Introduction
The determination of iron present in sediments as solid phases or dissolved in the pore-waters can, in principle, be performed using electroanalytical methods. By operating in neutral or alkaline pH conditions, Fe(II) is determined by taking advantage of its electrochemical reduction to Fe° at the hanging mercury drop electrode (HMDE). Such a method has been applied to the determination of iron for instance in anoxic lake waters [
1], in the pore-waters of marine [
2], estuarine [
3] or lagoon sediments [
4,
5], by operating at their natural pH. In such experimental conditions, the main peak at about –1.440 V vs. Ag/AgCl, relevant to the above mentioned Fe
2+ reduction, is often preceded by a peak at about –1.100 V, that is generally attributed to the reduction of the complex FeHS
+ [
6]. Such a complex species is formed as a consequence of the high concentration of HS
- produced in the porewaters of anoxic sediments by sulfate reducing bacteria [
3,
7,
8]. Note that in the pore-water of intertidal sediments the concentration of the reduced species such as Fe(II) and S(-II) increases with increasing bacterial activity.
From an analytical viewpoint, the coexistence in the voltammograms of signals both for labile Fe2+ and for the complex FeHS+ hinders the possibility to obtain accurate quantitative data. The complexity of the equilibria involving interchange of Fe(II) between free and complexed species prevents, in fact, the use of standard methods for quantifying signals.
Really, complexes between Fe(II) and S(-II) can be destroyed by simple acidification of the sample and consequent release of gaseous H
2S. In acidic conditions, however, the direct electrochemical reduction of Fe
2+ to Fe° is preceded and hindered by the reduction of the hydrogen ions to evolve gaseous hydrogen. An alternative is offered by the possibility of exploit the electrochemical oxidation of Fe
2+ to Fe
3+, which takes place at solid electrodes at about 0.5 V vs Ag/AgCl [
9]. Unfortunately, such an oxidation is usually of poor analytical usefulness since voltammetric responses for this process are scarcely reversible and depends dramatically on the surface state of the solid electrode as well as on the concentration and nature of the supporting electrolyte [
10,
11].
The possibility to develop electroanalytical methods for iron determinations at acidic pH values is of great relevancy not only for pore-waters analysis, but also for the analysis of sediments extracts. In the study of sediments toxicity, in fact, great interest is put in determining the chemicals extracted by simple acidification of the sample with a solution of diluted HCl [
12,
13,
14]. The sulphides extracted in this conditions are called AVS (Acid Volatile Sulfides) and they are composed mainly by insoluble iron sulphides different from pyrite, namely by amorphous FeS, mackinawite, and greigite. The concentration of trace metals dissolved by this treatment constitutes the SEM (Simultaneously Extracted Metals). Numerous studies showed that the SEM/AVS ratio can be used to quantify the toxicity of sediments [
15,
16] since, when this ratio is > 1, the sulphide present in the sediment is not high enough to warranty insolubilisation of the trace metals. Note that the fraction of metal ions of the sediment that are dissolved by hydrochloric acid is commonly considered as the fraction of metals potentially available for partitioning in biota and in surface waters.
It was shown few years ago that the voltammetric behaviour of iron in acidic solutions is dramatically improved when the solid electrode is coated with a thin film of the perfluorinated cation exchanger Nafion [
17]. The polymeric layer allows the preconcentration of cationic Fe
2+ (or Fe
3+ as well) at the polymer/electrode interface improving significantly the sensitivity and detection limits of the analysis [
18]. Moreover, the presence of Nafion improves the reversibility and reproducibility of the voltammetric signals, which correspond to Fe
2+ oxidation and/or Fe
3+ reduction [
18,
19]. Such methods, which take advantage of the peculiar characteristics of voltammetric signals recorded at solid electrodes coated with thin films of ion-exchange polymers, has given rise to an extremely sensitive voltammetric technique, named ion-exchange voltammetry (IEV) [
20,
21].
In the present paper, the application of IEV methods at Nafion coated electrodes (NCE) for sensing and determining iron in pore-waters and sediment extracts is examined and discussed.
Experimental Section
Chemicals
Milli-Q water was used throughout to prepare the solutions.
All chemicals used were of analytical grade quality apart HCl and NaCl which were of Suprapur® grade (Merck). Iron solutions were prepared by dilution of 1000 ppm standard solutions (Merck).
2.5% w/v Nafion
® solutions were prepared by 1:1 dilution with methanol of 5% w/v Nafion
® solutions (Aldrich). Nafion
® coated electrodes were prepared by microvolume evaporation of 3 µl of the 2.5% w/v Nafion
® (film coating 0.375mg cm
-2 and film thickness of 2 µm) [
18].
Instrumentation and procedures
All electroanalytical measurements were carried out at room temperature (22±1°C) under a nitrogen atmosphere, using a conventional single compartment cell equipped with a platinum coil counter electrode and a KCl saturated Ag/AgCl reference electrode, to which all potentials are referred. The working electrode was a PTFE shrouded glassy carbon disk (area 0.2 cm2) polished to a mirror finish with graded alumina powder, coated with Nafion as described above.
Cyclic voltammetric measurements were carried out with an EG&G PAR Model 366 potentiostat connected to a Yokogawa model 3023 YX recorder.
Ion-exchange preconcentration of iron at the NCE was carried out in open circuit condition in stirred solution, typically for 10 min. After stopping the stirrer, the voltammetric scan was started from 0 mV vs Ag/AgCl and the final potential was 0.8 V. Quantitative analyses in real samples were performed by the standard addition method, recording the ion-exchange voltammograms after equilibration of the NCE with the spiked solutions. Typically, a new modified electrode was used for each set of measurements in one sample and relevant standard additions.
Differential pulse cathodic stripping voltammetry of reduced sulphur, S(-II), present in raw porewaters or evolved as H
2S during the acid treatment of sediments, was performed by using the following experimental conditions [
5]: deposition and starting potential –0.4 V; deposition time 0 s; scan rate 20 mV s
-1; pulse height 70 mV; step time 0.2 s. The instrumentation used for cathodic stripping was composed by an EG&G PAR 384B polarographic analyser interfaced with an EG&G PAR 303 A hanging mercury drop electrode, HMDE.
Analyses of iron by flame atomic absorption spectroscopy, FAAS, were performed with a Varian Spectrometer 250 Plus.
Quantification of signals was achieved by standard additions for voltammetric measurements and by external calibration plots for FAAS.
Samples
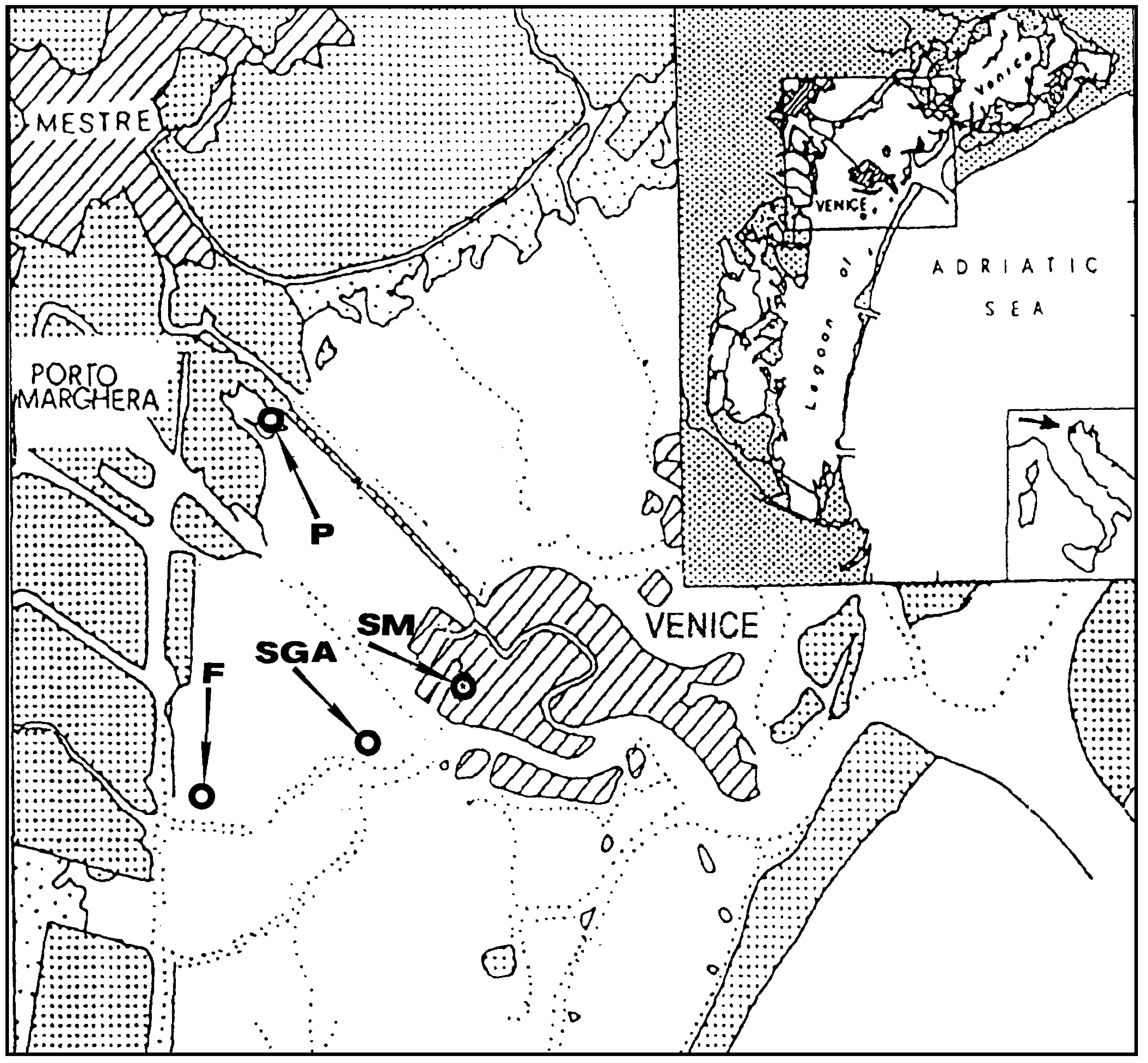
Sampling was performed in the Venice Lagoon, Venice (Italy), a shallow water basin with an average water depth of ca. 60 cm. Sampling sites, indicated in
Figure 1, were: an urban channel, rio S. Marta (SM), sampling date September’98; a mud-flat, in the vicinity of S. Giorgio in Alga Island (SGA), sampling date May’99, a two mud-flats in the industrial zone of Porto Marghera, near Fusina (F), sampling date May 99, and Pili, (P), sampling date October’00.
The pore-waters were sampled with an
in situ sampler and following the procedures described elsewhere [
22]. Before the analysis, the samples were diluted 1:100 with 0.03 M HCl.
Figure 1.
Sampling sites in the Venice Lagoon.
Figure 1.
Sampling sites in the Venice Lagoon.
Sediment samples were collected with a cylindrical corer, as previously described [
23] and were freeze-dried soon after the sampling. A known amount (typically, 2 g) of the homogenised freeze-dried sediment were added with 40 ml of HCl (1 M or 3 M, see below) operating under strictly anaerobic conditions under a nitrogen atmosphere. H
2S produced by the acid attack of the sediment was forced by the nitrogen flow to pass through a trap containing 20 ml of 1 M NaOH. A second trap containing 1 M Zn(CH
3COO)
2 was employed in order to have a visual check that no H
2S escaped from the first trap. If this was the case, some white ZnS should precipitate. In all our experiments no ZnS was detected. After 24 h digestion the NaOH trapping solution was diluted with 0.2 M borate buffer, pH 9, under anaerobic conditions up to a final volume of 200 ml. 10 ml of the diluted solution were introduced in the electrochemical cell for cathodic stripping determinations of S(-II). The sediment was separated by filtration from the HCl extract. The extract was diluted 1:100 with degassed Milli-Q water and introduced in the electrochemical cell for the IEV determination of iron.
Results and discussion
Sediments
One of the goals of the present study is the development of electrochemical sensing devices for the determination of iron in acidic extracts from sediments. In order to exploit such a possibility, at first, some samples of freeze-dried sediments of the lagoon were acid treated by using 1 M or 3 M HCl. In the literature, different authors [
12,
14,
15,
24,
25] used different concentrations of HCl in order to extract from the sediment the AVS and SEM. For this reason we compared the results obtained with two different extraction procedures, determining extracted iron by IEV at a NCE. For comparison also the AVS content of the sediment was quantified by cathodic stripping determination of evolved S(-II), using a HMDE.
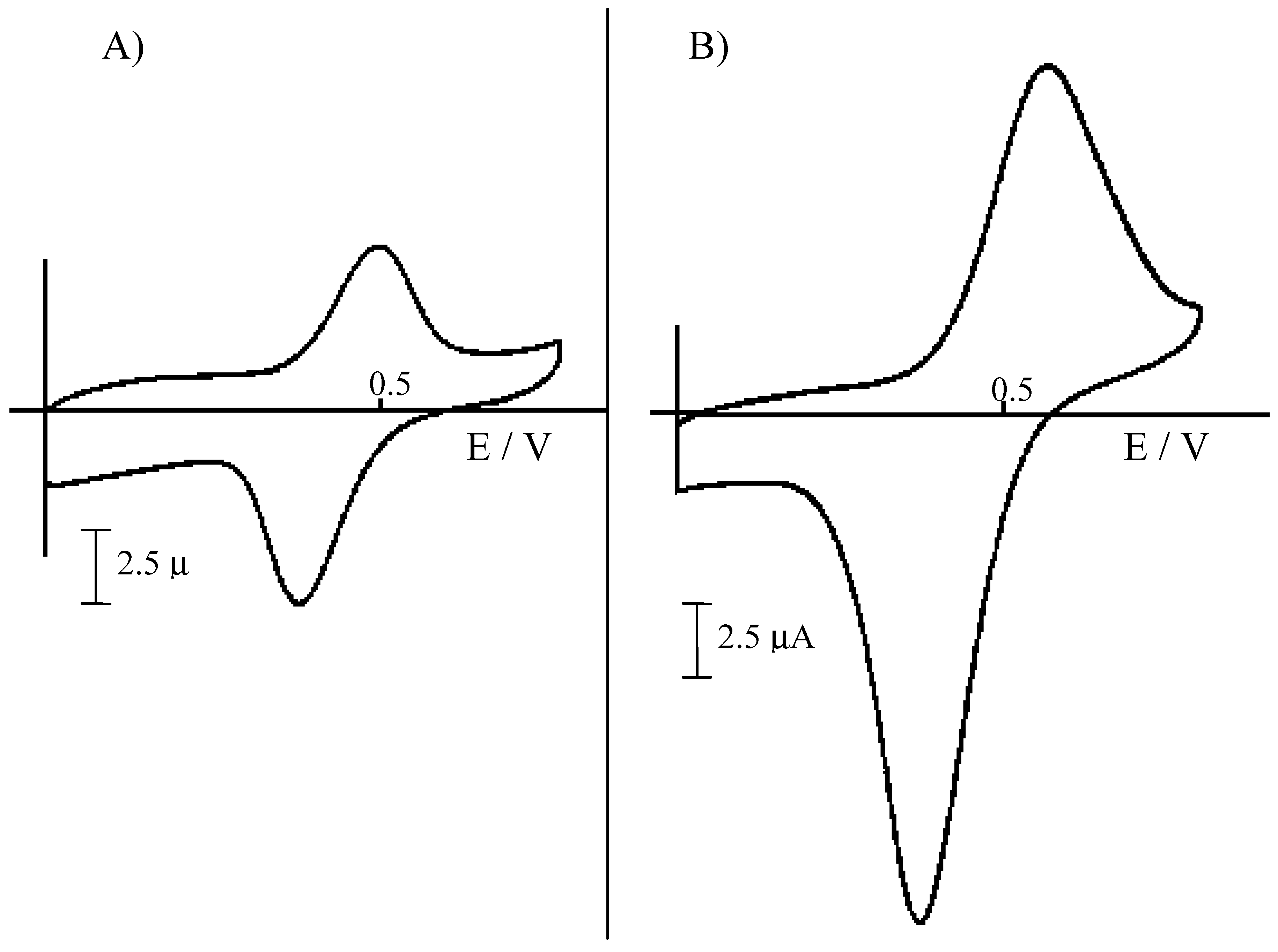
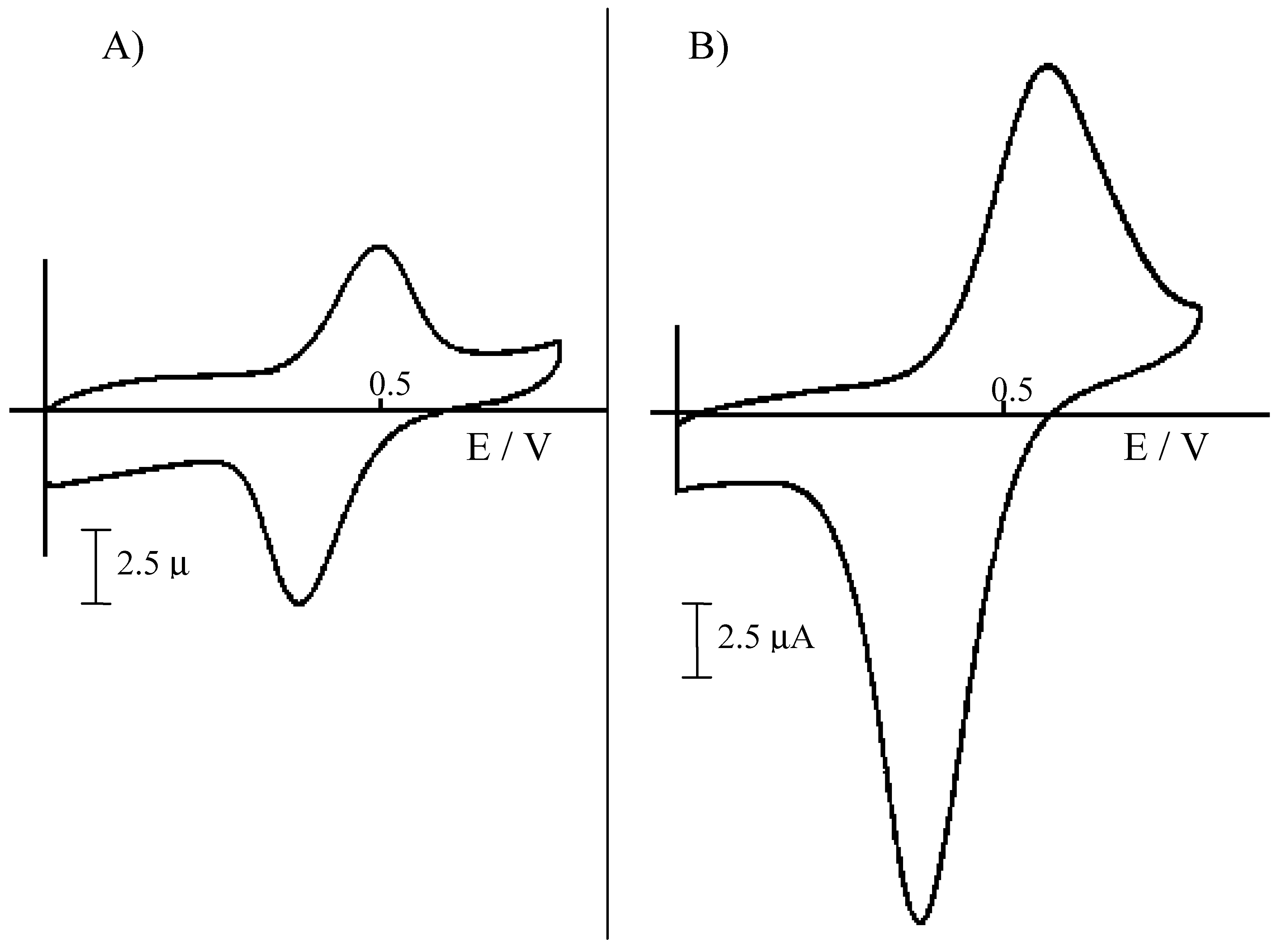
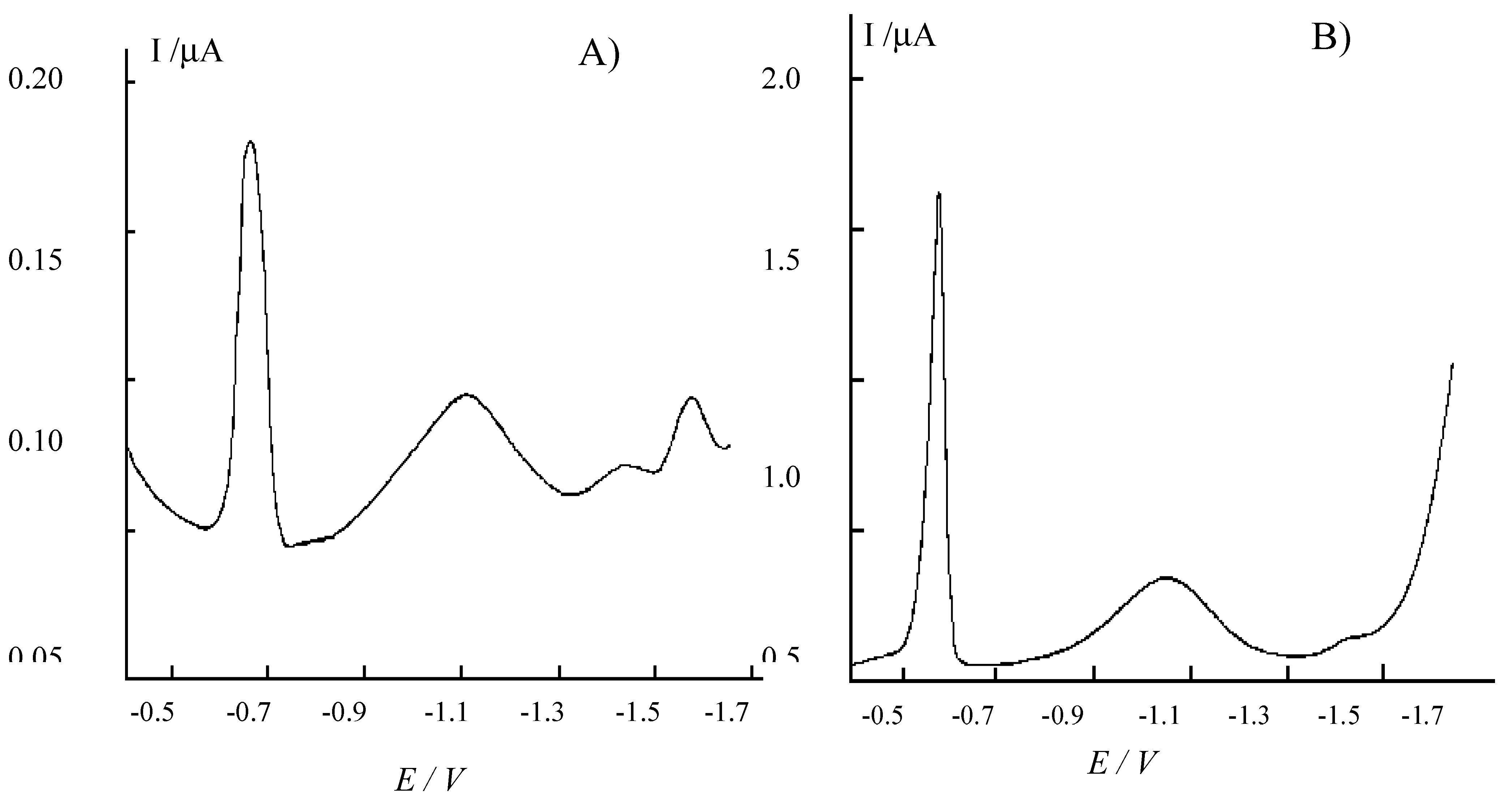
Figure 2.
Cyclic voltammograms recorded at 50 mV s-1 at a NCE, after ion-exchange equilibration of the Nafion coating with acid extracts from sediments, diluted 1:100 with water. A) Extraction for 24 h with 1 M HCl; B) extraction for 24 h with 3 M HCl.
Figure 2.
Cyclic voltammograms recorded at 50 mV s-1 at a NCE, after ion-exchange equilibration of the Nafion coating with acid extracts from sediments, diluted 1:100 with water. A) Extraction for 24 h with 1 M HCl; B) extraction for 24 h with 3 M HCl.
For iron determinations, both the 1 M and the 3 M HCl extracts were diluted 1:100 with water (after filtration); such a dilution allows one to lower the H
+ concentration as well as to lower the overall ionic strength of the solution, so pushing the ion-exchange competition between Fe
2+ and H
+ in favour of iron preconcentration. Note that in any case the distribution coefficient of iron in Nafion [
18] is much higher than that of H
+ or Na
+ [
26].
Fig. 2 compares the cyclic voltammetric signal obtained for Fe(II) at a NCE in the two different extracts. In both cases, the Fe
2+ oxidation signal appears well resolved from background currents. However, signals obtained in the 3M HCl extract (after 1:100 dilution) result higher, so indicating a significatively higher extraction yield. Note that extractions yields for Fe(II) parallel S(-II) extraction yields, quantified by DPV analysis of H
2S at the HMDE and shown in
Fig. 3.
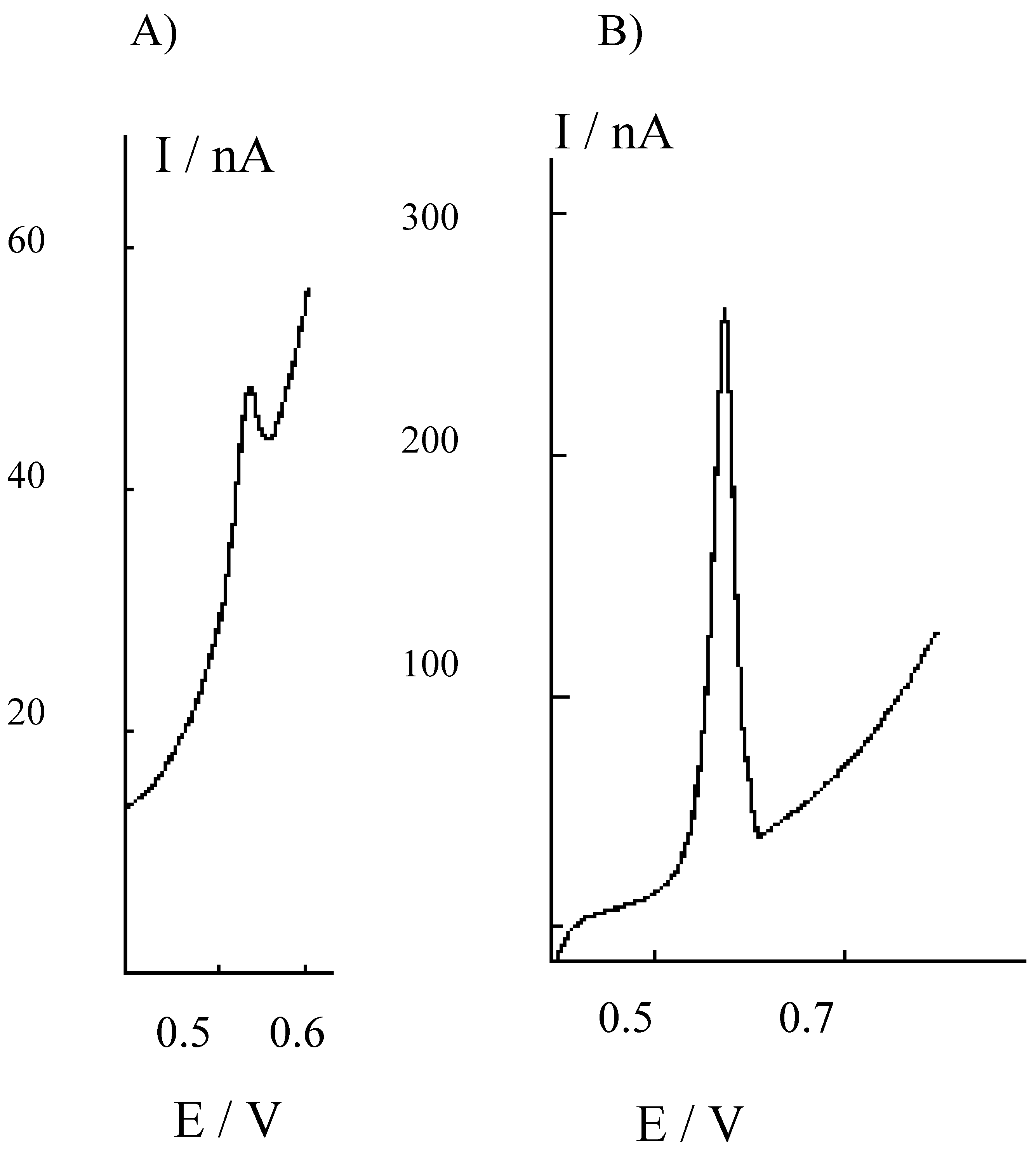
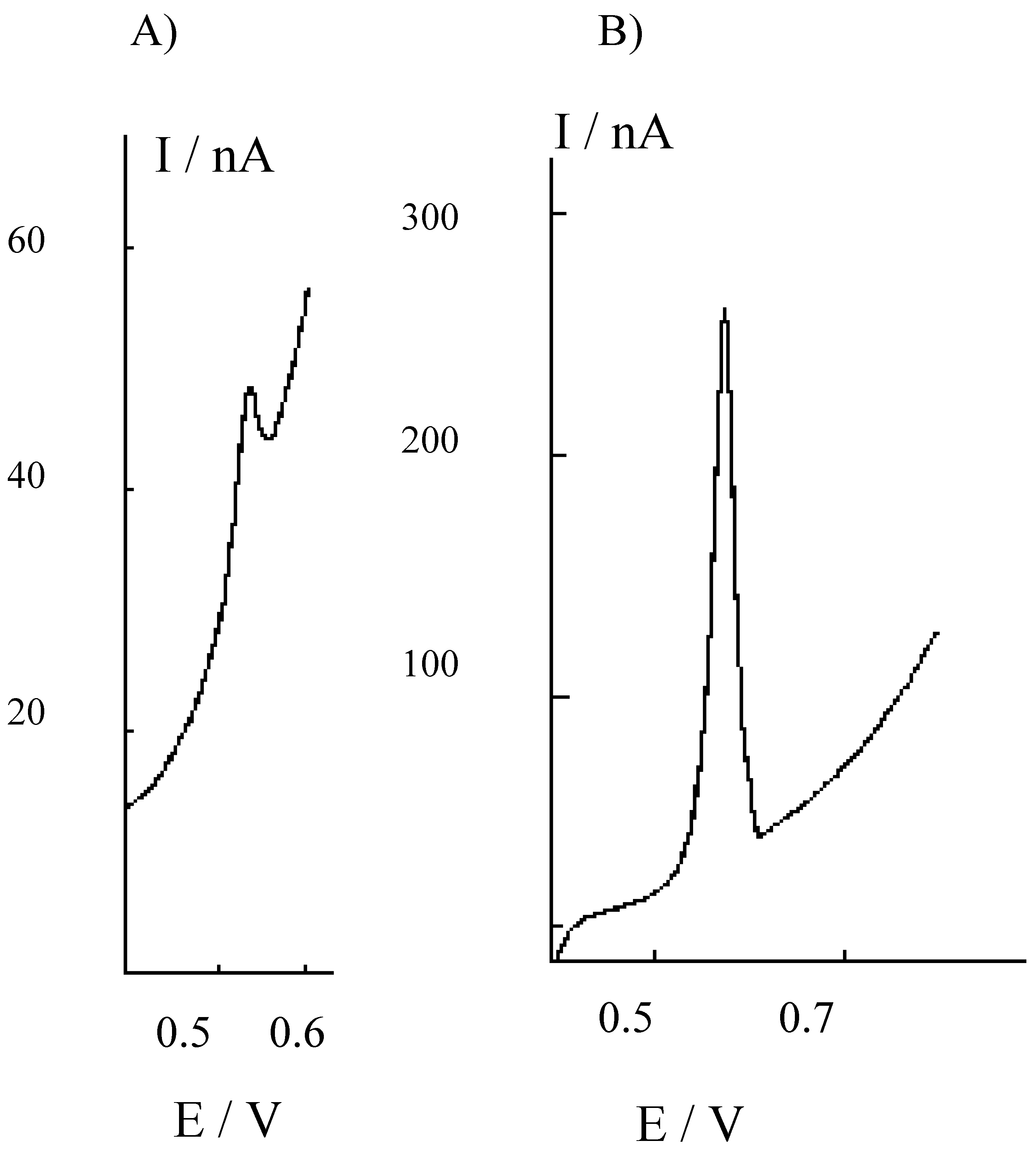
Figure 3.
Differential pulse cathodic stripping voltammograms recorded at the HMDE in the 1M NaOH solution used for trapping H2S evolved by acid attack of sediments, after buffering to pH 9 and 1:10 dilution with borate. A) Sediments treated for 24 h with 1M HCl; B) sediments treated for 24 h with 3M HCl.
Figure 3.
Differential pulse cathodic stripping voltammograms recorded at the HMDE in the 1M NaOH solution used for trapping H2S evolved by acid attack of sediments, after buffering to pH 9 and 1:10 dilution with borate. A) Sediments treated for 24 h with 1M HCl; B) sediments treated for 24 h with 3M HCl.
In this case, H2S vapours released during the acid treatment of the sediment were trapped in 1 M NaOH and then diluted 1:10 with 0.2 M borate buffer, pH 9, before the analysis. Again, higher H2S amounts are released and detected after treatment with the more concentrated acid solution. These evidences indicate that the use of 3 M HCl is more effective as far as higher extraction yields are concerned.
The question about the meaning of this extract with respect to the toxicity of sediments and to the quantification of the fraction of metals in sediment which is really bio-available still remains an open issue. Studies to correlate AVS, SEM and toxicity of sediments of the Venice Lagoon are presently in progress [
27]. However what is relevant to the aims of the present research is that NCEs show to be simple and powerful tools for the easy determination of iron in these acidic extracts, independently on the concentration of the acid used to obtain the extract.
Pore-waters
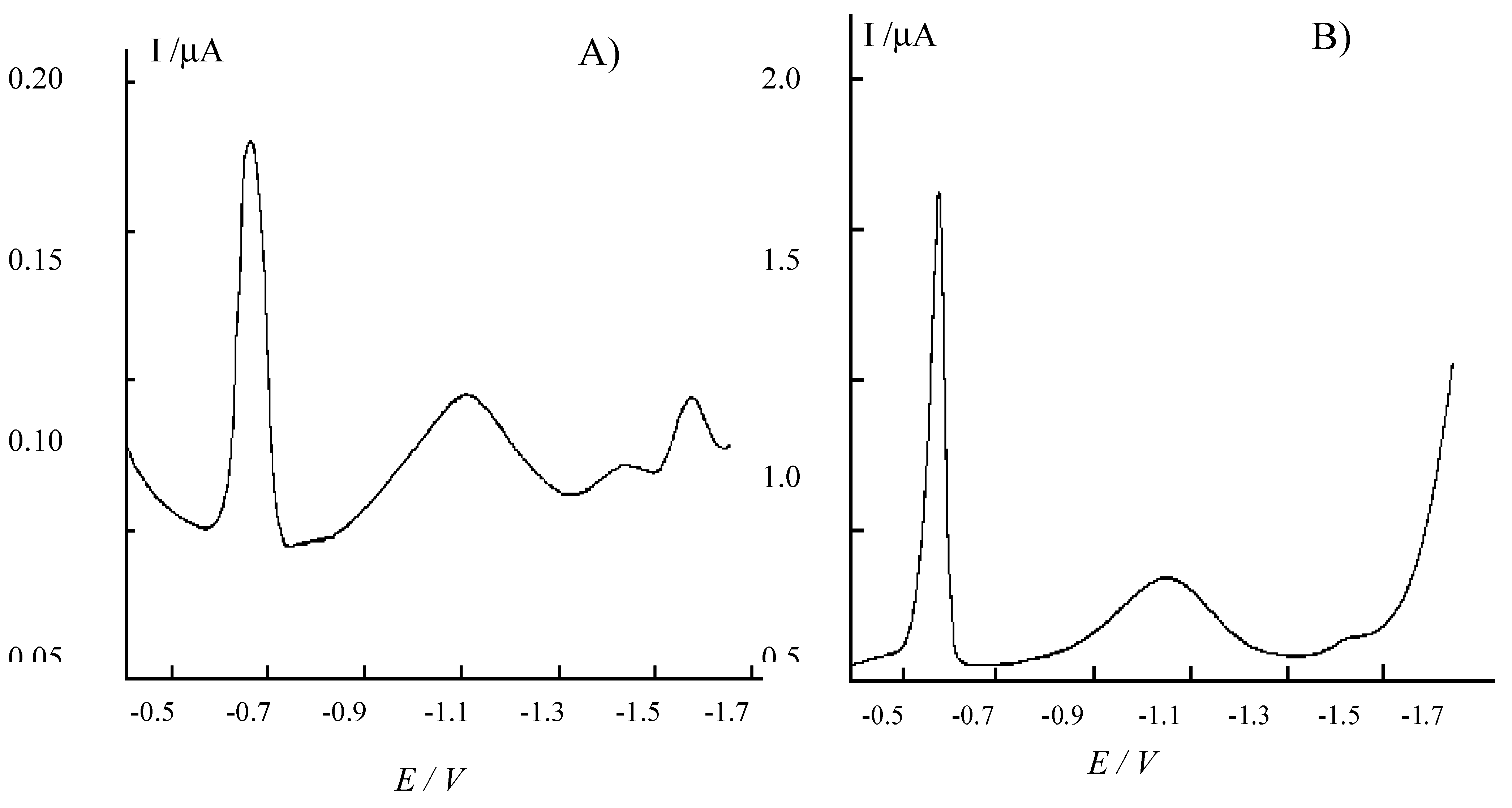
Figure 4 shows the typical cathodic scan differential pulse voltammograms recorded at the HMDE in raw pore-water samples from sediments of Venice channels. The peak at about -0.660 V corresponds to the reduction of HgS formed at the Hg/solution interface as a consequence of the presence of HS
- in the anoxic pore-water [
1,
2,
5]. It is followed by two peaks at more negative potential values. Comparison with the literature [
1,
2] indicates that the smaller peak at -1.460 V is related to the reduction of Fe
2+ to Fe°, while the larger peak at –1.058 V corresponds to the reduction of the FeHS
+ complex [
1]. In samples of this kind, the strongly anoxic conditions of the sediments make the FeHS
+ complex to be the prevailing soluble iron species. As discussed in the Introduction section, equilibria between free and complexed Fe(II) prevents the possibility to perform the quantitative determination of total Fe (i.e. labile Fe
2+ plus complexed FeHS
+) by voltammeric methods which employs the HMDE in this kind of samples.
Figure 4.
Differential pulse cathodic stripping voltammograms recorded at the HMDE in raw pore-water samples from sediments of the lagoon of Venice. A) Sample from Pili (P) station in
Fig. 1; B) sample from Santa Marta (SM) station in
Fig. 1.
Figure 4.
Differential pulse cathodic stripping voltammograms recorded at the HMDE in raw pore-water samples from sediments of the lagoon of Venice. A) Sample from Pili (P) station in
Fig. 1; B) sample from Santa Marta (SM) station in
Fig. 1.
The peak at very negative potential values (-1.570 V) detected in some of the samples (e.g.
Fig. 4A) corresponds to the reduction of Mn(II) to Mn° [
1]. Whenever detected, it can be used for monitoring the cycling of this species in pore-waters [
4,
5].
The cyclic voltammogram obtained at a NCE, in the same sample, after 1:100 dilution with 0.03 M HCl shows similar characteristics than the IEV voltammogram obtained in the sediment extract (see
Fig. 2). It is characterised by a well resolved oxidation-reduction peak system centred around an E
1/2 value of 0.450 V. In acidic conditions, all S(-II) species (including the complex FeHS
+) are protonated and can be eliminated from the sample solution by simple degassing with N
2 so that the prevailing signal recorded at the NCE after this treatment is due to the one-electron oxidation of Fe
2+ incorporated within the Nafion coating [
18]. This is confirmed by the evidence that the oxidation peak current increases linearly with iron standard additions. Really, in the same experimental conditions a small signal is obtained also at a bare GC electrode. However, the voltammetric peaks at the bare GC are broader and characterised by a much larger peak to peak separation (∆E
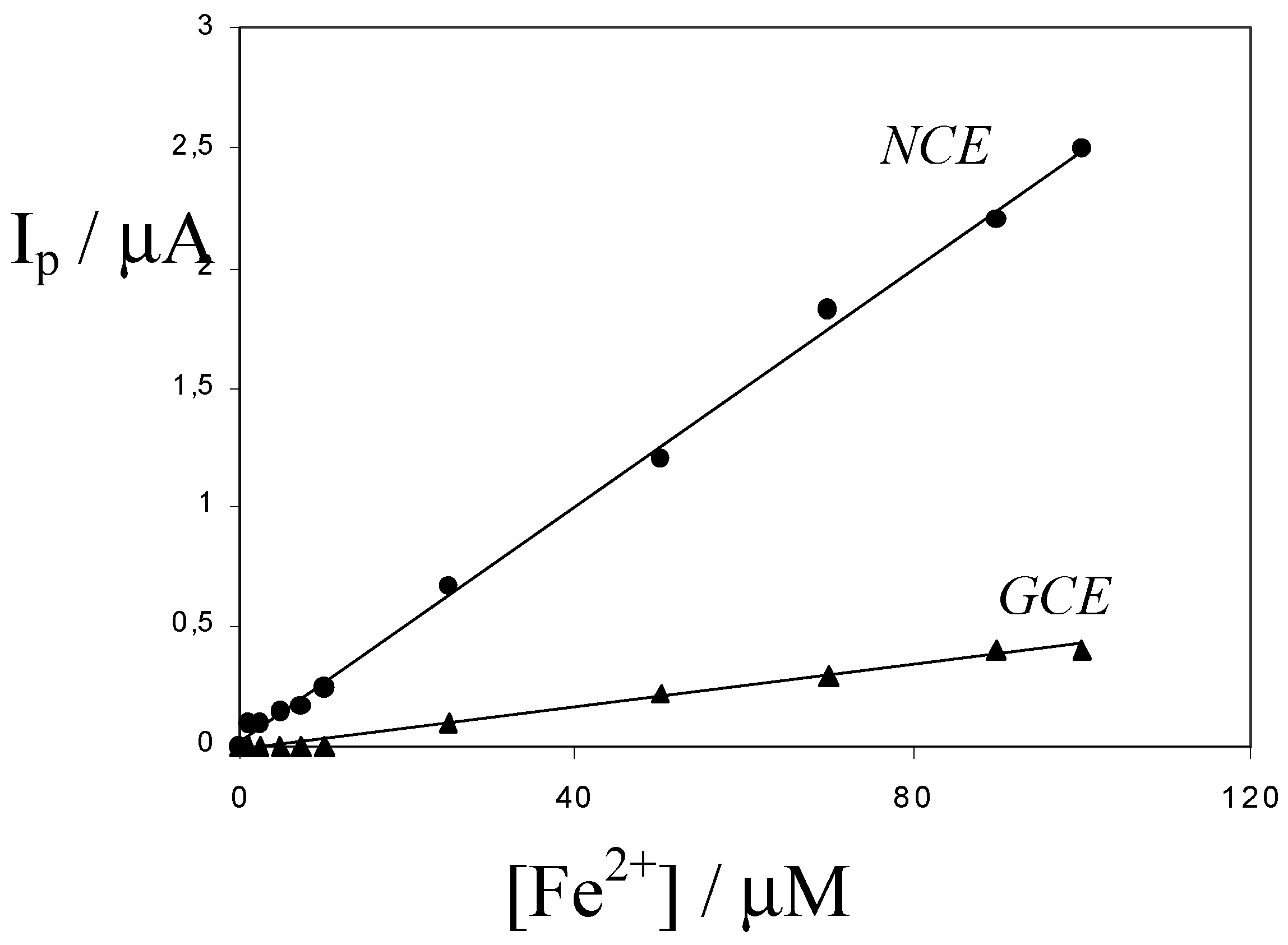
p ≈ 700 mV) with respect to the voltammograms obtained at the NCE. As shown by the calibration plots shown in
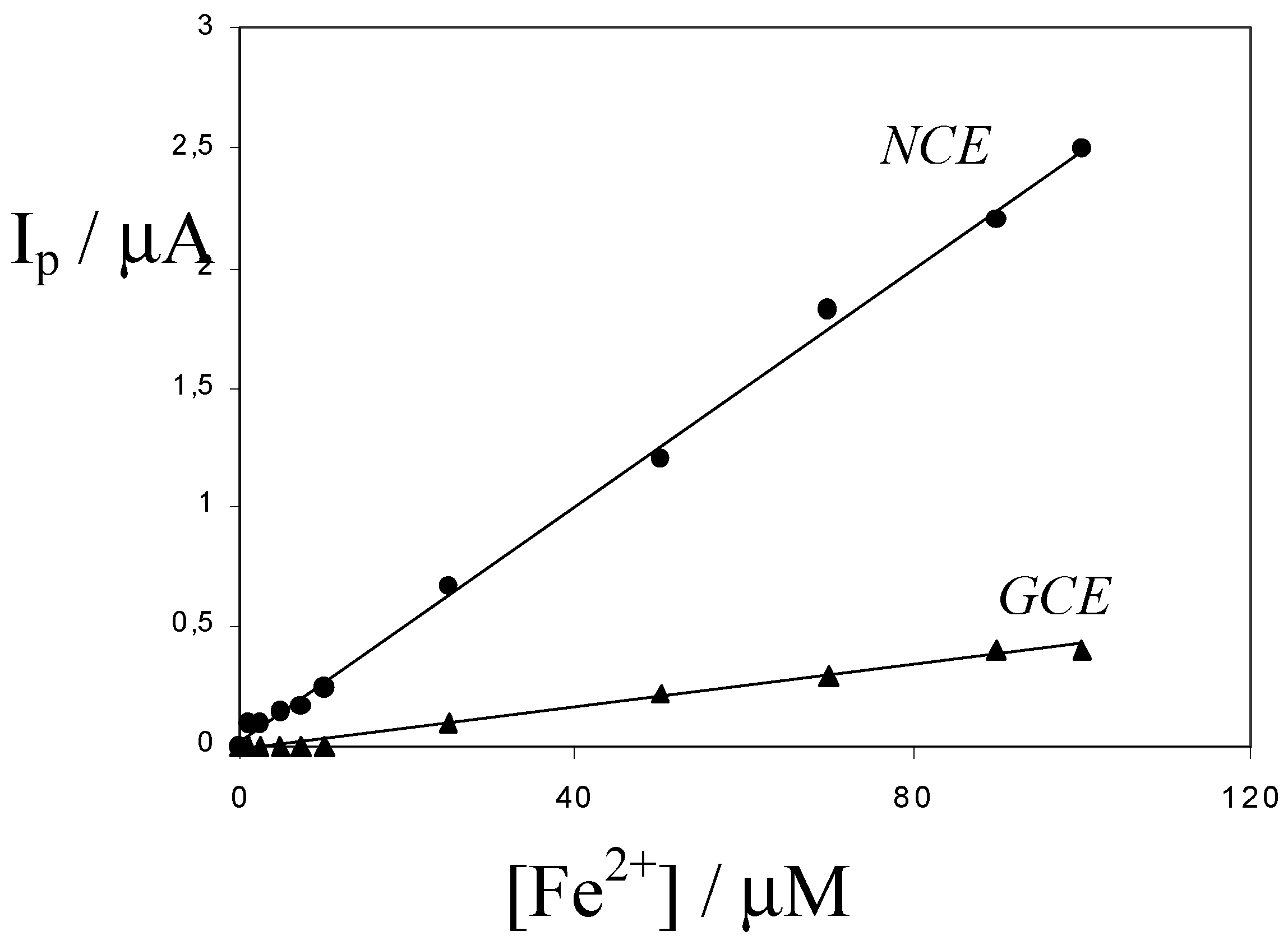
Fig. 5, obtained in synthetic samples containing 0.03 M HCl, the sensitivity at the NCE is dramatically higher than the sensitivity at a bare GCE.
In the pore-water sample examined by us, in which usually Fe 2+ > 5 M, the optimal procedure for iron determination at the NCE is to employ a 1:100 dilution with 0.03 M HCl followed by vigorous degassing to eliminate H2S. As for sediment extracts, the dilution improves the performances of the IEV method by lowering the effect of ion-exchange competition between H+ (or Na+) and Fe2+.
Figure 5.
Calibration plots obtained by cyclic voltammetry at 50 mV s-1, at the Nafion coated electrode (NCE) and at a bare glassy carbon electrode (GCE) of the same geometric area, in 0.03 M HCl.
Figure 5.
Calibration plots obtained by cyclic voltammetry at 50 mV s-1, at the Nafion coated electrode (NCE) and at a bare glassy carbon electrode (GCE) of the same geometric area, in 0.03 M HCl.
Application to Venice Lagoon samples
The methods above described for determining Fe(II) with the NCE have been tested and applied to a series of samples gained in the Venice Lagoon. Aim of this application was, on one side, testing the use of NCE in real samples and, on the other, the achievement of preliminary information on the iron(II) concentration levels in pore-waters and sediment extracts from different parts of the lagoon in order to plan more detailed sampling campaigns aimed to monitoring the environmental relevancy of iron cycling between waters and sediments of the lagoon of Venice.
On the basis of the above described interaction between Fe(II) and S(-II) which in many pore-water samples bring to the formation of the FeHS
+ complex, also the concentration of reduced S(-II) was determined.
Table 1 shows Fe(II) and S(-II) concentration values determined in pore-water samples gained in different parts of the lagoon.
Table 1.
Iron (II) and S(-II) concentrations determined in some pore-waters of sediments of the Lagoon of Venice. See
Fig. 1 for positions of the sampling stations.
Table 1.
Iron (II) and S(-II) concentrations determined in some pore-waters of sediments of the Lagoon of Venice. See Fig. 1 for positions of the sampling stations.
| Date | Sampling station | Depth / cm | Fe(II) /µM | S(-II) /µM |
|---|
| September 98 | SM | 10 | 230 ± 10 | 300 ± 12 |
| May 99 | SGA | 10 | 30 ± 4 | < DL (2) |
| October 00 | P | 10 | 15 ± 4 | < DL(2) |
| May 99 | F | 10 | < DL (1) | < DL (2) |
Data obtained for the SM sample (which refer to a municipal channel within the town of Venice) show the highest value for both analytes. This indicates that very strongly anoxic conditions occur in this sediment, even in the upper levels in proximity of the sediment/surface water interface. On the contrary, no Fe(II) and S(-II) were detected in the F site pore-waters. This is probably a consequence of the easier and more efficient water exchange between the lagoon and the open sea in this part of the lagoon. Upper pore water concentrations in the SGA site and P site show intermediate concentration values for Fe(II) with respect to upper data from the other two sites. It is interesting to note that high Fe(II) levels in SM site correlate with relatively high S(-II) levels. This confirms the relevant role of S(-II) in promoting Fe(II) formation and dissolution, suggesting that in the conditions of the pore-waters examined here, the formation of soluble complex species such as FeHS
+ can compete with FeS precipitation processes. Note that, as shown in
Fig. 4B, a high FeHS
+ reduction peak is detected at the HMDE.
Table 2 lists iron and reduced sulfur concentrations determined with the NCE and HMDE, respectively, in 3 M HCl sediments extracts. For some samples, also the AAS determination of Fe have been performed. Note that such a comparison between IEV and AAS data is possible only for sediment and not for pore-water samples; for the latter, in fact, both the lower Fe concentration and the high concentration of interfering dissolved salts hinder the reliable use of simple AAS methods (even if matrix correction procedures are used).
First of all, a satisfactory agreement between IEV and FAAS iron data is evidenced. Iron concentrations in the extracts are always much higher than reduced sulfur concentrations. Since the reduced sulphur determined by us corresponds to AVS (amorphous FeS, greigite and mackinawite and not to pyritic sulphur), the evidence that high Fe/AVS concentration ratios are determined in the extracts indicate that only a small fraction of HCl-soluble Fe is really in the form of AVS. Other fractions, such as the carbonatic or oxo/hydroxydes fractions, appear important in determining the level of the antrophogenic fraction of iron in the lagoon sediments. This observation agrees with data obtained in mud-flat sediments of neighboring sampling stations examined in previous researches [
28] and obtained by using more laborious and complex sequential extractions procedures. The increase of iron concentration with depth in the HCl-extracts from site P, parallels the trend observed for total iron concentrations in sediments of the same area of the lagoon [
24].
Table 2.
Iron and sulphides extracted from sediments of the lagoon of Venice after 24 h digestion with 3 M HCl. Iron was determined both by ion-exchange voltammetry (IEV) at -10.NCE and by flame atomic absorption spectroscopy (FAAS). Sulphides were determined by CSDPV at -10.HMDE.
Table 2.
Iron and sulphides extracted from sediments of the lagoon of Venice after 24 h digestion with 3 M HCl. Iron was determined both by ion-exchange voltammetry (IEV) at -10.NCE and by flame atomic absorption spectroscopy (FAAS). Sulphides were determined by CSDPV at -10.HMDE.
| Date | Sampling station | Depth / cm | Fe (SEM-IEV)
µmoles / g | Fe (FAAS)
µmoles / g | AVS
µmoles / g |
|---|
| May 99 | F | 5-10 | 49 ± 7 | 45±2 | 0.05 ± 0.01 |
| May 99 | SGA | 5-10 | 40 ± 80 | 53±2 | 0.04 ± 0.01 |
| October 00 | P | 5-10 | 394 ± 40 | n.d. | 1.9 ± 0.4 |
| October 00 | P | 30-40 | 652 ± 48 | 582 | 6.53 ± 0.5 |
Conclusions
Ion-exchange voltammetry at Nafion coated electrodes can be reliably used for determining the iron content of pore-waters and acid extracts of sediments.
In the latter case, the amounts determined correspond to the total iron extracted by the acid treatment; the possibility to determine simultaneously the sulphide and iron extracted allows one to speciate easily between acid volatile iron sulphides and other acid soluble iron forms.
The IEV iron determination in pore-waters after acidification is very sensitive; it gives information on the total iron content in the pore water. These data are complementary to those obtained by performing the electrochemical reduction of Fe(II) at the HMDE, which correlate with free ionic or labile complex/colloidal iron forms present at the natural pH of pore-waters.



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}