
A Quantitative PCR-Electrochemical Genosensor Test for the Screening of Biotech Crops


 ,
,  ,
, 
Abstract
1. Introduction
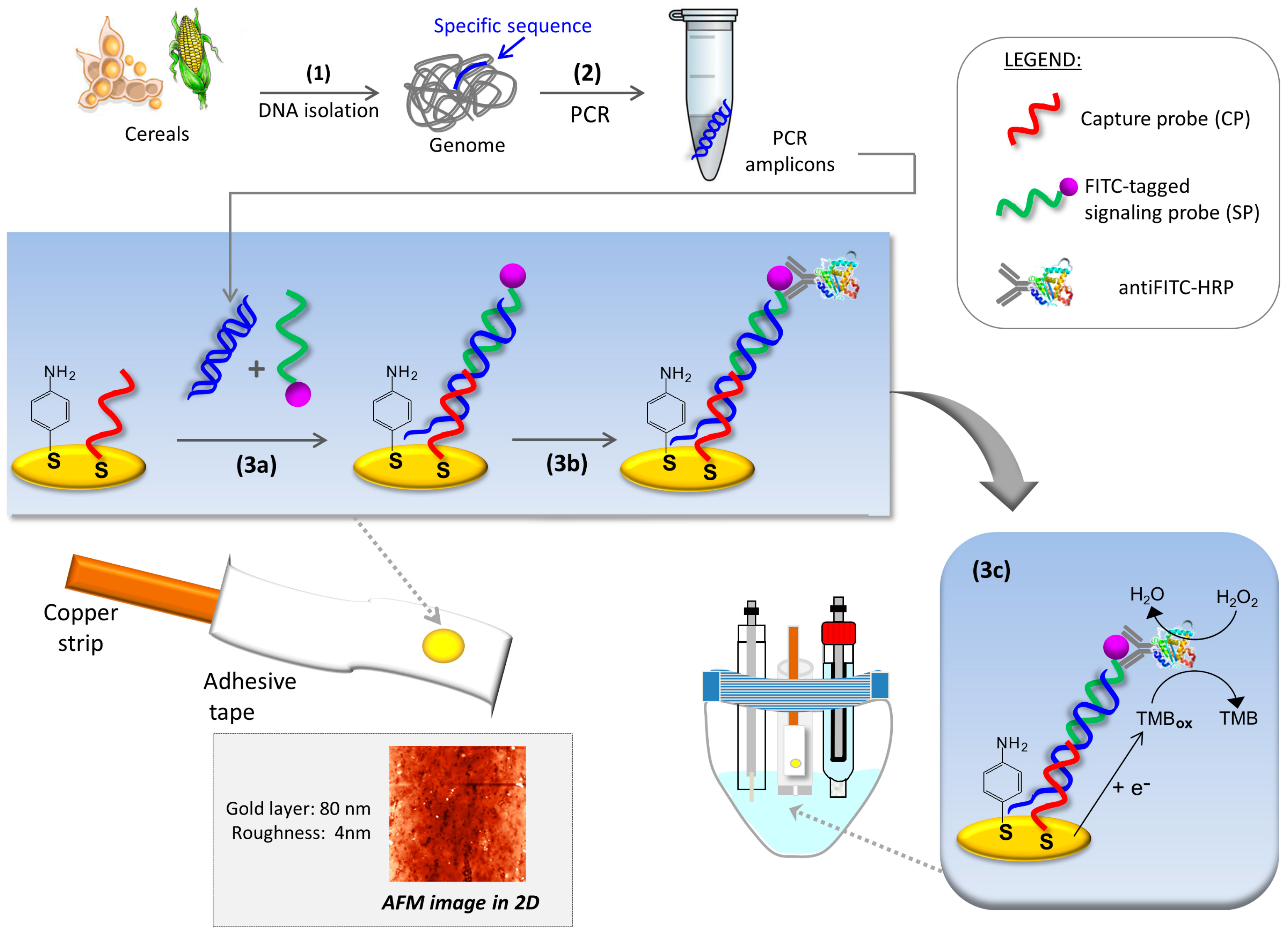
2. Materials and Methods
2.1. Instrumentation
2.2. Reagents
2.3. Sample Preparation and DNA Extraction
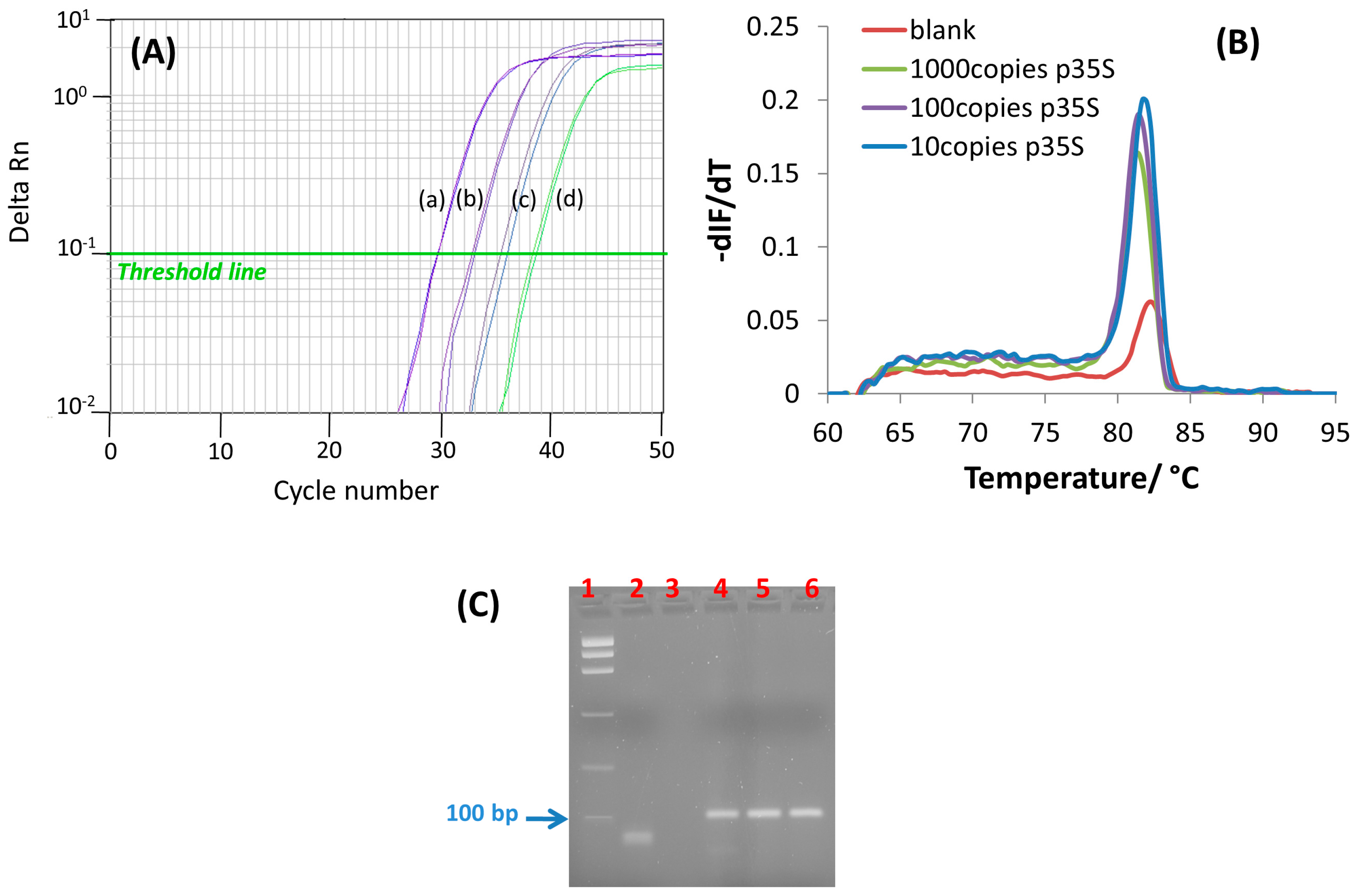
2.4. Real-Time Polymerase Chain Reaction (qPCR)
2.5. End-Point Polymerase Chain Reaction (PCR)
2.6. Agarose Gel Electrophoresis
2.7. Electrochemical Genosensor
3. Results and Discussion
3.1. PCR Amplification: Design and Evaluation
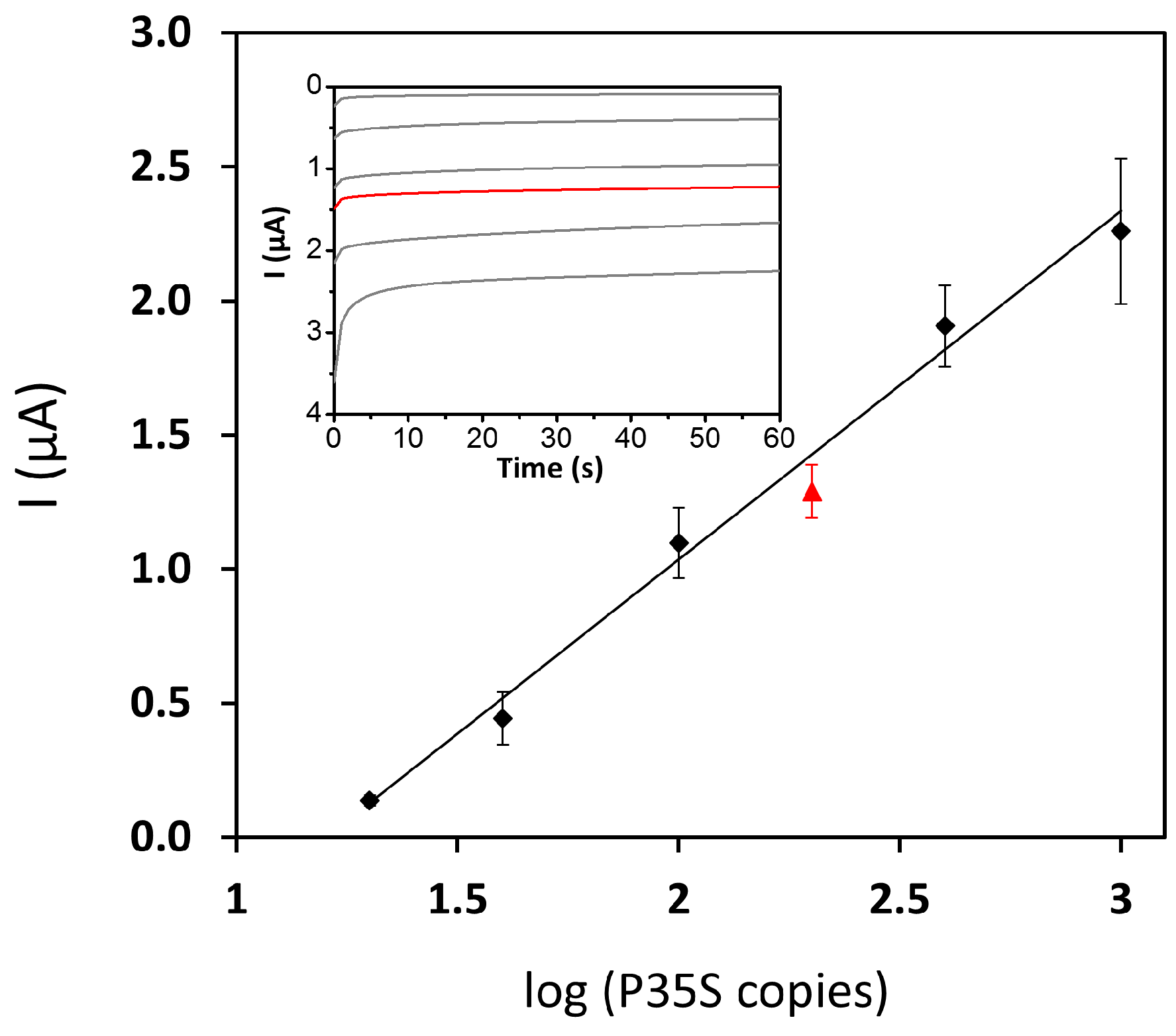
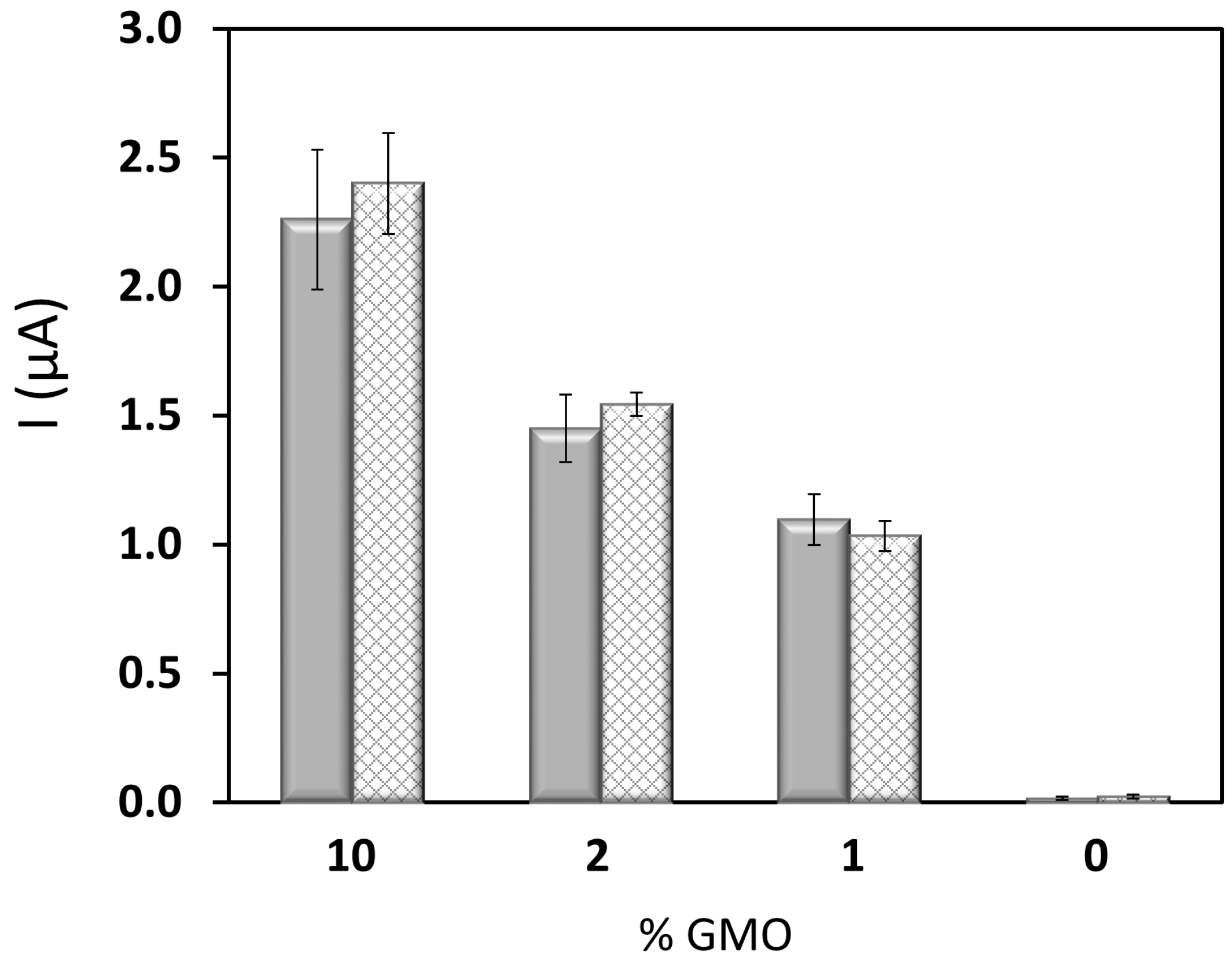
3.2. Combining PCR and Electrochemical Genosensor
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- James, C.; Brief, C. No. 49: Global Status of Commercialized Biotech/GM Crops: 2014; International Service for the Acquisition of Agribiotech Applications (ISAAA): Ithaca, NY, USA, 2014. [Google Scholar]
- Buiatti, M.; Christou, P.; Pastore, G. The application of GMOs in agriculture and in food production for a better nutrition: Two different scientific points of view. Genes Nutr. 2013, 8, 255–270. [Google Scholar] [CrossRef] [PubMed]
- European Commission. Regulation (EC) No 1829/2003 of the European Parliament and of the Council of 22 September 2003 on genetically modified food and feed. Off. J. Eur. Union 2003, 268, 1–23. [Google Scholar]
- European Commission. Regulation (EC) No 1830/2003 concerning the traceability and labelling of genetically modified organisms and the traceability of food and feed products produced from genetically modified organisms and amending Directive 2001/18/EC. Off. J. Eur. Union 2003, 268, 24–28. [Google Scholar]
- Center for Environmental Risk Assessment. Available online: http://cera-gmc.org (accessed on 13 March 2017).
- GMO Compass. Available online: http://www.gmo-compass.org (accessed on 13 March 2017).
- GMO Detection Laboratory in Shanghai Jiao Tong University (GMODL-SJTU): GMO Detection Method Database (GMDD). Available online: http://gmdd.shgmo.org (accessed on 13 March 2017).
- Wu, Y.; Wang, Y.; Li, J.; Li, W.; Zhang, L.; Li, Y.; Li, X.; Li, J.; Zhu, L.; Wu, G. Development of a general method for detection and quantification of the P35S promoter based on assessment of existing methods. Sci. Rep. 2014, 4, 7358. [Google Scholar] [CrossRef] [PubMed]
- Manzanares-Palenzuela, C.L.; Martín-Fernández, B.; Sánchez-Paniagua López, M.; López-Ruiz, B. Electrochemical genosensors as innovative tools for detection of genetically modified organisms. TrAC 2015, 66, 19–31. [Google Scholar] [CrossRef]
- Arugula, M.A.; Zhang, Y.; Simonian, A.L. Biosensors as 21st century technology for detecting genetically modified organisms in food and feed. Anal. Chem. 2014, 86, 119–129. [Google Scholar] [CrossRef] [PubMed]
- Bonanni, A.; Esplandiu, M.J.; del Valle, M. Impedimetric genosensors employing COOH-modified carbon nanotube screen-printed electrodes. Biosens. Bioelectron. 2009, 24, 2885–2891. [Google Scholar] [CrossRef] [PubMed]
- Guven, B.; Boyaci, I.H.; Tamer, U.; Acar-Soykut, E.; Dogan, U. Development of rolling circle amplification based surface-enhanced Raman spectroscopy method for 35S promoter gene detection. Talanta 2015, 136, 68–74. [Google Scholar] [CrossRef] [PubMed]
- Miranda-Castro, R.; de-los-Santos-Álvarez, P.; Lobo-Castañón, M.J.; Miranda-Ordieres, A.J.; Tuñón-Blanco, P. Hairpin-DNA probe for enzyme-amplified electrochemical detection of Legionella pneumophila. Anal. Chem. 2007, 79, 4050–4055. [Google Scholar] [CrossRef] [PubMed]
- Moura-Melo, S.; Miranda-Castro, R.; de-los-Santos-Álvarez, N.; Miranda-Ordieres, A.J.; Ribeiro-Dos-Santos-Junior, J.; da-Silva-Fonseca, R.A.; Lobo-Castañón, M.J. Targeting helicase-dependent amplification products with an electrochemical genosensor for reliable and sensitive screening of genetically modified organisms. Anal. Chem. 2015, 87, 8547–8554. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Wu, Y.; Huang, W.; Wang, B.; Sun, C.; Ge, Y.; Chen, Y. PCR and DHPLC methods used to detect juice ingredient for 7 fruits. Food Control 2012, 25, 696–703. [Google Scholar] [CrossRef]
- Miranda-Castro, R.; Sánchez-Salcedo, R.; Suárez-Álvarez, B.; de-los-Santos-Álvarez, N.; Miranda-Ordieres, A.J.; Lobo-Castañón, M.J. Thioaromatic DNA monolayers for target-amplification-free electrochemical sensing of environmental pathogenic bacteria. Biosens. Bioelectron. 2017, 92, 162–170. [Google Scholar] [CrossRef] [PubMed]
- Barreda-García, S.; Miranda-Castro, R.; de-los-Santos-Álvarez, N.; Miranda-Ordieres, A.J.; Lobo-Castañón, M.J. Comparison of isothermal helicase-dependent amplification and PCR for the detection of Mycobacterium tuberculosis by an electrochemical genomagnetic assay. Anal. Bioanal. Chem. 2016, 408, 8603–8610. [Google Scholar] [CrossRef] [PubMed]
- Bennett, M.D.; Leitch, I.J. Plant DNA C-Values Database (Release 6.0, December 2012). Available online: http://www.kew.org/cvalues (accessed on 13 March 2017).
- Definition of Minimum Performance Requirements for Analytical Methods of GMO Testing. European Network of GMO Laboratories (ENGL), 20 October 2015. Available online: http://gmo-crl.jrc.ec.europa.eu (accessed on 17 April 2017).
- Bubner, B.; Gase, K.; Baldwin, I.T. Two-fold differences are the detection limit for determining transgene copy numbers in plants by real-time PCR. BMC Biotechnol. 2004, 4, 14. [Google Scholar] [CrossRef] [PubMed]
- Liao, W.C.; Chuang, M.-C.; Ho, J.-A. Electrochemical sensor for multiplex screening of genetically modified DNA: Identification of biotech crops by logic-based biomolecular analysis. Biosens. Bioelectron. 2013, 50, 414–420. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Zheng, L.; Chen, Y.; Xue, F.; Cheng, L.; Adelojua, S.B.; Chen, W. A novel GMO biosensor for rapid ultrasensitive and simultaneous detection of multiple DNA components in GMO products. Biosens. Bioelectron. 2015, 66, 431–437. [Google Scholar] [CrossRef] [PubMed]
- Zhan, F.; Liao, X.; Gao, F.; Qiu, W.; Wang, Q. Electroactive crown ester-Cu2+ complex with in-situ modification at molecular beacon probe serving as a facile electrochemical DNA biosensor for the detection of CaMV 35s. Biosens. Bioelectron. 2017, 92, 589–595. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sequence Name | Length | Oligonucleotide Sequences 5′ → 3′ |
|---|---|---|
| Capture probe (CP) | 27 nt | AGA GGA AGG GTC TTG CGA AGG ATA GTG-(CH2)6SH |
| Signaling probe (SP) | 53 nt | 1FITC-CTA GAG TCA GCT TGT CAG CGT GTC CTC TCC AAA TGA AAT GAA CTT CCT TAT AT |
| P35S-Forward primer (P35S-FP) | 27 nt | GTA AGG GAT GAC GCA CAA TCC CAC TAT |
| P35S-Reverse primer (P35S-RP) | 27 nt | TCT GCT AGA GTC AGC TTG TCA GCG TGT |
| Plant-Forward primer (Plant-FP) | 23 nt | CTT GAT TTT ACC AAA GAT GAT GA |
| Plant-Reverse primer (Plant-RP) | 20 nt | TTC TTC GCA TGT ACC CGC AG |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moura-Melo, S.; Miranda-Castro, R.; De-los-Santos-Álvarez, N.; Miranda-Ordieres, A.J.; Dos Santos Junior, J.R.; Da Silva Fonseca, R.A.; Lobo-Castañón, M.J. A Quantitative PCR-Electrochemical Genosensor Test for the Screening of Biotech Crops. Sensors 2017, 17, 881. https://doi.org/10.3390/s17040881
Moura-Melo S, Miranda-Castro R, De-los-Santos-Álvarez N, Miranda-Ordieres AJ, Dos Santos Junior JR, Da Silva Fonseca RA, Lobo-Castañón MJ. A Quantitative PCR-Electrochemical Genosensor Test for the Screening of Biotech Crops. Sensors. 2017; 17(4):881. https://doi.org/10.3390/s17040881
Chicago/Turabian StyleMoura-Melo, Suely, Rebeca Miranda-Castro, Noemí De-los-Santos-Álvarez, Arturo J. Miranda-Ordieres, José Ribeiro Dos Santos Junior, Rosana A. Da Silva Fonseca, and María Jesús Lobo-Castañón. 2017. "A Quantitative PCR-Electrochemical Genosensor Test for the Screening of Biotech Crops" Sensors 17, no. 4: 881. https://doi.org/10.3390/s17040881
APA StyleMoura-Melo, S., Miranda-Castro, R., De-los-Santos-Álvarez, N., Miranda-Ordieres, A. J., Dos Santos Junior, J. R., Da Silva Fonseca, R. A., & Lobo-Castañón, M. J. (2017). A Quantitative PCR-Electrochemical Genosensor Test for the Screening of Biotech Crops. Sensors, 17(4), 881. https://doi.org/10.3390/s17040881