The Effects of Graphene Stacking on the Performance of Methane Sensor: A First-Principles Study on the Adsorption, Band Gap and Doping of Graphene

 ,
,
Abstract
:1. Introduction
2. Theory and Simulations
3. Results and Discussion
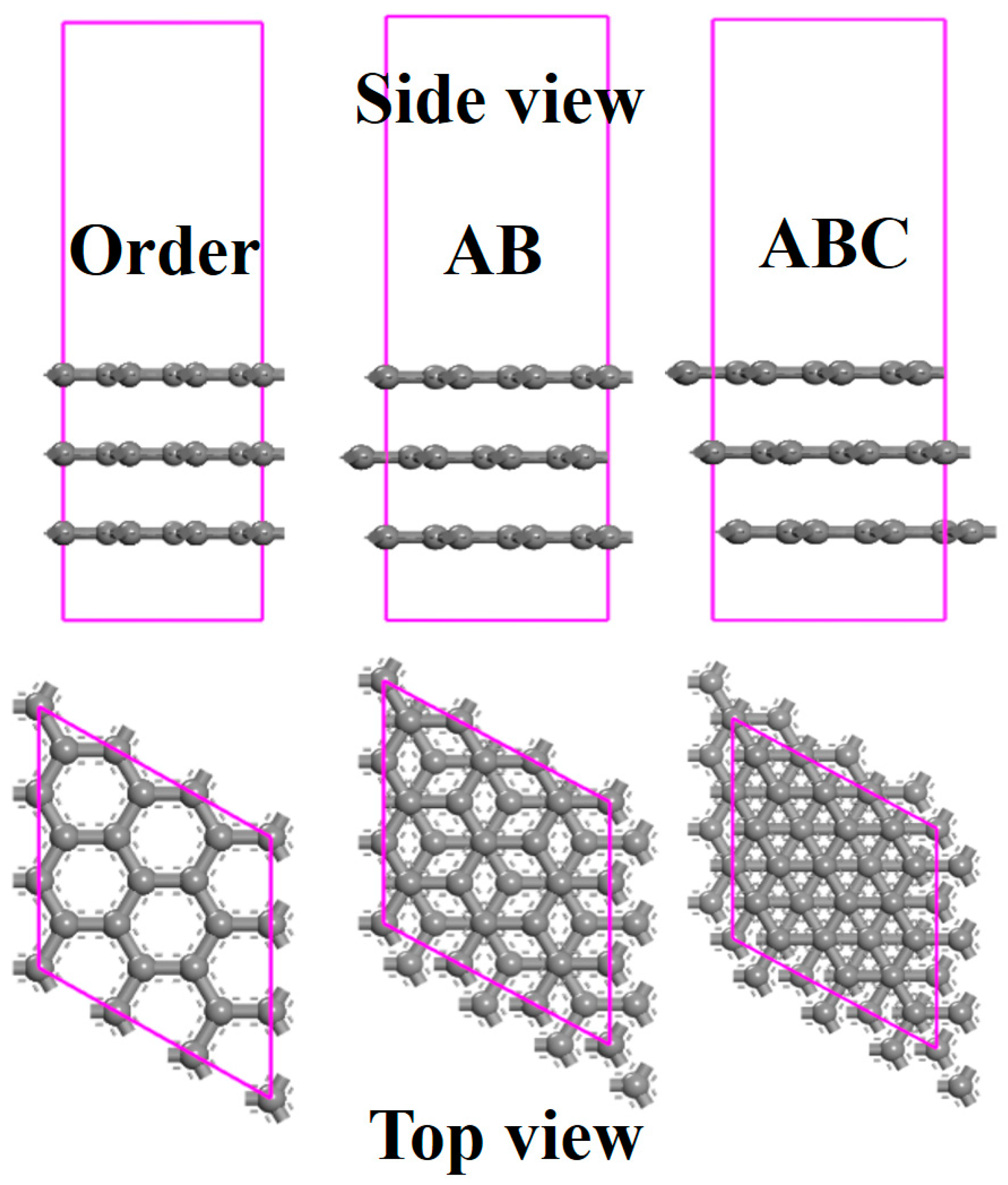
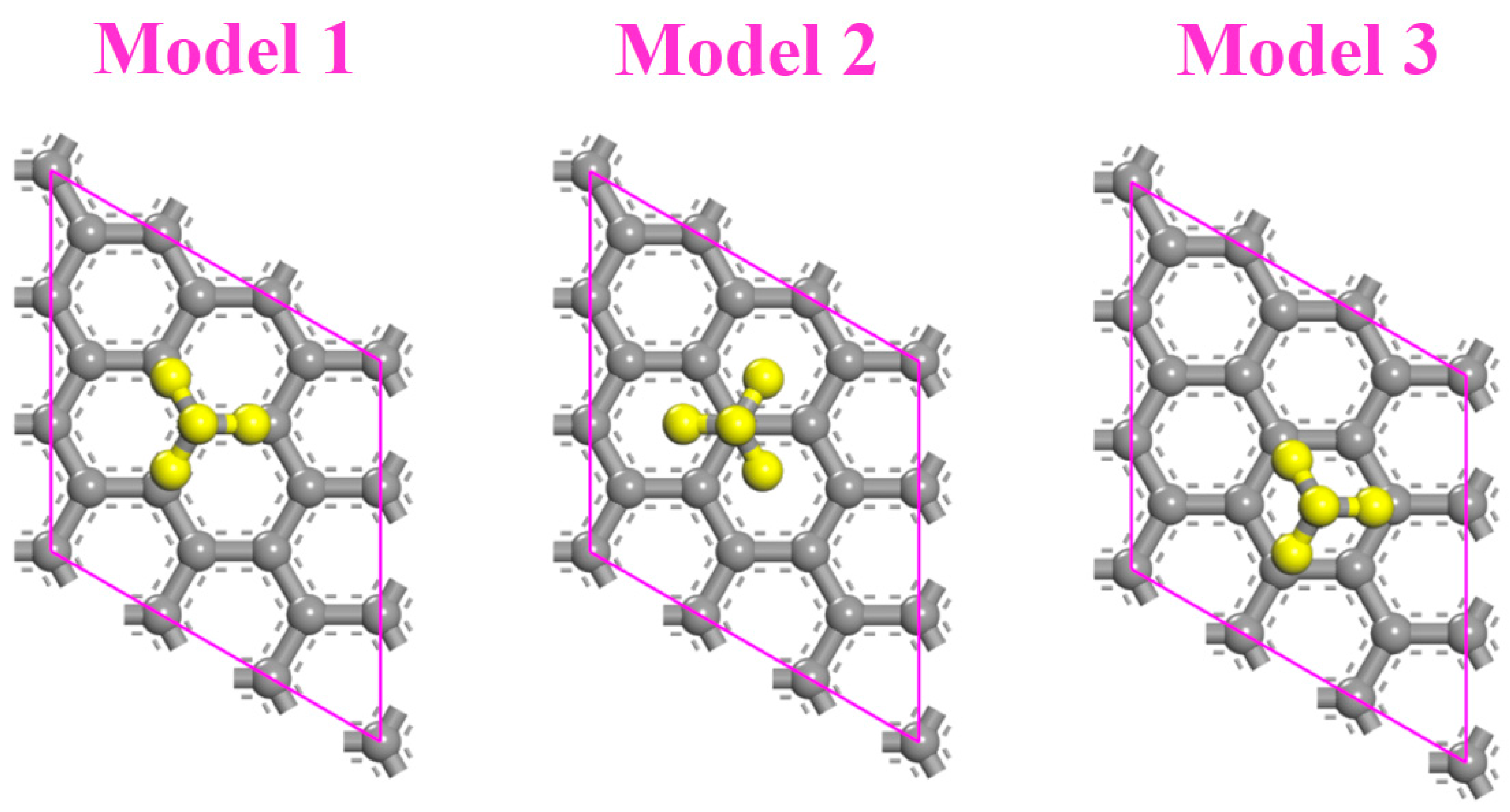
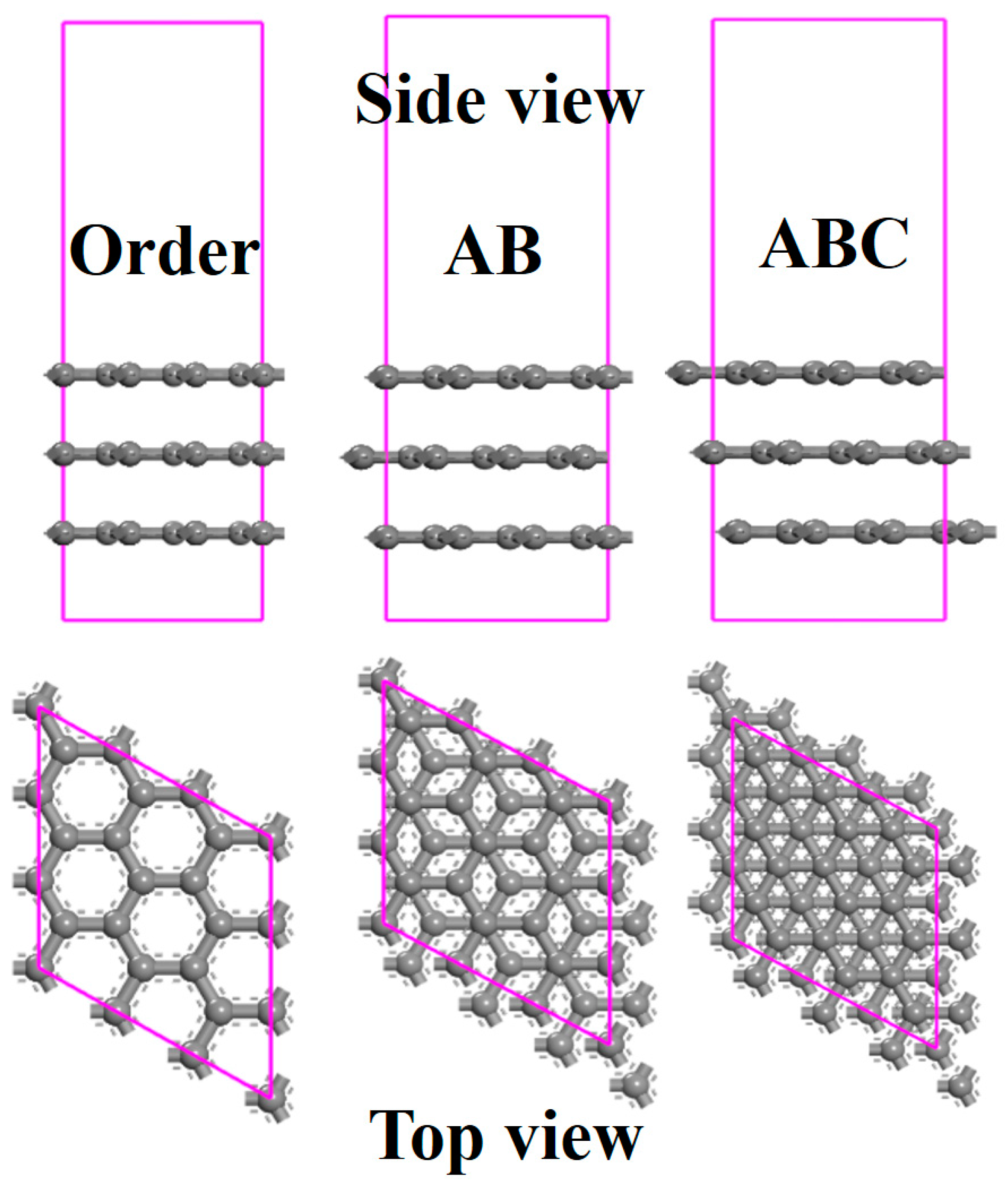
3.1. The Effects of Stacking on Adsorption
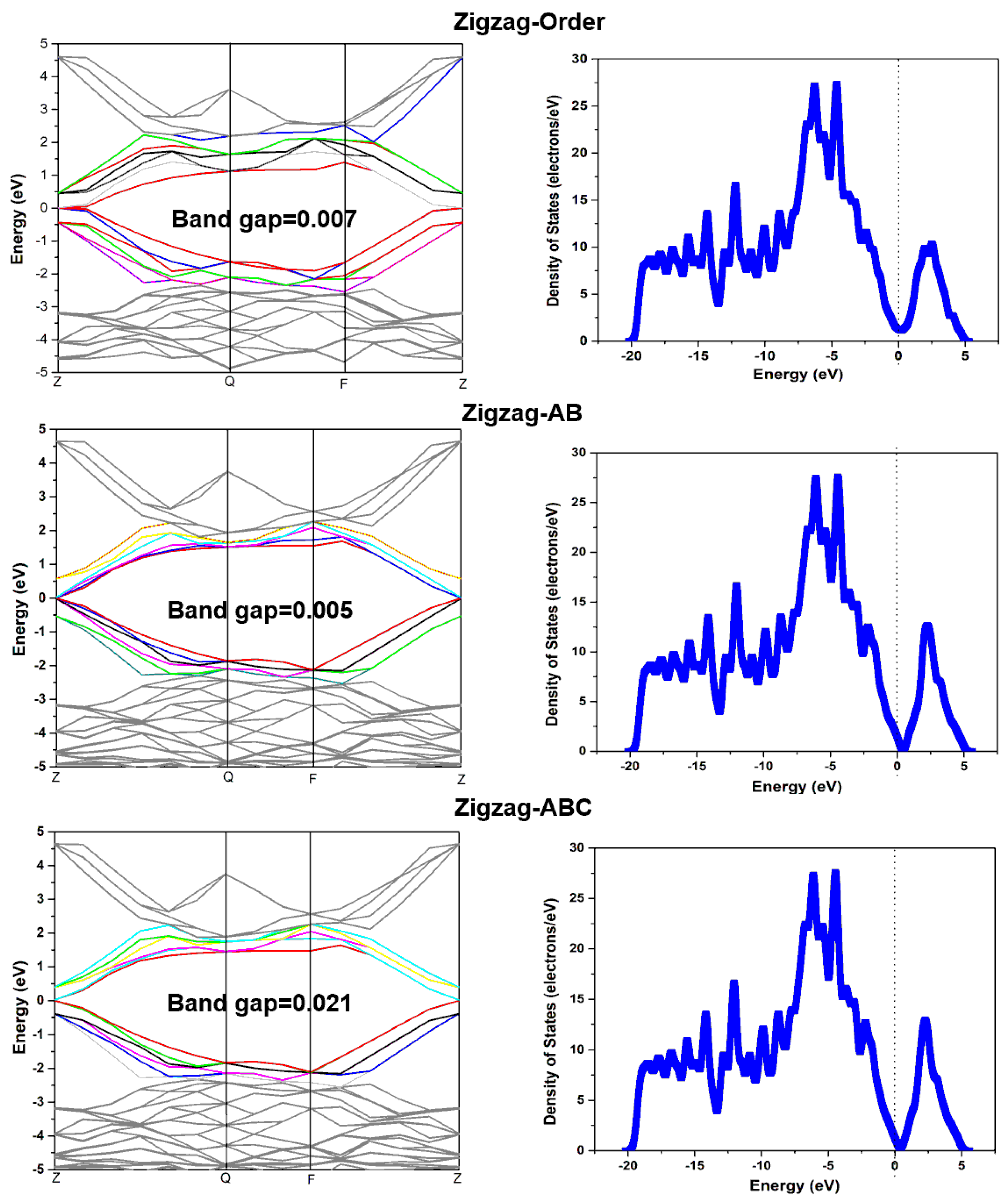
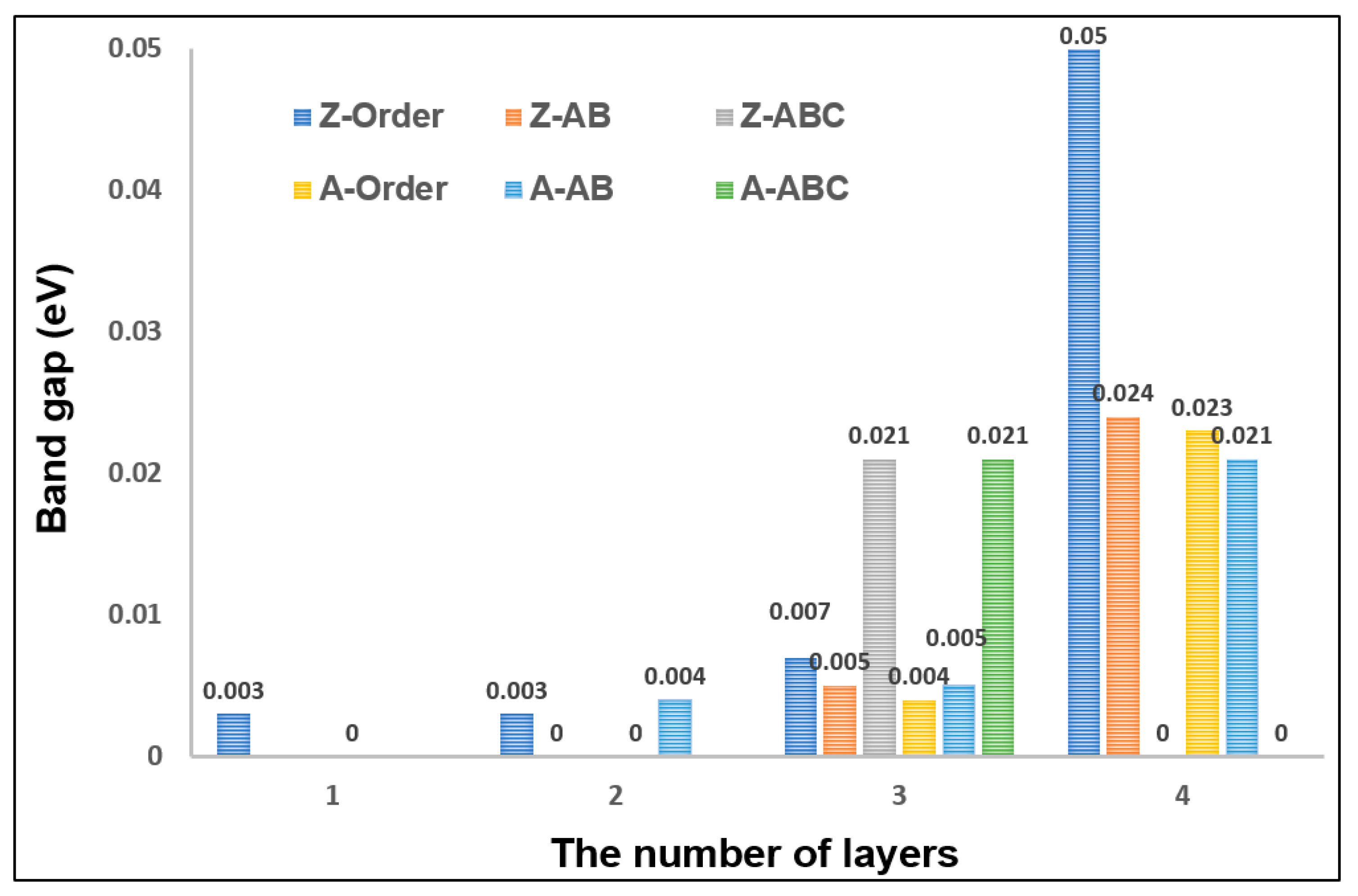
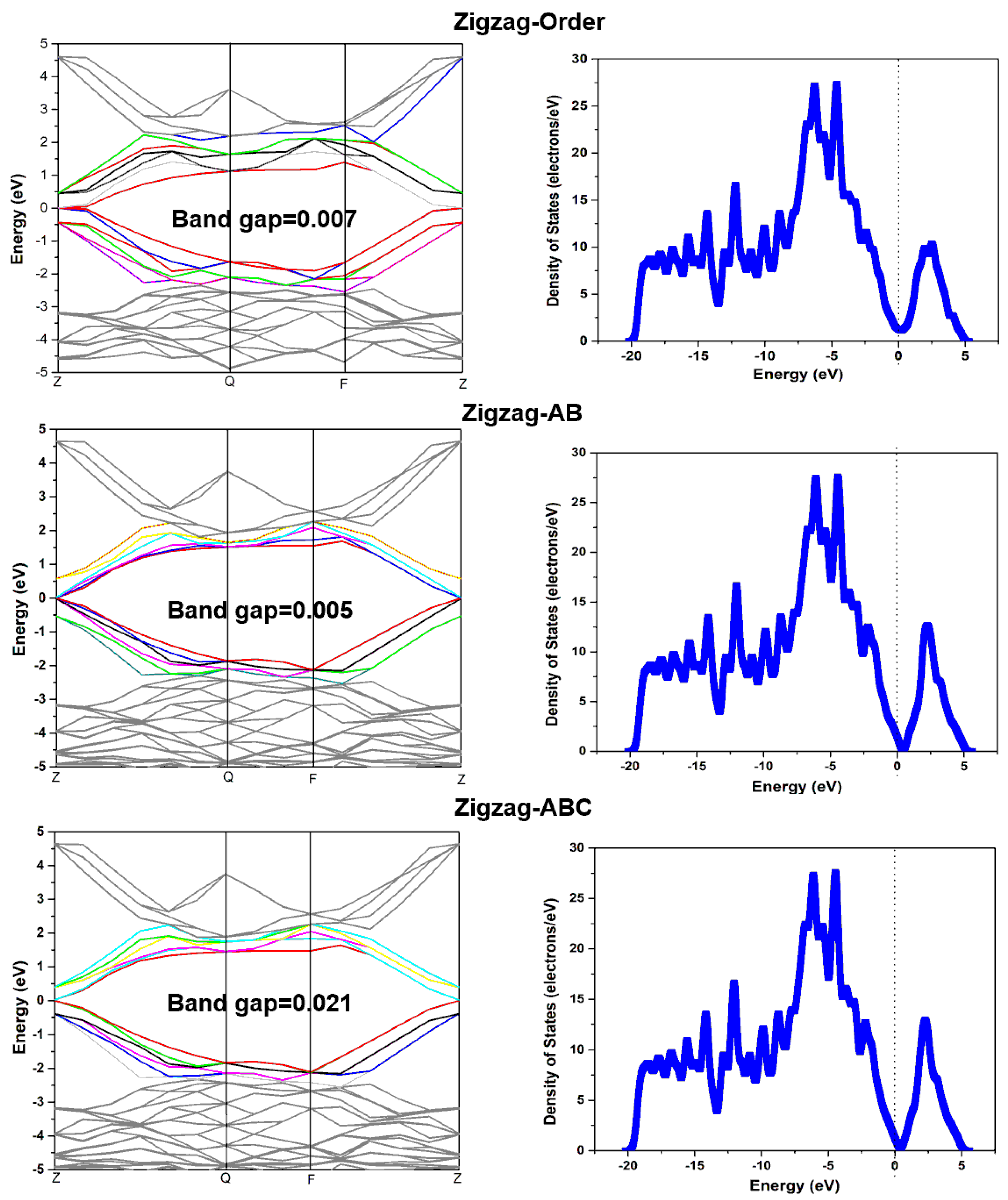
3.2. The Effects of Stacking on Electronic Properties, Such as Band Gap and Density of States
3.3. The Effects of Stacking on the Distance of Average Interlamellar and the Distance between Graphene and Methane
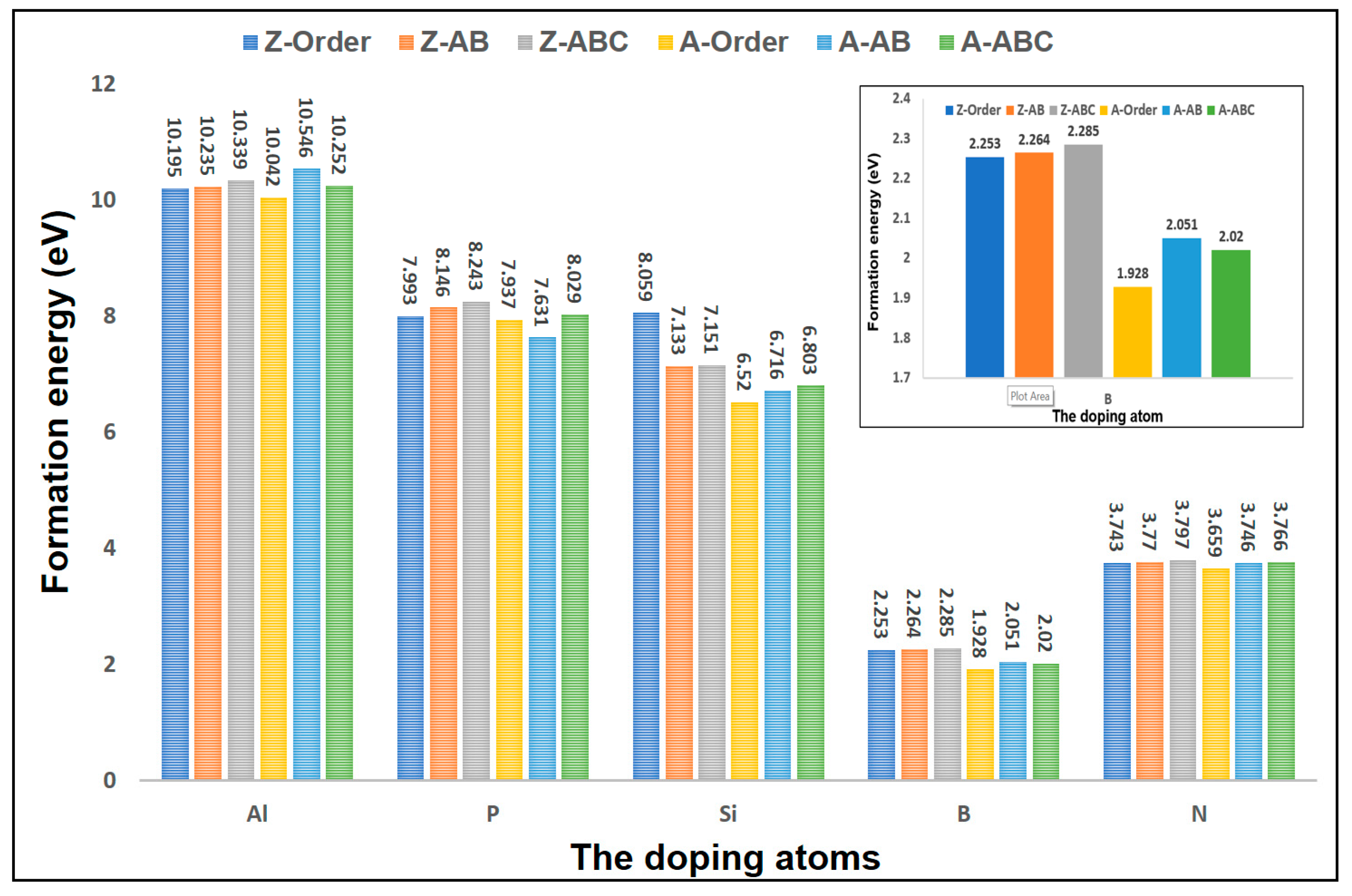
3.4. The Effects of Stacking on Doping
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Huang, X.; Qi, X.; Boey, F.; Zhang, H. Graphene-based composites. Chem. Soc. Rev. 2012, 41, 666–686. [Google Scholar] [CrossRef] [PubMed]
- Geim, A.K. Graphene: Status and prospects. Science 2009, 324, 1530–1534. [Google Scholar] [CrossRef] [PubMed]
- Geim, A.K.; Novoselov, K.S. The rise of graphene. Nat. Mater. 2007, 6, 183–191. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; Meng, Q.Y. Adsorption of methane on pristine and Al-doped graphene: A comparative study via first-principles calculation. Adv. Mater. Res. 2013, 602–604, 870–873. [Google Scholar] [CrossRef]
- Bhattacharyya, P.; Basu, P.; Saha, H.; Basu, S. Fast response methane sensor using nanocrystalline zinc oxide thin films derived by sol–gel method. Sens. Actuators B Chem. 2007, 124, 62–67. [Google Scholar] [CrossRef]
- Matveev, B.; Aidaraliev, M.; Gavrilov, G.; Zotova, N.; Karandashov, S.; Sotnikova, G.; Stus, N.; Talalakin, G.; Il’inskaya, N.; Aleksandrov, S. Room temperature InAs photodiode–InGaAs LED pairs for methane detection in the mid-IR. Sens. Actuators. B Chem. 1998, 51, 233–237. [Google Scholar] [CrossRef]
- Hussain, T.; Hankel, M.; Searles, D.J. Improving Sensing of Sulfur-Containing Gas Molecules with ZnO Monolayers by Implanting Dopants and Defects. J. Phys. Chem. C 2017, 121, 24365–24375. [Google Scholar] [CrossRef]
- Liu, L.; Yang, Q.; Ye, H.Y.; Chen, X.P.; Zhang, G.Q. Adsorption of gases on monolayer GeSe: A first principle study. In Proceedings of the 18th International Conference on IEEE Thermal, Mechanical and Multi-Physics Simulation and Experiments in Microelectronics and Microsystems (EuroSimE), Dresden, Germany, 3–5 April 2017; pp. 1–5. [Google Scholar]
- Meng, R.; Huang, Y.; Yang, Q.; Chen, X.P. A first-principle study of H2, CO, CH4, H2S and SO2 gas molecules on antimonene. In Proceedings of the 17th International Conference on International Conference on IEEE Electronic Packaging Technology (ICEPT), Wuhan, China, 16–19 August 2016; pp. 741–744. [Google Scholar]
- Hussain, T.; Kaewmaraya, T.; Chakraborty, S.; Rajeev, A. Defect and substitution-induced silicene sensor to probe toxic gases. J. Phys. Chem. C 2016, 120, 25256–25262. [Google Scholar] [CrossRef]
- Hu, W.; Wu, X.J.; Li, Z.; Yang, J.L. Porous silicene as a hydrogen purification membrane. Phys. Chem. Chem. Phys. 2013, 15, 5753–5757. [Google Scholar] [CrossRef] [PubMed]
- Mahabal, M.S.; Deshpande, M.D.; Hussain, T.; Rajeev, A. Sensing characteristics of phosphorene monolayers toward PH3 and AsH3 gases upon the introduction of vacancy defects. J. Phys. Chem. C 2016, 120, 20428–20436. [Google Scholar] [CrossRef]
- Wu, Z.; Chen, X.; Zhu, S.; Zhou, Z.; Yao, Y.; Quan, W.; Liu, B. Room temperature methane sensor based on graphene nanosheets/polyaniline nanocomposite thin film. IEEE Sens. J. 2013, 13, 777–782. [Google Scholar] [CrossRef]
- Yang, D.; Yang, N.; Ni, J.; Xiao, J.; Jiang, J.; Liang, Q.; Ren, T.; Chen, X. First-principles approach to design and evaluation of graphene as methane sensors. Mater. Des. 2017, 119, 397–405. [Google Scholar] [CrossRef]
- Jhang, S.H.; Craciun, M.F.; Schmidmeier, S.; Tokumitsu, S.; Russo, S.; Yamamoto, M.; Skourski, Y.; Wosnitza, J.; Tarucha, S.; Eroms, J.; et al. Stacking-order dependent transport properties of trilayer graphene. Phys. Rev. B 2011, 84, 1401–1408. [Google Scholar] [CrossRef]
- Koshino, M. Interlayer screening effect in graphene multilayers with ABA and ABC stacking. Phys. Rev. B 2010, 81, 760–762. [Google Scholar] [CrossRef]
- Lui, C.H.; Li, Z.Q.; Chen, Z.Y.; Klimov, P.V.; Brus, L.E.; Heinz, T.F. Imaging Stacking Order in Few-Layer Graphene. Nano Lett. 2011, 11, 164–169. [Google Scholar] [CrossRef] [PubMed]
- Dreyer, D.R.; Park, S.; Bielawski, C.W.; Ruoff, R.S. The chemistry of graphene oxide. Chem. Soc. Rev. 2010, 39, 228–240. [Google Scholar] [CrossRef] [PubMed]
- Latil, S.; Henrard, L. Charge carriers in few-layer graphene films. Phys. Rev. Lett. 2006, 97, 036803. [Google Scholar] [CrossRef] [PubMed]
- Menezes, M.G.; Capaz, R.B.; Louie, S.G. Ab initio quasiparticle band structure of ABA and ABC-stacked graphene trilayers. Phys. Rev. B 2014, 89, 407–411. [Google Scholar] [CrossRef]
- Kasry, A.; Kuroda, M.A.; Martyna, G.J.; Tulevski, G.S.; Bol, A.A. Chemical Doping of Large-Area Stacked Graphene Films for Use as Transparent, Conducting Electrodes. ACS Nano 2010, 4, 3839–3844. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.W.; Li, F.; Wu, Z.S.; Ren, W.C.; Cheng, H.M. Electrochemical interfacial capacitance in multilayer graphene sheets: Dependence on number of stacking layers. Electrochem. Commun. 2009, 11, 1729–1732. [Google Scholar] [CrossRef]
- Bjork, J.; Hanke, F.; Palma, C.A.; Samori, P.; Cecchini, M.; Persson, M. Adsorption of Aromatic and Anti-Aromatic Systems on Graphene through π–π Stacking. J. Phys. Chem. Lett. 2010, 1, 3407–3412. [Google Scholar] [CrossRef]
- Lu, C.L.; Chang, C.P.; Huang, Y.C.; Chen, R.B.; Lin, M.L. Influence of an electric field on the optical properties of few-layer graphene with AB stacking. Phys. Rev. B 2006, 73, 144427. [Google Scholar] [CrossRef]
- Delley, B. An all-electron numerical-method for solving the local density functional for polyatomic-molecules. J. Chem. Phys. 1990, 92, 508–517. [Google Scholar] [CrossRef]
- Lazzeri, M.; Attaccalite, C.; Wirtz, L.; Mauri, F. Impact of the electron-electron correlation on phonon dispersion: Failure of LDA and GGA DFT functionals in graphene and graphite. Phys. Rev. B 2008, 78, 081406. [Google Scholar] [CrossRef]
- Chen, X.P.; Yang, N.; Jiang, J.K.; Liang, Q.H.; Yang, D.G.; Zhang, G.Q.; Ren, T.L. Ab initio Study of Temperature, Humidity and Covalent Functionalization Induced Band Gap Change of Single-Walled Carbon Nanotubes. IEEE Electron Device Lett. 2015, 36, 606–608. [Google Scholar] [CrossRef]
- Hohenberg, P.; Kohn, W. Inhomogeneous electron gas. Phys. Rev. 1964, 136, B864. [Google Scholar] [CrossRef]
- Monkhorst, H.J.; Pack, J.D. Special points for Brillouin-zone integrations. Phys. Rev. B 1976, 13, 5188–5192. [Google Scholar] [CrossRef]
- Methfessel, M.; Paxton, A. High-precision sampling for Brillouin-zone integration in metals. Phys. Rev. B 1989, 40, 3616–3621. [Google Scholar] [CrossRef]
- Dispersion Correction for DFT. Available online: http://www.iucr.org/news/research-news/dispersion-corrected-density-functional-theory-dft-d (accessed on 1 December 2017).
- Dappe, Y.; Basanta, M.A.; Flores, F.; Ortega, J. Weak chemical interaction and van der Waals forces between graphene layers: A combined density functional and intermolecular perturbation theory approach. Phys. Rev. B 2006, 74, 205434. [Google Scholar] [CrossRef]
- Vanin, M.; Mortensen, J.J.; Kelkkanen, A.; Garcia-Lastra, J.M.; Thygesen, K.S.; Jacobsen, K.W. Graphene on metals: A van der Waals density functional study. Phys. Rev. B 2010, 81, 081408. [Google Scholar] [CrossRef]
- Zheng, Z.Y.; Zhao, J.J. Lattice energies and elastic properties of solid methane: Assessment of different density functionals. Acta Phys. Chim. Sin. 2012, 28, 1809–1814. [Google Scholar]
- Le, D.; Kara, A.; Schröder, E.; Hyldgaard, P.; Rahman, T.S. Physisorption of nucleobases on graphene: A comparative van der Waals study. J. Phys. Condens. Matter 2012, 24, 424210. [Google Scholar] [CrossRef] [PubMed]
- Thierfelder, C.; Witte, M.; Blankenburg, S.; Rauls, E.; Schmidt, W. Methane adsorption on graphene from first principles including dispersion interaction. Surf. Sci. 2011, 605, 746–749. [Google Scholar] [CrossRef]
- Dai, J.; Yuan, J.; Giannozzi, P. Gas adsorption on graphene doped with B, N, Al, and S: A theoretical study. Appl. Phys. Lett. 2009, 95, 232105. [Google Scholar] [CrossRef]
- Ao, Z.; Jiang, Q.; Zhang, R.; Tan, T.; Li, S. Al doped graphene: A promising material for hydrogen storage at room temperature. J. Appl. Phys. 2009, 105, 074307. [Google Scholar] [CrossRef]
- Chen, X.P.; Yang, N.; Ni, J.M.; Cai, M.; Ye, H.Y.; Wong, C.K.; Leung, S.Y.; Ren, T.L. Density-functional calculation of methane adsorption on graphenes. IEEE Electron Device Lett. 2015, 36, 1366–1368. [Google Scholar] [CrossRef]
- Denis, P.A.; Faccio, R.; Mombru, A.W. Is It Possible to Dope Single-Walled Carbon Nanotubes and Graphene with Sulfur? ChemPhysChem 2009, 10, 715–722. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| The Number of Layers (DFT) | Adsorption Energy (eV) | The Average Distance of Graphene (Å) | The Distance between Graphene and Methane (Å) | |
| - | d1 | d2 | d3 | |
| 1 | 50 | - | - | 3.658 |
| 2 | 62 | 4.147 | 3.929 | 4.192 |
| 3 | 99 | 4.151 | 4.133 | 4.149 |
| 4 | 138 | 4.306 | 4.077 | 3.737 |
| The Number of Layers (DFT-D) | Adsorption Energy (eV) | The Average Distance of Graphene (Å) | The Distance between Graphene and Methane (Å) | |
| - | d1 | d2 | d3 | |
| 1 | 255 | - | - | 3.337 |
| 2 | 277 | 3.422 | 3.420 | 3.354 |
| 3 | 308 | 3.418 | 3.415 | 3.352 |
| 4 | 351 | 3.416 | 3.415 | 3.360 |
| Model | Adsorption Energy | Distance | Band Gap | |||||
|---|---|---|---|---|---|---|---|---|
| Zigzag (meV) | Armchair (meV) | Zigzag (Å) | Armchair (Å) | Zigzag | Armchair | |||
| Graphene (eV) | m-Graphene (eV) | Graphene (eV) | m-Graphene (eV) | |||||
| 1 | 242 | 217 | 3.363 | 3.362 | 0 | 0.006 | 0 | 0 |
| 2 | 255 | 230 | 3.337 | 3.336 | 0 | 0.007 | 0 | 0 |
| 3 | 243 | 218 | 3.352 | 3.355 | 0 | 0 | 0 | 0 |
| The Number of Layers in Order-Graphene | The Average Distance of Z-Graphene (Å) | The Distance between Z-Graphene and Methane (Å) | The Distance between A-Graphene and Methane (Å) | The Average Distance of A-Graphene (Å) | ||
| d1 | d2 | d3 | d1 | d2 | d3 | |
| 1 | - | - | 3.337 | - | - | 3.336 |
| 2 | 3.422 | 3.420 | 3.354 | 3.450 | 3.454 | 3.355 |
| 3 | 3.418 | 3.415 | 3.352 | 3.435 | 3.434 | 3.350 |
| 4 | 3.416 | 3.415 | 3.360 | 3.435 | 3.434 | 3.370 |
| The Number of Layers in AB-Graphene | The Average Distance of Z-Graphene (Å) | The Distance between Z-Graphene and Methane (Å) | The Average Distance of A-Graphene (Å) | The Distance between A-Graphene and Methane (Å) | ||
| d1 | d2 | d3 | d1 | d2 | d3 | |
| 1 | - | - | 3.337 | - | - | 3.336 |
| 2 | 3.301 | 3.290 | 3.351 | 3.308 | 3.298 | 3.353 |
| 3 | 3.288 | 3.286 | 3.356 | 3.293 | 3.292 | 3.358 |
| 4 | 3.283 | 3.282 | 3.366 | 3.292 | 3.291 | 3.371 |
| The Number of Layers in ABC-Graphene | The Average Distance of Z-Graphene (Å) | The Distance between Z-Graphene and Methane (Å) | The Average Distance of A-Graphene (Å) | The Distance between A-Graphene and Methane (Å) | ||
| d1 | d2 | d3 | d1 | d2 | d3 | |
| 1 | - | - | - | - | - | - |
| 2 | - | - | - | - | - | - |
| 3 | 3.289 | 3.288 | 3.356 | 3.295 | 3.296 | 3.329 |
| 4 | 3.285 | 3.287 | 3.340 | 3.291 | 3.299 | 3.367 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, N.; Yang, D.; Zhang, G.; Chen, L.; Liu, D.; Cai, M.; Fan, X. The Effects of Graphene Stacking on the Performance of Methane Sensor: A First-Principles Study on the Adsorption, Band Gap and Doping of Graphene. Sensors 2018, 18, 422. https://doi.org/10.3390/s18020422
Yang N, Yang D, Zhang G, Chen L, Liu D, Cai M, Fan X. The Effects of Graphene Stacking on the Performance of Methane Sensor: A First-Principles Study on the Adsorption, Band Gap and Doping of Graphene. Sensors. 2018; 18(2):422. https://doi.org/10.3390/s18020422
Chicago/Turabian StyleYang, Ning, Daoguo Yang, Guoqi Zhang, Liangbiao Chen, Dongjing Liu, Miao Cai, and Xuejun Fan. 2018. "The Effects of Graphene Stacking on the Performance of Methane Sensor: A First-Principles Study on the Adsorption, Band Gap and Doping of Graphene" Sensors 18, no. 2: 422. https://doi.org/10.3390/s18020422
APA StyleYang, N., Yang, D., Zhang, G., Chen, L., Liu, D., Cai, M., & Fan, X. (2018). The Effects of Graphene Stacking on the Performance of Methane Sensor: A First-Principles Study on the Adsorption, Band Gap and Doping of Graphene. Sensors, 18(2), 422. https://doi.org/10.3390/s18020422