
Molecular Techniques for the Detection of Organisms in Aquatic Environments, with Emphasis on Harmful Algal Bloom Species



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Molecular—Whole Cell Methods
3. Molecular—Cell-Free Format
3.1. Sandwich Hybridization Assay (SHA)
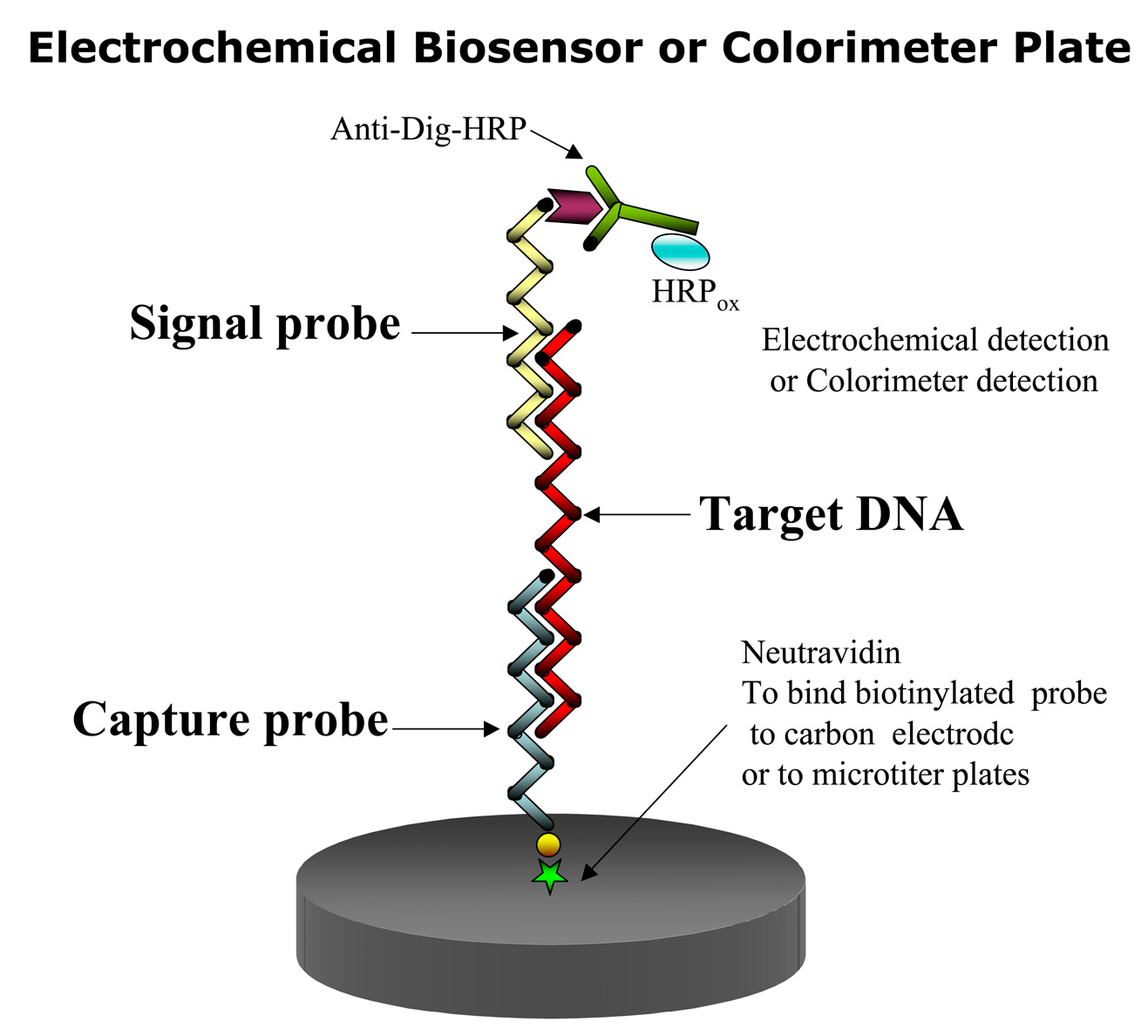
3.2. Biosensors
3.3. Microarrays
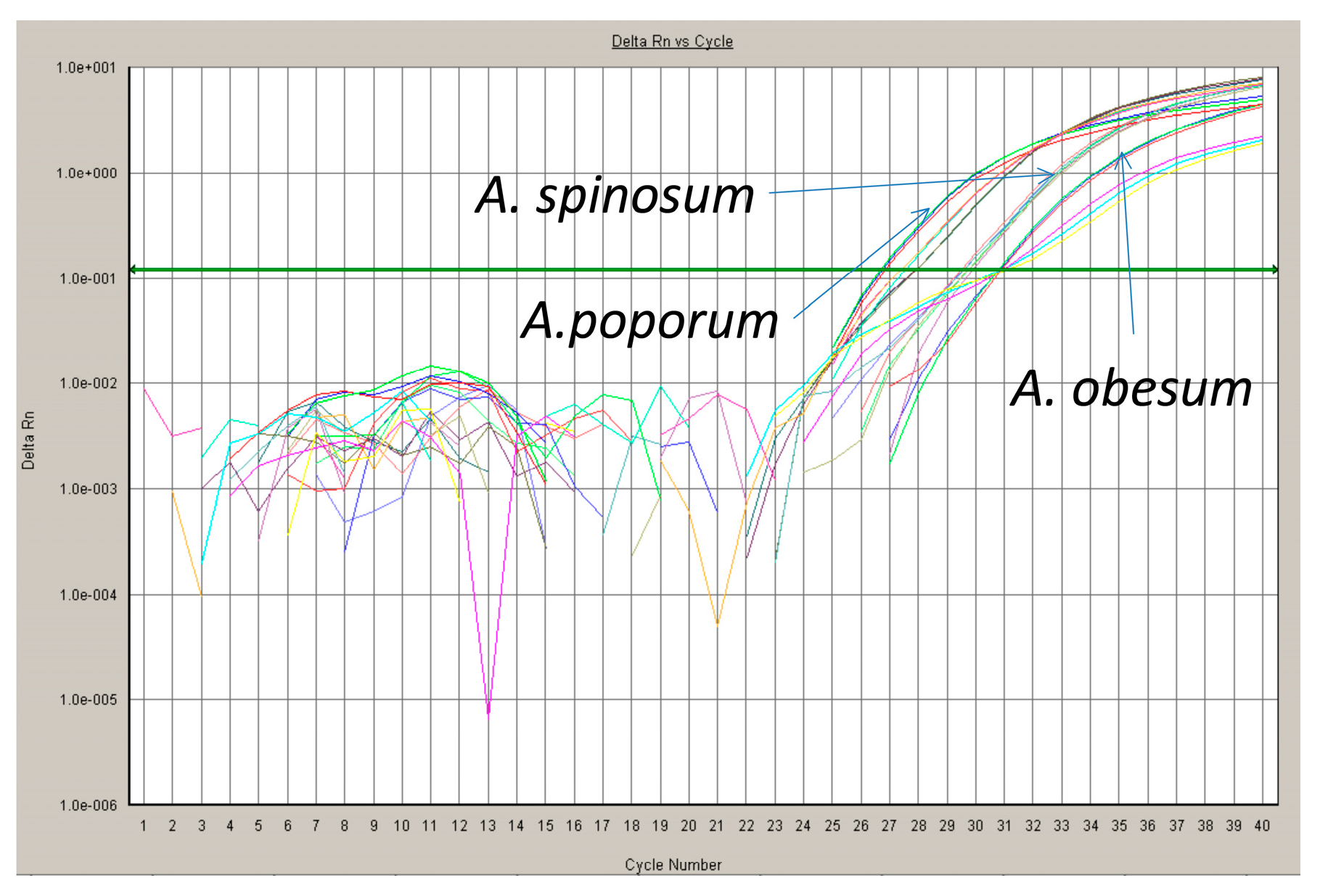
3.4. qPCR
3.5. RT-qPCR
3.6. Lab-on-a-Chip
3.7. Next-Generation Sequencing or High Throughput Sequencing (NGS or HTS)
4. Enhancing Performance
4.1. Nano-Bioengineered Probes and Platforms
4.2. Pre-Concentration and Magnetic Separation Systems
4.3. Solid-Phase Hybridization
4.4. Isothermal Amplification
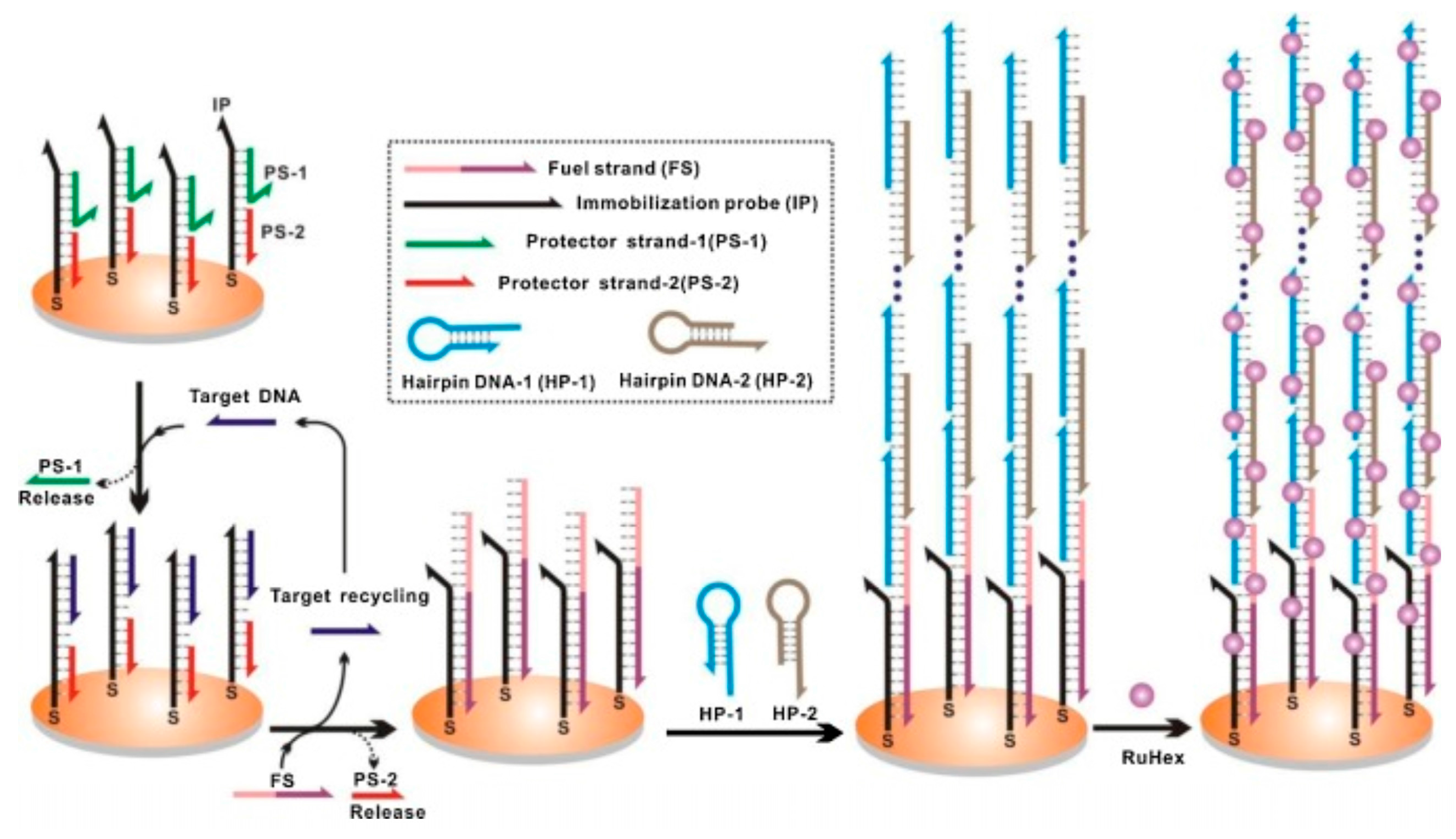
4.5. Hybridization Chain Reaction
5. In-Situ Remote Sensing, HAB Monitoring in Buoys as Study Case
6. Future Directions and Concluding Remarks
Acknowledgments
Conflicts of Interest
References
- Census of Marine Life. Available online: http://www.coml.org/investigating/identifying/molecular_techniques (accessed on 18 May 2017).
- Amann, R.I.; Binder, B.J.; Olson, R.J.; Chisholm, S.W.; Devereux, R.; Stahl, D.A. Combination of 16S rRNA-targeted oligonucleotide probes with flow cytometry for analyzing mixed microbial populations. Appl. Environ. Microbiol. 1990, 56, 1919–1925. [Google Scholar] [PubMed]
- López-Garcia, P.; Rodriguez-Valera, F.; Pedros-Alio, C.; Moreira, D. Unexpected diversity of small eukaryotes in deep-sea Antarctic plankton. Nature 2001, 409, 603–607. [Google Scholar] [CrossRef] [PubMed]
- Sogin, M.L.; Morrison, H.G.; Huber, J.A.; Welch, D.M.; Huse, S.M.; Neal, P.R.; Arrieta, J.M.; Herndl, G.J. Microbial diversity in the deep sea and the underexplored “rare biosphere”. Proc. Natl. Acad. Sci. USA 2006, 103, 12115–12120. [Google Scholar] [CrossRef] [PubMed]
- Guillou, L.; Chrétiennot-Dinet, M.-J.; Medlin, L.K.; Claustre, H.; Loiseaux-de Goer, S.; Vaulot, D. Bolidomonas, a new genus with two species belonging to new algal class, the Bolidophyceae Heterokonta. J. Phycol. 1999, 35, 368–381. [Google Scholar] [CrossRef]
- Groben, R.; John, U.; Eller, G.; Lange, M.; Medlin, L.K. Using fluorescently labelled rRNA probes for hierarchical estimation of phytoplankton diversity, a mini review. Nova Hedwig. 2004, 79, 313–320. [Google Scholar] [CrossRef]
- Kumar, Y.; Westram, R.; Behrens, S.; Fuchs, B.; Glöckner, F.O.; Amann, R.; Meier, H.; Ludwig, W. Graphical representation of ribosomal RNA probe accessibility data using ARB software package. BMC Bioinform. 2005, 6, 61. [Google Scholar] [CrossRef] [PubMed]
- Karlsen, B.; Cusack, C.; Beensen, E. Microscopic and molecular methods for quantitative phytoplankton analysis. In IOC Manuals and Guides, No. 55; UNESCO: Paris, France, 2010. [Google Scholar]
- Lewis, J.; Medlin, L.K.; Raine, R. MIDTAL (Microarrays for the Detection of Toxic Algae), a Protocol for a Successful Microarray Hybridisation and Analysis; Koeltz: Hermann Hesse, Germany, 2012. [Google Scholar]
- Amann, R.I. In situ identification of micro-organisms by whole cell hybridisation with rRNA-targeted nucleic acid probes. In Molecular Microbial Ecology Manual 336; Akkermans, A.D.L., van Elsas, J.D., de Bruijn, F.J., Eds.; Kluwer Academic Publishers: Dordrecht, NL, USA, 1995; pp. 1–15. [Google Scholar]
- Groben, R.; Medlin, L.K. In-situ hybridization of phytoplankton using fluorescently-labelled rRNA Probes. Methods Enzym. 2005, 395, 299–310. [Google Scholar] [PubMed]
- Miller, P.E.; Scholin, C.A. Identification and enumeration of cultured and wild Pseudo-nitzschia Bacillariophyceae. Using species specific LSU rRNA-targeted fluorescent probes and filter-based whole cell hybridization. J. Phycol. 1998, 34, 371–382. [Google Scholar] [CrossRef]
- Miller, P.E.; Scholin, C.A. On detection of Pseudo-nitzschia Bacillariophyceae. Species using whole cell hybridization, Sample fixation and stability. J. Phycol. 2000, 36, 238–250. [Google Scholar] [CrossRef]
- Simon, N.; Brenner, J.; Edvardsen, B.; Medlin, L.K. The identification of Chrysochromulina and Prymnesium species Haptophyta, Prymnesiophyceae using fluorescent or chemiluminescent oligonucleotide probes, a means of improving studies on toxic algae. Eur. J. Phycol. 1997, 32, 393–401. [Google Scholar] [CrossRef]
- Simon, N.; Campbell, L.; Örnólfsdóttir, E.; Groben, R.; Guillou, L.; Lange, M.; Medlin, L.K. Oligonucleotide probes for the identification of three algal groups by dot blot and fluorescent whole-cell hybridisation. J. Eukaryot. Microbiol. 2000, 47, 76–84. [Google Scholar] [CrossRef]
- John, U.; Cembella, A.; Hummert, C.; Ellbrächter, M.; Groben, R.; Medlin, L.K. Discrimination of the toxigenic dinoflagellates Alexandrium tamarense and A. ostenfeldii in co-occurring natural populations from Scottish coastal waters. Eur. J. Phycol. 2003, 38, 25–40. [Google Scholar] [CrossRef]
- Töbe, K.; Tullis, D.; Gladstone, M.; Anderson, D.; Medlin, L.K. Detecting intact algal cells with whole cell hybridisation assays. In Microscopic and Molecular Methods for Quantitative Phytoplankton Analysis. IOC Manuals and Guides, No. 50; Karlson, B., Cusack, C., Bresnan, E., Eds.; Intergovernmental Oceanographic Commission of UNESCO: Paris, France, 2010; pp. 55–66. [Google Scholar]
- Medlin, L.K.; Strieben, S. Refining cryptophyte identification, matching cell fixation methods to FISH hybridisation of cryptomonads. J. Appl. Phycol. 2010, 22, 725–731. [Google Scholar] [CrossRef]
- Scholin, C.A.; Marin, R., III; Miller, P.E.; Doucette, G.J.; Powell, C.L.; Haydock, P.; Howard, J.; Ray, J. DNA probes and a receptor binding assay for detection of Pseudo-nitzschia (Bacillariophyceae) species and domoic acid activity in cultured and natural samples. J. Phycol. 1999, 35, 1356–1367. [Google Scholar] [CrossRef]
- Scholin, C.; Miller, P.; Buck, K.; Chavez, F.; Harris, P.; Haydock, P.; Howard, J.; Cangelosi, G. Detection and quantification of Pseudo-nitzschia australis in cultured and natural populations using LSU rRNA-targeted probes. Limnol. Oceanogr. 1997, 42, 1265–1272. [Google Scholar] [CrossRef]
- Scholin, C.A.; Vrieling, E.; Peperzak, L.; Rhodes, L.; Rublee, P. Detection of HAB species using lectin, antibody and DNA probes. In Manual on Harmful Marine Microalgae; Hallegraeff, G.M., Anderson, D.M., Cembella, A.D., Eds.; UNESCO: Paris, France, 2003; pp. 131–164. [Google Scholar]
- Veldhuis, M.J.W.; Kraay, G. Application of flow cytometry in marine phytoplankton research, current applications and future perspectives. In Aquatic Flow Cytometry, Achievements and Prospects; Reckermann, M., Colijn, F., Eds.; CSIC: Madrid, Spain, 2000; pp. 121–134. [Google Scholar]
- Shapiro, H.M. Excitation and emission spectra of common dyes. In Current Protocols in Cytometry; Robinson, J.P., Darzynkiewicz, Z., Hyun, W., Orfao, A., Rabinovitch, P.S., Eds.; Wiley: New York, NY, USA, 2003. [Google Scholar]
- Biegala, I.C.; Not, F.; Vaulot, D.; Simon, N. Quantitative assessment of picoeukaryotes in the natural environment by using taxon-specific oligonucleotide probes in association with tyramide signal amplification, fluorescence-in-situ hybridization and flow cytometry. Appl. Environ. Microbiol. 2003, 69, 5519–5529. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, B.M.; Wallner, G.; Beisker, W.; Schwippl, I.; Ludwig, W.; Amann, R. Flow cytometric analysis of the in situ accessibility of Escherichia coli 16S rRNA for fluorescently labeled oligonucleotide probes. Appl. Environ. Microbiol. 1998, 64, 4973–4982. [Google Scholar] [PubMed]
- Schönhuber, W.; Fuchs, B.; Juretschko, S.; Amann, R. Improved sensitivity of whole cell hybridization by the combination of horseradish peroxidase-labeled oligonucleotides and tyramide signal amplification. Appl. Environ. Microbiol. 1997, 63, 3268–3273. [Google Scholar] [PubMed]
- Schönhuber, W.; Zarda, B.; Eix, S.; Rippka, R.; Herdmann, M.; Ludwig, W.; Amann, R. In situ identification of cyanobacteria with horseradish peroxidase-labeled, rRNA-targeted oligonucleotide probes. Appl. Environ. Microbiol. 1999, 65, 1259–1267. [Google Scholar] [PubMed]
- Pernthaler, A.; Pernthaler, J.; Amann, R. Fluorescence in situ hybridization hybridization and catalyzed reporter deposition for the identification of marine bacteria. Appl. Environ. Microbiol. 2002, 68, 3094–3101. [Google Scholar] [CrossRef] [PubMed]
- West, N.; Schoenhuber, W.; Fuller, N.; Amann, R.; Rippka, R.; Post, A.; Scanlan, D. Closely related Prochlorococcus genotypes show remarkably different depth distributions in two oceanic regions as revealed by in situ hybridization using 16S rRNA-targeted oligonucleotides. Microbiology 2001, 1477, 1731–1744. [Google Scholar] [CrossRef] [PubMed]
- Not, F.; Simon, N.; Biegala, I.C.; Vaulot, D. Application of fluorescent in situ hybridization coupled with tyramide signal amplification FISH-TSA to assess eukaryotic picoplankton composition. Aquat. Microb. Ecol. 2002, 28, 157–166. [Google Scholar] [CrossRef]
- Biegala, I.C.; Kennaway, G.; Alverca, E.; Lennon, J.F.; Vaulot, D.; Simon, N. Identification of bacteria associated with dinoflagellates Dinophyceae Alexandrium spp. using tyramide signal amplification-fluorescent in situ hybridization and confocal microscopy. J. Phycol. 2002, 38, 404–411. [Google Scholar] [CrossRef]
- Alverca, E.; Biegala, I.C.; Kennaway, G.M.; Lewis, J.; Franca, S. In situ identification and localization of bacteria associated with Gyrodinium instriatum (Gymnodiniales, Dinophyceae) by electron and confocal microscopy. Eur. J. Phycol. 2002, 37, 523–530. [Google Scholar] [CrossRef]
- Vives-Rego, J.; Lebaron, P.; von Nebe, C. Current and future applications of flow cytometry in aquatic Microbiology. FEMS Microbiol. Rev. 2000, 24, 429–448. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, D.T.; Fricker, C.R. Application of laser scanning for the rapid and automated detection of bacteria in water samples. J. Appl. Microbiol. 1999, 86, 785–795. [Google Scholar] [CrossRef] [PubMed]
- Mignon-Godefroy, K.; Guillet, J.-C.; Butor, C. Laser scanning cytometry for the detection of rare events. Cytometry 1997, 27 Pt A, 336–344. [Google Scholar] [CrossRef]
- West, N.J.R.; Bacchieri, R.; Hansen, G.; Tomas, C.; Lebaron, P.; Moreau, H. Rapid quantification of the toxic alga Prymnesium parvum in natural samples by use of a specific monoclonal antibody and solid-phase cytometry. Appl. Environ. Microbiol. 2006, 72, 860–868. [Google Scholar] [CrossRef] [PubMed]
- Töbe, K.; Eller, G.; Medlin, L.K. Automated detection and enumeration for toxic algae by solid-phase cytometry and the introduction of a new probe for Prymnesium parvum Haptophyta, Prymnesiophyceae. J. Plankton Res. 2006, 7, 643–657. [Google Scholar] [CrossRef]
- Töbe, K.; Karlson, B.; Cusack, C.; Bresnan, E. Tyramide signal amplification in combination with fluorescence in situ hybridization. In Microscopic and Molecular Methods for Quantitative Phytoplankton Analysis; IOC Manuals and Guides, No. 55; Karlson, B., Cusack, C., Bresnan, E., Eds.; UNESCO: Paris, France, 2010; pp. 103–108. [Google Scholar]
- Schauer, S.; Sommer, R.; Farnleitner, A.H.; Kirschner, A.K.T. Rapid and sensitive quantification of Vibrio cholerae and Vibrio mimicus cells in water samples by use of catalyzed reporter deposition fluorescence in situ hybridization combined with solid-phase cytometry. Appl. Environ. Microbiol. 2012, 78, 7369–7375. [Google Scholar] [CrossRef] [PubMed]
- Lemarchand, K.; Parthuisot, N.; Catala, P.; Lebaron, P. Comparative assessment of epifluorescence microscopy, flow cytometry and solid-phase cytometry used in the enumeration of specific bacteria in water. Aquat. Microb. Ecol. 2001, 25, 301–309. [Google Scholar] [CrossRef]
- Kamentsky, L.A. Laser scanning cytometry. Methods Mol. Cell. Biol. 2001, 63, 51–87. [Google Scholar]
- Darynkiewicz, Z.; Smolewski, P.; Bedner, E. Use of flow and laser scanning cytometry to study mechanisms regulating cell cycle and controlling cell death. Clin. Chem. Lab. Med. 2001, 21, 857–873. [Google Scholar]
- Viticulture and Oneology. Available online: http://wineserver.ucdavis.edu/industry/enology/methods_and_techniques/techniques/antibody-based_identification.html (accessed on 18 May 2017).
- Aguilera, A.; Gonzalez-Gil, S.; Keafer, B.A.; Anderson, D.M. Immunomagnetic separation of cells of the toxic dinoflagellate Alexandrium fundyense from natural plankton samples. Mar. Ecol. Prog. Ser. 1996, 143, 255–269. [Google Scholar] [CrossRef]
- Anderson, D.M.; Kulis, D.M.; Keafer, B.A.; Berdalet, E. Detection of the toxic dinoflagellate Alexandrium fundyense Dinophyceae with oligonucleotide and antibody probes, variability in labeling intensity with physiological condition. J. Phycol. 1999, 35, 870–883. [Google Scholar] [CrossRef]
- Anderson, D.M.; Kulis, D.; Keafer, B.A.; Gribble, K.E.; Marin, R.; Scholin, C.A. Identification and enumeration of Alexandrium from the Gulf of Maine using molecular probes. Deep-Sea Res. II 2005, 52, 2467–2490. [Google Scholar] [CrossRef]
- Rautio, J.B.K.B.; Lahdenpera, J.; Breinstein, A.; Molin, S.; Neubaure, P. Sandwich hybridisation assay for quantitative detection of yeast RNAs in crude cell lysates. Microb. Cell Fact. 2003, 2, 4. [Google Scholar] [CrossRef]
- Zamyadi, A.; McQuaid, N.; Prevost, M.; Dorner, S. Monitoring of potentially toxic cyanobacteria using an online multi-probe in drinking water sources. J. Environ. Monit. 2012, 14, 579–588. [Google Scholar] [CrossRef] [PubMed]
- Orozco, J.; Medlin, L.K. Electrochemical performance of a DNA-based sensor device for detecting toxic algae. Sens. Actuators B Chem. 2011, 153, 71–77. [Google Scholar] [CrossRef]
- Orozco, J.; Medlin, LK. Review, advances in electrochemical genosensors-based methods for monitoring blooms of toxic algae. Environ. Sci. Pollut. Res. 2012, 20, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Orozco, J.; Baudart, J.; Medlin, L.K. Evaluation of probe orientation and effect of the digoxigenin-enzymatic label in a sandwich hybridization format to develop toxic algae biosensors. Harmful Algae 2011, 10, 489–494. [Google Scholar] [CrossRef]
- Orozco, J.; Villa, E.; Manes, C.-L.; Medlin, L.K.; Guillebault, D. Electrochemical RNA genosensors for toxic algal species, enhancing selectivity and sensitivity. Talanta 2016, 161, 560–566. [Google Scholar] [CrossRef] [PubMed]
- Diercks, S.; Medlin, L.K.; Metfies, K. Colorimetric detection of the toxic dinoflagellate Alexandrium minutum using sandwich hybridization in a microtiter plate assay. Harmful Algae 2008, 7, 137–145. [Google Scholar] [CrossRef]
- Metfies, K.; Huljic, S.; Lange, M.; Medlin, L.K. Electrochemical detection of the toxic dinoflagellate Alexandrium ostenfeldii with a DNA-biosensor. Biosens. Bioelectron. 2005, 207, 1349–1357. [Google Scholar] [CrossRef] [PubMed]
- Marin, R., III; Scholin, C.A. Toxic algal detection using rRNA-targeted probes in a semi-automated sandwich hybridization format. In Microscopic and Molecular Methods for Quantitative Phytoplankton Analysis; IOC Manuals and Guides, No. 55; Karlson, B., Cusack, C., Bresnan, E., Eds.; UNESCO: Paris, France, 2010; pp. 87–94. [Google Scholar]
- Liao, J.C.; Mastali, M.; Li, Y.; Gau, V.; Suchard, M.A.; Babbitt, J.; Gombein, J.; Landaw, E.M.; McCabe, R.R.B.; Churchill, B.M.; et al. Development of an advanced electrochemical DNA biosensor for bacterial pathogen detection. J. Mol. Diagn. 2007, 9, 158–168. [Google Scholar] [CrossRef] [PubMed]
- Anderson, D.M.; Kulis, D.; Erdner, D.; Ahn, S.; Walt, D. Fiber optic microarrays for the detection and enumeration of harmful algal bloom species. Afr. J. Mar. Sci. 2006, 28, 231–235. [Google Scholar] [CrossRef]
- Ahn, S.; Kulis, D.; Erdner, D.; Anderson, D.M. Fiber optic microarrays for the simultaneous detection of multiple harmful algal bloom species. Appl. Environ. Microbiol. 2006, 72, 5742–5749. [Google Scholar] [CrossRef] [PubMed]
- Gentry, T.J.; Wickham, G.S.; Schadt, C.W.; He, Z.; Zhou, J. Microarray applications in microbial ecology research. Microb. Ecol. 2006, 52, 159–175. [Google Scholar] [CrossRef] [PubMed]
- Ye, R.W.; Wang, T.; Bedzyk, L.; Croker, K.M. Applications of DNA microarrays in microbial systems. J. Microbiol. Methods 2001, 47, 257–272. [Google Scholar] [CrossRef]
- Cheung, V.G.; Morley, M.; Aguilar, F.; Massimi, A.; Kucherlapati, R.; Childs, G. Making and reading microarrays. Nat. Genet. 1999, 21 (Suppl. S1), 15–19. [Google Scholar] [CrossRef] [PubMed]
- Southern, E.; Mir, K.; Shchepinov, M. Molecular interactions on microarrays. Nat. Genet. 1999, 21 (Suppl. 1), 5–9. [Google Scholar] [CrossRef] [PubMed]
- Metfies, K.; Medlin, L.K. DNA-microchips for phytoplankton. The fluorescent wave of the future. Nova Hedwig. 2004, 79, 321–327. [Google Scholar] [CrossRef]
- Gescher, G.; Metfies, K.; Frickenhaus, S.; Knelfelkamp, B.; Wiltshire, K.; Medlin, L.K. Feasibility of assessing community composition of Prasinophytes at the Helgoland Reede Sampling Site with a DNA-microarray. Appl. Environ. Microbiol. 2008, 74, 5305–5316. [Google Scholar] [CrossRef] [PubMed]
- Metfies, K.; Gescher, C.; Frickenhaus, S.; Niestroy, R.; Wichels, A.; Gerdts, G.; Knefelkamp, B.; Wiltshire, K.; Medlin, L.K. Contribution of the Class Cryptophyceae to phytoplankton structure in the German Bight. J. Phycol. 2010, 46, 1152–1160. [Google Scholar] [CrossRef]
- Barra, L.; Ruggiero, M.V.; Sarno, D.; Montresor, M.; Kooistra, W.H.C.F. Strengths and weaknesses of microarray approaches to detect Pseudo-nitzschia species in the field. Environ. Sci. Pollut. Res. 2013, 2010, 6705–6718. [Google Scholar] [CrossRef] [PubMed]
- Edvardsen, B.; Dittami, S.M.; Groben, R.; Brubak, S.; Escalera, L.; Rodríguez, F.; Reguera, B.; Chen, J.; Medlin, L.K. Molecular probes and microarrays for the detection of toxic algae in the genera Dinophysis and Phalacroma Dinophyta. Environ. Sci. Pollut. Res. 2013, 2010, 6733–6750. [Google Scholar] [CrossRef] [PubMed]
- Dittami, S.M.; Hostyeva, V.; Egge, E.S.; Kegel, J.U.; Eikrem, W.; Edvardsen, B. Seasonal dynamics of Harmful Algae in outer Oslofjorden monitored by microarray, qPCR, and microscopy. Environ. Sci. Pollut. Res. 2013, 2010, 6719–6732. [Google Scholar] [CrossRef] [PubMed]
- Dittami, S.M.; Pazos, Y.; Laspra, M.; Medlin, L.K. Microarray testing for the presence of toxic algae Monitoring Programme in Galicia SW Spain. Environ. Sci. Pollut. Res 2013, 2010, 6778–6793. [Google Scholar] [CrossRef] [PubMed]
- Kegel, J.U.; Del Amo, Y.; Costes, L.; Medlin, L.K. Testing a microarray to detect and monitor toxic microalgae in Arcachon Bay in France. Microarrays 2013, 2, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Kegel, J.U.; Del Amo, Y.; Medlin, L.K. Introduction to project MIDTAL, its methods and samples from Arcachon Bay, France. Environ. Sci. Pollut. Res. 2013, 2010, 6690–6704. [Google Scholar] [CrossRef] [PubMed]
- Ki, J.-S.; Han, M.-S. A low-density oligonucleotide array study for parallel detection of harmful algal species using hybridization of consensus PCR products of LSU rDNA D2 domain. Biosens. Bioelectron. 2006, 21, 1812–1821. [Google Scholar] [CrossRef] [PubMed]
- McCoy, G.R.; Kegel, J.U.; Touzet, N.; Fleming, G.T.A.; Medlin, L.K.; Raine, R. An assessment of RNA content in Prymnesium parvum, Prymnesium polylepis, cf. Chattonella sp. and Karlodinium veneficum under varying environmental conditions for calibrating an RNA microarray for species detection. FEMS Microbiol. Ecol. 2014, 881, 140–159. [Google Scholar]
- McCoy, G.R.; Raine, R.; Medlin, L.K.; Chen, J.; Kooistra, W.H.C.F.; Barra, L.; Ruggiero, M.V.; Graneli, E.; Hagström, J.A.; Salomon, P.S.; et al. Field testing for toxic algae with a microarray, initial results from the MIDTAL project. In Proceedings of the 15th International Conference on Harmful Algae, Changwon, Korea, 29 October–2 November 2012; pp. 210–212. [Google Scholar]
- Medlin, L.K.; Metfies, K.; Mehl, H.; Wiltshire, K.; Valentin, K. Picoplankton diversity at the Helgoland Time Series Site as assessed by three molecular methods. Microb. Ecol. 2006, 67, 1432–1451. [Google Scholar]
- Gescher, G.; Metfies, K.; Medlin, L.K. The ALEX Chip—Development of a DNA chip for identification and monitoring of Alexandrium. Harmful Algae 2008, 7, 485–494. [Google Scholar] [CrossRef]
- Taylor, J.D.; Kegel, J.U.; Lewis, J.M.; Medlin, L.K. Validation of the detection of Alexandrium species using specific RNA probes tested in a microarray format, Calibration of signal using variability of RNA content with environmental conditions. Harmful Algae 2014, 37, 17–27. [Google Scholar] [CrossRef]
- Loy, A.; Lehner, A.; Lee, N.; Adamczyk, J.; Meier, H.; Ernst, J.; Schleifer, K.-H.; Wagner, M. Oligonucleotide microarray for 16S rRNA gene-based detection of all recognized lineages of sulfate reducing prokaryotes in the environment. Appl. Environ. Microbiol. 2002, 68, 5064–5081. [Google Scholar] [CrossRef] [PubMed]
- Loy, A.; Schulz, C.; Lucker, S.; Schopfer-Wendels, A.; Stoecker, K.; Baranyi, C.; Lehner, A.; Wagner, M. 16S rRNA gene-based oligonucleotide microarray for environmental monitoring of the betaproteobacterial order “Rhodocyclales”. Appl. Environ. Microbiol. 2005, 71, 1373–1386. [Google Scholar] [CrossRef] [PubMed]
- Peplies, J.; Glockner, F.O.; Amann, R. Optimization strategies for DNA microarray-based detection of bacteria with 16S rRNA-targeting oligonucleotide probes. Appl. Environ. Microbiol. 2003, 69, 1397–1407. [Google Scholar] [CrossRef] [PubMed]
- Peplies, J.; Glockner, F.O.; Amann, R.; Ludwig, W. Comparative sequence analysis and oligonucleotide probe design based on 23S rRNA genes of Alphaproteobacteria from North Sea bacterioplankton. Syst. Appl. Microbiol. 2004, 27, 573–580. [Google Scholar] [CrossRef] [PubMed]
- Peplies, J.; Lachmund, C.; Glockner, F.O.; Manz, W. A DNA microarray platform based on direct detection of rRNA for characterization of freshwater sediment-related prokaryotic communities. Appl. Environ. Microbiol. 2006, 72, 4829–4838. [Google Scholar] [CrossRef] [PubMed]
- Peplies, J.; Lau, S.C.K.; Pernthaler, J.; Amann, R.; Glockner, F.O. Application and validation of DNA microarrays for the 16S rRNA-based analysis of marine bacterioplankton. Environ. Microbiol. 2004, 6, 638–645. [Google Scholar] [CrossRef] [PubMed]
- Lehner, A.; Loy, A.; Behr, T.; Gaenge, H.; Ludwig, W.; Wagner, M.; Schleifer, K.-H. Oligonucleotide microarray for identification of Enterococcus species. FEMS Microbiol. Lett. 2005, 246, 133–142. [Google Scholar] [CrossRef] [PubMed]
- Kappel, K.; Westernhagen, H.V.; Blohm, D.H. Microarray-based identification of eggs and larvae from fish species common in the North Sea. In Proceedings of the Dechema Chip-Technology Meeting, Frankfurt, Germany, 24–25 February 2003. [Google Scholar]
- MIDTAL: Microarrays for the Detection of Toxic Algae. Available online: http://www.midtal.com (accessed on 18 May 2017).
- Baudart, J.; Guillebault, D.; Mielke, E.; Meyer, T.; Tqandon, N.; Fischer, S.; Weigel, W.; Medlin, L.K. Microarray (phylochip) analysis of freshwater pathogens at several sites along the Northern German coast transecting both estuarine and freshwaters. Appl. Microbiol. Biotechnol. 2016, 101, 871–886. [Google Scholar] [CrossRef] [PubMed]
- Van Der Waal, D.; Guillebault, D.; Alfonso, A.; Rodríguez, I.; Botana, L.; Medlin, L.K. µAqua Microarrays for Phylogenetic and Toxin Expression of Cyanobacteria with Validation by Cell counts and UPLC/MS-MS. Harmful Algae 2017. under review. [Google Scholar]
- Kegel, J.U.; Guillebault, D.; Medlin, L.K. Application of microarrays (phylochips) for analysis of community diversity by species identification. Perspect. Phycol. 2016. [Google Scholar] [CrossRef]
- Scorzetti, G.; Brand, L.E.; Hitchcock, G.L.; Rein, K.S.; Sinigalliano, C.D.; Fell, J.W. Multiple simultaneous detection of Harmful Algal Blooms (HABs) through a high throughput bead array technology, with potential use in phytoplankton community analysis. Harmful Algae 2000, 8, 196–211. [Google Scholar] [CrossRef] [PubMed]
- Diaz, M.R.; Jacobson, J.W.; Goodwin, K.D.; Dunbar, S.A.; Fell, J.W. Molecular detection of harmful algal blooms (HABs) using locked nucleic acids and bead array technology. Limnol. Oceanogr. Methods 2010, 8, 269–284. [Google Scholar] [CrossRef] [PubMed]
- Saunders, N.A. Introduction to Real-Time PCR. In Real-Time PCR: An Essential Guide; Edwards, K., Logan, J., Saunders, N., Eds.; Horizon Bioscience: Norfolk, UK, 2004; pp. 1–11. [Google Scholar]
- Handy, S.M.; Hutchins, D.A.; Cary, S.C.; Coyne, K.J. Simultaneous enumeration of multiple raphidophyte species by quantitative real-time PCR, capabilities and limitations. Limnol. Oceanogr. Methods 2006, 4, 193–204. [Google Scholar] [CrossRef]
- Erdner, D.L.; Percy, L.; Keafer, B.; Lewis, J.; Anderson, D.M. A quantitative real-time PCR assay for the identification and enumeration of Alexandrium cysts in the marine sediments. Deep Sea Res. II 2010, 57, 279–287. [Google Scholar] [CrossRef] [PubMed]
- Nolen, R.S. Endangered sea otters threatened by toxic algae. JAVMA J. Am. Vet. Med. Assoc. 2010, 237, 1116–1117. [Google Scholar]
- Cardullo, R.A.; Agrawal, S.; Flores, C.; Zamecnik, P.C.; Wolf, D.E. Detection of nucleic acid hybridization by nonradioactive fluorescence resonance energy transfer. Proc. Natl. Acad. Sci. USA 1988, 85, 8790–8794. [Google Scholar] [CrossRef] [PubMed]
- Kudela, R.M.; Berdalet, E.; Bernard, S.; Burford, M.; Fernand, L.; Lu, S.; Roy, S.; Tester, P.; Usup, G.; Magnien, R.; et al. Harmful Algal Blooms. A Scientific Summary for Policy Makers; IOC/INF-1320; IOC/UNESCO: Paris, France, 2015; pp. 1–20. [Google Scholar]
- Sykes, P.J.; Neoh, S.H.; Brisco, S.H.; Hughes, E.; Condon, J.; Morley, A.A. Quantitation of targets for PCR by use of limiting dilution. Biotechniques 1992, 13, 444–449. [Google Scholar] [PubMed]
- Tewhey, R.; Warner, J.B.; Nakano, M.; Libby, B.; Medkova, M.; David, P.H.; Kotsopoulos, S.K.; Samuels, M.L.; Hutchison, J.B.; Larson, J.W.; et al. Microdroplet-based PCR enrichment for large-scale targeted sequencing. Nat. Biotechnol. 2000, 27, 1025–1031. [Google Scholar] [CrossRef] [PubMed]
- Dong, L.; Meng, Y.; Sui, Z.; Wang, J.; Wu, L.; Fu, B. Comparison of four digital PCR platforms for accurate quantification of DNA copy number of a certified plasmid DNA reference material. Sci. Rep. 2015, 5, 13174. [Google Scholar] [CrossRef] [PubMed]
- Te, S.H.; Chen, E.Y.; Gin, K.Y.-H. Comparison of quantitative PCR and droplet digital PCR multiplex assays for two genera of bloom-forming cyanobacteria, Cylindrospermopsis and Microcystis. Appl. Environ. Microbiol. 2015, 81, 5203–5211. [Google Scholar] [CrossRef] [PubMed]
- Hosoi-Tanabe, S.; Sako, Y. Species-specific detection and quantification of toxic marine dinoflagellates Alexandrium tamarense and A. catenella by real-time PCR assay. Mar. Biotechnol. 2005, 7, 506–514. [Google Scholar] [PubMed]
- Bowers, H.A.; Tengs, T.; Glasgow, H.B.; Burkholder, J.M.; Rublee, P.A.; Oldach, D.W. Development of real-time PCR assays for rapid detection of Pfiesteria piscicida and related dinoflagellates. Appl. Environ. Microbiol. 2000, 66, 4641–4648. [Google Scholar] [CrossRef] [PubMed]
- Kavanagh, S.; Brennan, C.; O’Connor, L.; Moran, S.; Salas, R.; Lyons, J.; Silke, J.; Maher, M. Real-time PCR detection of Dinophysis species in Irish coastal waters. Mar. Biotechnol. 2010, 12, 534–542. [Google Scholar] [CrossRef] [PubMed]
- IDT: Integrated DNA Technologies. Available online: http://www.idtdna.com/pages/decoded/decoded-articles/core-concepts/decoded/2011/09/12/one-step-two-step (accessed on 18 May 2017).
- Mark, D.; Haeberle, S.; Roth, G.; von Stetten, F.; Zengerle, R. Microfluidic lab-on-a-chip platforms, requirements, characteristics and applications. Chem. Soc. Rev. 2010, 39, 1153–1182. [Google Scholar] [CrossRef] [PubMed]
- Pedrero, M.; Susana Campuzano, S.; Pingarrón, J.M. Electroanalytical sensors and devices for multiplexed detection of foodborne pathogen microorganisms. Sensors 2000, 9, 5503–5520. [Google Scholar] [CrossRef] [PubMed]
- Hadar, B.Y.; Dykstra, P.H.; Bentley, W.E.; Ghodssi, R. A controlled microfluidic electrochemical lab-on-a-chip for label-free diffusion-restricted DNA hybridization analysis. Biosens. Bioelectron. 2015, 64, 579–585. [Google Scholar]
- Ebenezer, V.; Medlin, L.K.; Kei, J.-S. Molecular detection, quantification, and diversity evaluation of microalgae. Mar. Biotechnol. 2011, 14, 129–142. [Google Scholar] [CrossRef] [PubMed]
- Oxford Nanopore. Available online: https://nanoporetech.com/ (accessed on 18 May 2017).
- Merkoçi, A. Electrochemical biosensing with nanoparticles. FEBS J. 2007, 274, 310–316. [Google Scholar]
- Wang, J. Nanomaterial-based electrochemical biosensors. Analyst 2005, 130, 421–426. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Xiong, E.; Zhang, X.; Zhang, X.; Chen, J. Nanomaterials as signal amplification elements in DNA-based electrochemical sensing. Nano Today 2014, 9, 197–211. [Google Scholar] [CrossRef]
- Mirkin, C.A.; Letsinger, R.L.; Mucic, R.C.; Storhoff, J.J. A DNA-based method for rationally. Assembling nanoparticles into macroscopic materials. Nature 1996, 382, 607–609. [Google Scholar] [CrossRef] [PubMed]
- Kwon, S.J.; Bard, A.J. DNA Analysis by Application of Pt Nanoparticle Electrochemical Amplification with Single Label Response. J. Am. Chem. Soc. 2012, 134, 10777–10779. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Xu, D.; Kawde, A.N.; Polsky, R. Metal Nanoparticle-Based Electrochemical Stripping Potentiometric Detection of DNA Hybridization. Anal. Chem. 2001, 73, 5576–5581. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Song, S.; Zhang, L.; Wang, L.; Wu, H.; Pan, D.; Fan, C. Sequence-Specific Detection of Femtomolar DNA via a Chronocoulometric DNA Sensor (CDS), Effects of Nanoparticle-Mediated Amplification and Nanoscale Control of DNA Assembly at Electrodes. J. Am. Chem. Soc. 2006, 128, 8575–8580. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Xu, D.; Polsky, R. Magnetically-Induced Solid-State Electrochemical Detection of DNA Hybridization. J. Am. Chem. Soc. 2002, 124, 4208–4209. [Google Scholar] [CrossRef] [PubMed]
- Kuana, G.C.; Shenga, L.P.; Rijiravanichb, P.; Marimuthua, K.; Ravichandrana, M.; Yina, L.S.; Lertanantawonga, B.; Surareungchaid, W. Gold-nanoparticle based electrochemical DNA sensor for the detection of fish pathogen Aphanomyces invadans. Talanta 2013, 117, 312–317. [Google Scholar] [CrossRef] [PubMed]
- Vijian, D.; Chinni, S.V.; Yin, L.S.; Lertanantawong, B.; Surareungchai, W. Non-protein coding RNA-based genosensor with quantum dots as electrochemical labels for attomolar detection of multiple pathogens. Biosens. Bioelectron. 2016, 77, 805–811. [Google Scholar] [CrossRef] [PubMed]
- Jafari, S.; Faridbod, F.; Norouzi, P.; Dezfuli, A.S.; Ajloo, D.; Mohammadipanah, F.; Ganjali, R.S. Detection of Aeromonas hydrophila DNA oligonucleotide sequence using a biosensor design based on Ceria nanoparticles decorated reduced graphene oxide and Fast Fourier transform square wave voltammetry. Ana. Chim. Acta 2015, 895, 80–88. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Deng, J.; Fang, L.; Yu, K.; Huang, H.; Jiang, L.; Liang, W.; Zheng, J. A novel electrochemical DNA biosensor based on HRP-mimicking hemin/G-quadruplex wrapped GOx nanocomposites as tag for detection of Escherichia coli O157, H7. Biosens. Bioelectron. 2015, 63, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Rodiger, S.; Liebsch, C.; Schmidt, C.; Lehmann, W.; Resch-Genger, U.; Schedler, U.; Schierack, P. Nucleic acid detection based on the use of microbeads, a review. Microchim. Acta 2014, 181, 1151–1168. [Google Scholar] [CrossRef]
- Ganesh, I.; Buu Minh Tran, B.M.; Kim, Y.; Kim, J.; Chen, H.; Lee, N.L.; Park, S. An integrated microfluidic PCR system with immunomagnetic nanoparticles for the detection of bacterial pathogens. Biomed. Microdevices 2016, 18, 116. [Google Scholar] [CrossRef] [PubMed]
- Lermo, S.; Campoy, J.; Barbe, S.; Hernandez, S.; Alegret, A.; Pividori, M.I. In situ DNA amplification with magnetic primers for the electrochemical detection of food pathogens. Biosens. Bioelectron. 2007, 22, 2010–2017. [Google Scholar] [CrossRef] [PubMed]
- Olsvik, O.; Opovic, T.; Skjerve, E.; Cudjoe, K.S.; Hornes, E.; Ugelstad, J.; Uhlen, M. Magnetic Separation Techniques in Diagnostic Microbiology. Clin. Microbial. Rev. 1994, 7, 43–54. [Google Scholar] [CrossRef]
- Wilson, T.; Carson, J. Development of sensitive, high-throughput one-tube RT-PCR-enzyme hybridisation assay to detect selected bacterial fish pathogens. Dis. Aquat. Org. 2003, 54, 127–134. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Dai, T.; Serwadda, A.; Shen, H. Detecting a novel Eriocheir sinensis reovirus by reverse transcription loop-mediated isothermal amplification assay. Lett. Appl. Microbiol. 2016, 63, 363–368. [Google Scholar] [CrossRef] [PubMed]
- Suebsing, R.; Kampeera, J.; Tookdee, B.; Withyachumnarnkul, B.; Turner, W.; Kiatpathomchai, W. Evaluation of colorimetric loop-mediated isothermal amplification assay for visual detection of Streptococcus agalactiae and Streptococcus iniae in tilapia. Lett. App. Microbiol. 2013, 57, 1–8. [Google Scholar]
- Xie, G.S.; Zhang, Q.L.; Han, N.N.; Shi, C.Y.; Wang, X.H.; Liu, Q.H.; Huang, J. An improved method for detection of Edwardsiella tarda by loop-mediated isothermal amplification by targeting the EsrB gene. Chin. J. Ocean. Limnol. 2012, 30, 595–603. [Google Scholar] [CrossRef]
- Gao, H.W.; Li, F.H.; Zhang, X.J.; Wang, B.; Xiang, J.H. Rapid, sensitive detection of Vibrio anguillarum using loop-mediated isothermal amplification. Chin. J. Ocean. Limnol. 2015, 28, 62–66. [Google Scholar] [CrossRef]
- Ren, C.H.; Hu, C.Q.; Luo, P.; Wang, Q.B. Sensitive and rapid identification of Vibrio vulnificus by loop-mediated isothermal amplification. Microbiol. Res. 2000, 164, 514–521. [Google Scholar] [CrossRef] [PubMed]
- Tsai, M.A.; Wang, P.C.; Yoshida, T.; Liaw, L.L.; Chen, S.C. Development of a sensitive and specific LAMP PCR assay for detection of fish pathogen Lactococcus garvieae. Dis. Aquat. Organ. 2013, 102, 225–235. [Google Scholar] [CrossRef] [PubMed]
- Dirkst, R.M.; Pierce, N.A. Triggered amplification by hybridization chain reaction. Proc. Natl. Acad. Sci. USA 2004, 101, 15275–15278. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, T.; Kawakami, S.; Hatamoto, M.; Imachi, H.; Takahashi, M.; Araki, N.; Yamaguchi, T.; Kubota, K. In situ DNA-hybridization chain reaction (HCR), a facilitated in situ HCR system for the detection of environmental microorganisms. Environ. Microbiol. 2015, 17, 2532–2541. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, T.; Fuchs, B.M.; Amann, R.; Kawakami, S.; Kubota, K.; Hatamoto, M.; Yamaguchi, T. Rapid and sensitive identification of marine bacteria by an improved in situ DNA hybridization chain reaction (quickHCR-FISH). Syst. Appl. Microbiol. 2015, 38, 400–405. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Fang, L.; Tian, Y.; Wei, W.; Wang, L. Label-free, non-enzymatic and ultrasensitive electrochemical nucleic acid biosensing by tandem DNA-fueled target recycling and hybridization chain reaction. Sens. Actuators B Chem. 2017, 244, 450–457. [Google Scholar] [CrossRef]
- Scholin, C.; Doucette, G.; Jensen, S.; Roman, B.; Pargett, D.; Marin, R., III; Preston, C.; Jones, W.; Feldman, J.; Everlove, C.; et al. Remote detection of marine microbes, small invertebrates, Harmful Algae and biotoxins using the Environmental Sample Processor (ESP). Oceanography 2000, 22, 158–167. [Google Scholar] [CrossRef]
- Preston, C.M.; Harris, A.; Ryan, J.P.; Roman, B.; Marin, R., III; Jensen, S.; Everlove, C.; Birch, J.; Dzenitis, J.M.; Pargett, D.; et al. Underwater application of quantitative PCR on an ocean mooring. PLoS ONE 2011, 6, e22522. [Google Scholar] [CrossRef] [PubMed]
- Greenfield, D.I.; Marin, R., III; Doucette, G.J.; Mikulski, C.; Jones, K.; Jensen, S.; Roman, B.; Alvarado, J.; Feldman, J.; Scholin, C. Field applications of the second-generation Environmental Sample Processor (ESP) for remote detection of harmful algae, 2006–2007. Limnol. Oceanogr. Methods. 2008, 6, 667–679. [Google Scholar] [CrossRef]
- Doucette, G.J.; Mikulski, C.M.; Jones, K.L.; King, K.L.; Greenfield, D.I.; Marin, R., III; Jensen, S.; Roman, B.; Elliott, C.T.; Scholin, C.A. Remote, subsurface detection of the algal toxin domoic acid onboard the Environmental Sample Processor, assay development and field trials. Harmful Algae 2000, 8, 880–888. [Google Scholar] [CrossRef]
- Ryan, J.; Greenfield, D.; Marin, R., III; Preston, C.; Roman, B.; Jensen, S.; Pargett, D.; Birch, Mikulski, C.; Doucette, G.; Scholin, C. Harmful phytoplankton ecology studies using an autonomous molecular analytical and ocean observing network. Limnol. Oceanogr. 2011, 56, 1255–1272. [Google Scholar] [CrossRef]
- Medlin, L.K. Mini Review: Molecular Techniques for Identification and Characterization of Marine Biodiversity. Ann. Mar. Biol. Res. 2016, 3, 1015. [Google Scholar]


© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Medlin, L.K.; Orozco, J. Molecular Techniques for the Detection of Organisms in Aquatic Environments, with Emphasis on Harmful Algal Bloom Species. Sensors 2017, 17, 1184. https://doi.org/10.3390/s17051184
Medlin LK, Orozco J. Molecular Techniques for the Detection of Organisms in Aquatic Environments, with Emphasis on Harmful Algal Bloom Species. Sensors. 2017; 17(5):1184. https://doi.org/10.3390/s17051184
Chicago/Turabian StyleMedlin, Linda K., and Jahir Orozco. 2017. "Molecular Techniques for the Detection of Organisms in Aquatic Environments, with Emphasis on Harmful Algal Bloom Species" Sensors 17, no. 5: 1184. https://doi.org/10.3390/s17051184
APA StyleMedlin, L. K., & Orozco, J. (2017). Molecular Techniques for the Detection of Organisms in Aquatic Environments, with Emphasis on Harmful Algal Bloom Species. Sensors, 17(5), 1184. https://doi.org/10.3390/s17051184