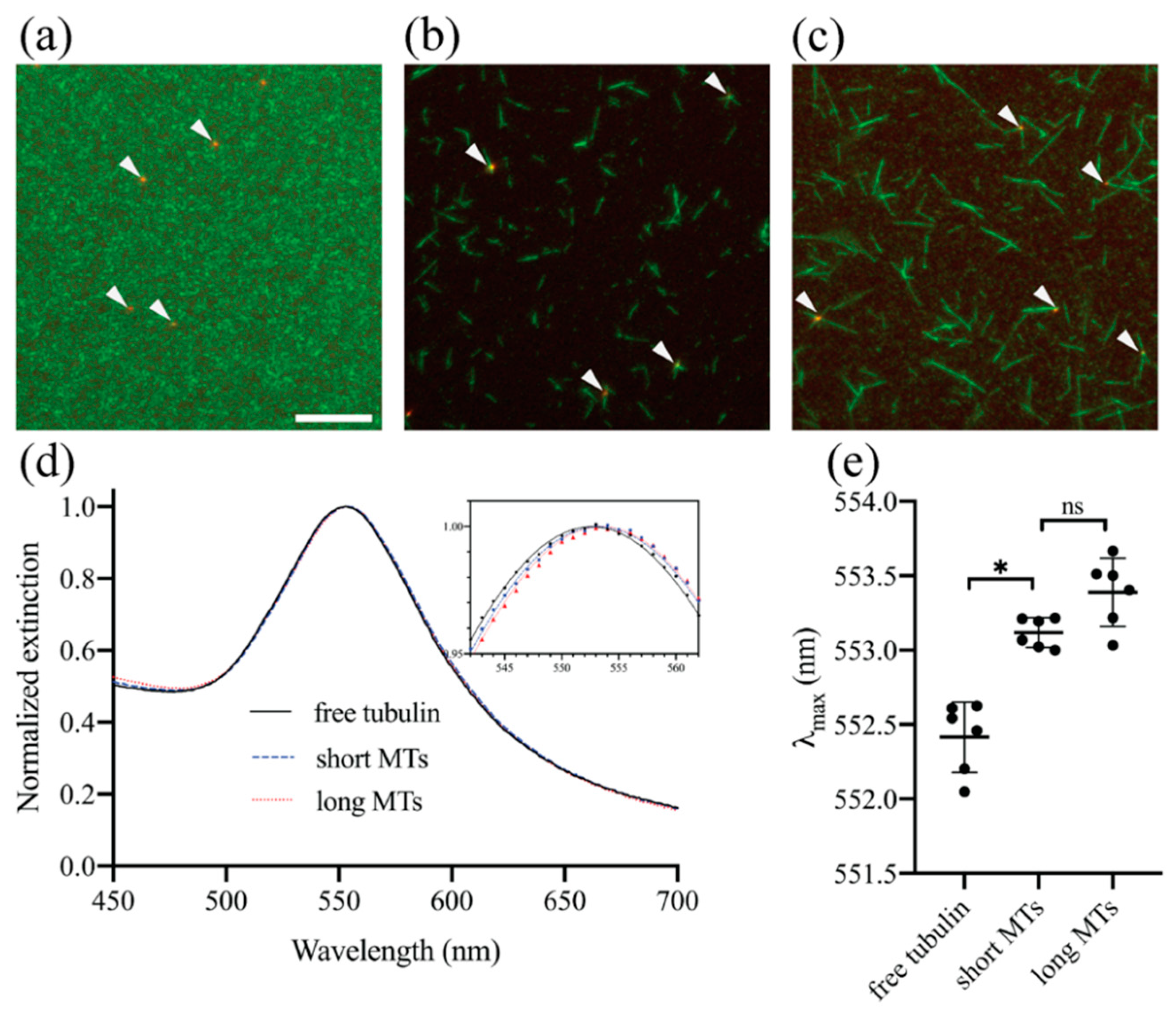
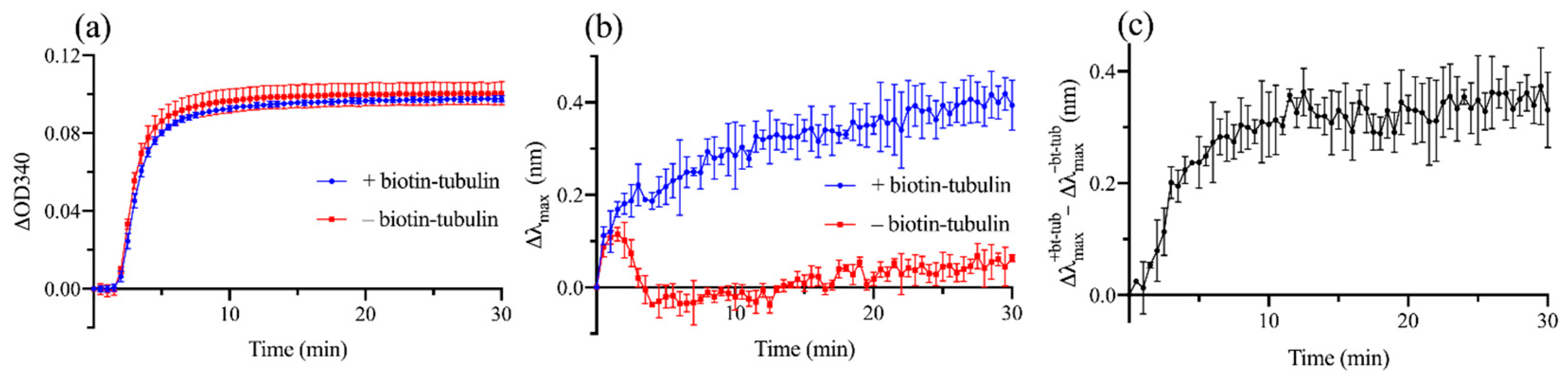
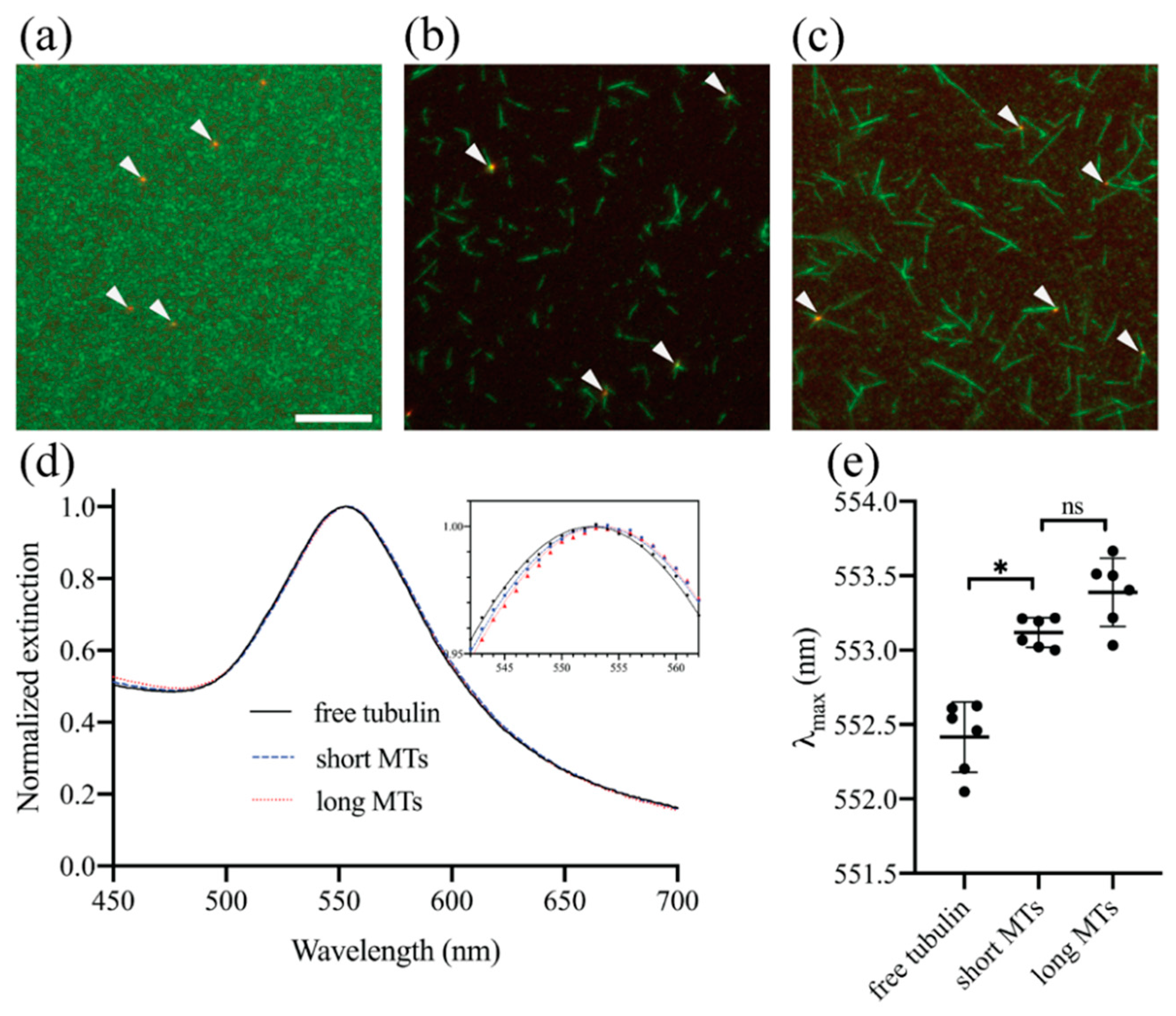
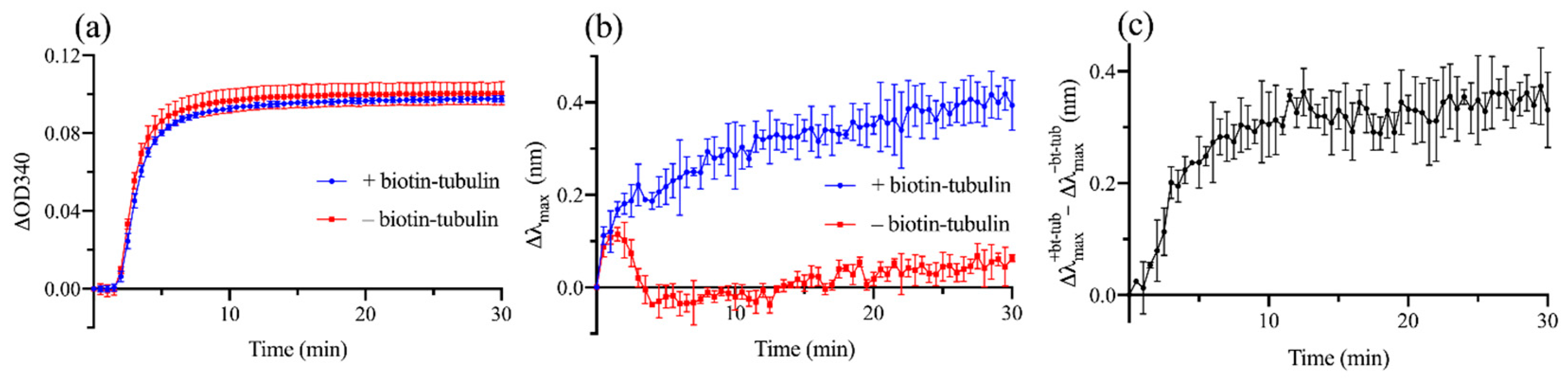
3.2. Experimental Demonstration of Localized Surface Plasmon Resonance (LSPR) Biosensing Approach for Detecting Microtubule (MT) Nucleation
4. Conclusions
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
Appendix A
References
- Mitchison, T.; Kirschner, M. Dynamic instability of microtubule growth. Nature 1984, 312, 237–242. [Google Scholar] [CrossRef] [PubMed]
- Gaskin, F.; Cantor, C.R.; Shelanski, M.L. Turbidimetric studies of the in vitro assembly and disassembly of porcine neurotubules. J. Mol. Biol. 1974, 89, 737–755. [Google Scholar] [CrossRef]
- Bonne, D.; Heuséle, C.; Simon, C.; Pantaloni, D. 4′,6-Diamidino-2-phenylindole, a fluorescent probe for tubulin and microtubules. J. Biol. Chem. 1985, 260, 2819–2825. [Google Scholar]
- Gaskin, F. Techniques for the study of microtubule assembly in vitro. Methods Enzymol. 1982, 85 Pt B, 433–439. [Google Scholar]
- Bailey, M.; Conway, L.; Gramlich, M.W.; Hawkins, T.L.; Ross, J.L. Modern methods to interrogate microtubule dynamics. Integr. Biol. Quant. Biosci. Nano Macro 2013, 5, 1324–1333. [Google Scholar] [CrossRef]
- Zwetsloot, A.J.; Tut, G.; Straube, A. Measuring microtubule dynamics. Essays Biochem. 2018, 62, 725–735. [Google Scholar] [CrossRef] [PubMed]
- Kaye, B.; Yoo, T.Y.; Foster, P.J.; Yu, C.-H.; Needleman, D.J. Bridging length scales to measure polymer assembly. Mol. Biol. Cell 2017, 28, 1379–1388. [Google Scholar] [CrossRef] [Green Version]
- Roostalu, J.; Surrey, T. Microtubule nucleation: Beyond the template. Nat. Rev. Mol. Cell Biol. 2017, 18, 702–710. [Google Scholar] [CrossRef]
- Voter, W.A.; Erickson, H.P. The kinetics of microtubule assembly. Evidence for a two-stage nucleation mechanism. J. Biol. Chem. 1984, 259, 10430–10438. [Google Scholar]
- Flyvbjerg, H.; Jobs, E. Microtubule dynamics. II. Kinetics of self-assembly. Phys. Rev. E 1997, 56, 7083–7099. [Google Scholar] [CrossRef]
- Portran, D.; Schaedel, L.; Xu, Z.; Théry, M.; Nachury, M.V. Tubulin acetylation protects long-lived microtubules against mechanical ageing. Nat. Cell Biol. 2017, 19, 391–398. [Google Scholar] [CrossRef]
- Wieczorek, M.; Bechstedt, S.; Chaaban, S.; Brouhard, G.J. Microtubule-associated proteins control the kinetics of microtubule nucleation. Nat. Cell Biol. 2015, 17, 907–916. [Google Scholar] [CrossRef] [PubMed]
- Thawani, A.; Kadzik, R.S.; Petry, S. XMAP215 is a microtubule nucleation factor that functions synergistically with the γ-tubulin ring complex. Nat. Cell Biol. 2018, 20, 575–585. [Google Scholar] [CrossRef] [PubMed]
- Piehl, M.; Tulu, U.S.; Wadsworth, P.; Cassimeris, L. Centrosome maturation: Measurement of microtubule nucleation throughout the cell cycle by using GFP-tagged EB1. Proc. Natl. Acad. Sci. USA 2004, 101, 1584–1588. [Google Scholar] [CrossRef] [Green Version]
- Gopinath, S.C.B. Biosensing applications of surface plasmon resonance-based Biacore technology. Sens. Actuators B Chem. 2010, 150, 722–733. [Google Scholar] [CrossRef]
- Nguyen, H.H.; Park, J.; Kang, S.; Kim, M. Surface plasmon resonance: A versatile technique for biosensor applications. Sensors 2015, 15, 10481–10510. [Google Scholar] [CrossRef] [PubMed]
- Homola, J. Surface plasmon resonance sensors for detection of chemical and biological species. Chem. Rev. 2008, 108, 462–493. [Google Scholar] [CrossRef]
- Mayer, K.M.; Hafner, J.H. Localized surface plasmon resonance sensors. Chem. Rev. 2011, 111, 3828–3857. [Google Scholar] [CrossRef]
- Hill, R.T. Plasmonic biosensors. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2015, 7, 152–168. [Google Scholar] [CrossRef]
- Unser, S.; Bruzas, I.; He, J.; Sagle, L. Localized surface plasmon resonance biosensing: Current challenges and approaches. Sensors 2015, 15, 15684–15716. [Google Scholar] [CrossRef] [PubMed]
- Kang, M.K.; Lee, J.; Nguyen, A.H.; Sim, S.J. Label-free detection of ApoE4-mediated β-amyloid aggregation on single nanoparticle uncovering Alzheimer’s disease. Biosens. Bioelectron. 2015, 72, 197–204. [Google Scholar] [CrossRef]
- Haes, A.J.; Zou, S.; Schatz, G.C.; Van Duyne, R.P. A nanoscale optical biosensor: The long range distance dependence of the localized surface plasmon resonance of noble metal nanoparticles. J. Phys. Chem. B 2004, 108, 109–116. [Google Scholar] [CrossRef]
- Marinakos, S.M.; Chen, S.; Chilkoti, A. Plasmonic detection of a model analyte in serum by a gold nanorod sensor. Anal. Chem. 2007, 79, 5278–5283. [Google Scholar] [CrossRef]
- Eck, D.; Helm, C.A.; Wagner, N.J.; Vaynberg, K.A. Plasmon resonance measurements of the adsorption and adsorption kinetics of a biopolymer onto gold nanocolloids. Langmuir 2001, 17, 957–960. [Google Scholar] [CrossRef]
- Khlebtsov, N.G.; Bogatyrev, V.A.; Khlebtsov, B.N.; Dykman, L.A.; Englebienne, P. A multilayer model for gold nanoparticle bioconjugates: Application to study of gelatin and human IgG adsorption using extinction and light scattering Spectra and the dynamic light scattering method. Colloid J. 2003, 65, 622–635. [Google Scholar] [CrossRef]
- Raschke, G.; Kowarik, S.; Franzl, T.; Sönnichsen, C.; Klar, T.A.; Feldmann, J.; Nichtl, A.; Kürzinger, K. Biomolecular recognition based on single gold nanoparticle light scattering. Nano Lett. 2003, 3, 935–938. [Google Scholar] [CrossRef]
- Yonzon, C.R.; Jeoung, E.; Zou, S.; Schatz, G.C.; Mrksich, M.; Van Duyne, R.P. A comparative analysis of localized and propagating surface plasmon resonance sensors: The binding of concanavalin a to a monosaccharide functionalized self-assembled monolayer. J. Am. Chem. Soc. 2004, 126, 12669–12676. [Google Scholar] [CrossRef]
- Johnson, P.B.; Christy, R.W. Optical constants of the noble metals. Phys. Rev. B 1972, 6, 4370–4379. [Google Scholar] [CrossRef]
- Gittes, F.; Mickey, B.; Nettleton, J.; Howard, J. Flexural rigidity of microtubules and actin filaments measured from thermal fluctuations in shape. J. Cell Biol. 1993, 120, 923–934. [Google Scholar] [CrossRef] [Green Version]
- Barer, R.; Joseph, S. Refractometry of living cells: Part I. basic principles. J. Cell Sci. 1954, s3-95, 399–423. [Google Scholar]
- Petry, S.; Groen, A.C.; Ishihara, K.; Mitchison, T.J.; Vale, R.D. Branching microtubule nucleation in xenopus egg extracts mediated by augmin and TPX2. Cell 2013, 152, 768–777. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Brown, P.H.; Schuck, P. On the distribution of protein refractive index increments. Biophys. J. 2011, 100, 2309–2317. [Google Scholar] [CrossRef]
- Perelman, A.Y. Scattering by particles with radially variable refractive indices. Appl. Opt. 1996, 35, 5452–5460. [Google Scholar] [CrossRef] [PubMed]
- Bohren, C.F.; Huffman, D.R. Absorption and Scattering of Light by Small Particles; Wiley-VCH: Weinheim, Germany, 1998; ISBN 978-0-471-29340-8. [Google Scholar]
- Toon, O.B.; Ackerman, T.P. Algorithms for the calculation of scattering by stratified spheres. Appl. Opt. 1981, 20, 3657–3660. [Google Scholar] [CrossRef] [PubMed]
- Castoldi, M.; Popov, A.V. Purification of brain tubulin through two cycles of polymerization-depolymerization in a high-molarity buffer. Protein Expr. Purif. 2003, 32, 83–88. [Google Scholar] [CrossRef]
- Dahlin, A.B.; Tegenfeldt, J.O.; Höök, F. Improving the instrumental resolution of sensors based on localized surface plasmon resonance. Anal. Chem. 2006, 78, 4416–4423. [Google Scholar] [CrossRef] [PubMed]
- Messersmith, R.E.; Nusz, G.J.; Reed, S.M. Using the localized surface plasmon resonance of gold nanoparticles to monitor lipid membrane assembly and protein binding. J. Phys. Chem. C 2013, 117, 26725–26733. [Google Scholar] [CrossRef]
- Nenninger, G.G.; Piliarik, M.; Homola, J. Data analysis for optical sensors based on spectroscopy of surface plasmons. Meas. Sci. Technol. 2002, 13, 2038. [Google Scholar] [CrossRef]
- Rosano, C.; Arosio, P.; Bolognesi, M. The X-ray three-dimensional structure of avidin. Biomol. Eng. 1999, 16, 5–12. [Google Scholar] [CrossRef]
- Ling, K.; Jiang, H.; Zhang, Q. A colorimetric method for the molecular weight determination of polyethylene glycol using gold nanoparticles. Nanoscale Res. Lett. 2013, 8, 538. [Google Scholar] [CrossRef] [PubMed]
- Wiscombe, W.J. Improved Mie scattering algorithms. Appl. Opt. 1980, 19, 1505–1509. [Google Scholar] [CrossRef] [PubMed]


© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hasegawa, K.; Nazarov, O.; Porter, E. LSPR Biosensing Approach for the Detection of Microtubule Nucleation. Sensors 2019, 19, 1436. https://doi.org/10.3390/s19061436
Hasegawa K, Nazarov O, Porter E. LSPR Biosensing Approach for the Detection of Microtubule Nucleation. Sensors. 2019; 19(6):1436. https://doi.org/10.3390/s19061436
Chicago/Turabian StyleHasegawa, Keisuke, Otabek Nazarov, and Evan Porter. 2019. "LSPR Biosensing Approach for the Detection of Microtubule Nucleation" Sensors 19, no. 6: 1436. https://doi.org/10.3390/s19061436
APA StyleHasegawa, K., Nazarov, O., & Porter, E. (2019). LSPR Biosensing Approach for the Detection of Microtubule Nucleation. Sensors, 19(6), 1436. https://doi.org/10.3390/s19061436