Abstract
Various types of errors during the measurements of ion-selective electrodes, ion-sensitive field effect transistors, and fibre optic chemical sensors are described. The errors were divided according to their nature and place of origin into chemical, instrumental and non-chemical. The influence of interfering ions, leakage of the membrane components, liquid junction potential as well as sensor wiring, ambient light and temperature is presented.
Introduction
Any kind of measurement carried out in a laboratory, environment, industry etc. is not errorless. The data obtained from the measurements are only estimations of the true values of the measurand i.e. in the case of analytical chemistry – the analyte. In analysis of the measurement data, uncertainty and error have to be distinguished. Error is defined as the difference between a result obtained from the measurement and the true value of the analyte concentration. The value of a known error is a single number and can be used to correct the measurement result whereas uncertainty is expressed as a range and cannot be applied as a correction to the result. According to [1] uncertainty is a parameter which characterises the dispersion of the values obtained from the measurement and this parameter can be attributed to the concentration or the activity of the analyte.
The problem of the evaluation of uncertainty in analytical chemistry measurements has been pointed out by chemists for many years [2,3,4,5]. Some general rules for evaluation and expressing uncertainty in a variety of measurements are given in ref. [6]. On this basis EURACHEM document [1] explains how ISO Guide can be adapted for chemical measurements giving a number of practical examples. Some sources of systematic errors in chemical sensor metrology were described in ref. [7].
The aim of this paper is to point out various types of errors, which occur during the chemical sensor measurements. Sources of errors in measurements of ion-selective electrodes (ISE), ion-sensitive field effect transistors (ISFET), and fibre optic chemical sensors (FOCS) are presented as well as some advice how to avoid or at least diminished them. The described errors are classified according to their origins not to their random or systematic character.
Sources of errors in chemical sensors
The analytical signal in a chemical sensor ought to be produced by the analyte but this is only the ideal case. During the sensor measurements this signal is influenced by a variety of chemical and non-chemical causes. Primary assumption in chemical sensor metrology is that the concentration of the analyte applied during the calibration process is known precisely. Obviously this is not true but in many cases such an error is smaller than the error in the measured signal. This error can be neglected because the standard calibration solutions are prepared by careful calculation and appropriate dilutions of the stock solution. The situation changes when solutions with very low concentration of the analyte should be prepared i.e. in the order of ppm and below.
The work of any sensor can be described by a very general equation (the exact expressions describing theory of sensors can be found in monographs [8,9]):
where:
S = f (c) + ε
- S – analytical signal
- f(c) – the response function of the sensor
- c – concentration or activity of the analyte
- ε – error in the measuring system response values
The error depicted as ε in equation (1) consists of two components: a random error and a constant systematic error. A random error is caused by the variations of influence quantities that cannot be predicted. So its value is unknown and the only one remedy to reduce it, is to increase the number of measurements. In contrary to this error, a systematic error remains constant or its variations can be predicted. More details on the error classification and origin can be found in the literature [10,11,12,13].
In this article, different approach to the error classification is proposed. Since the chemical sensor metrology involves chemistry, electronics, computer science etc., the errors are divided according to their origin into errors caused by chemical and non-chemical reasons. Both types of the errors can be schematically depicted as it is shown in Fig. 1.
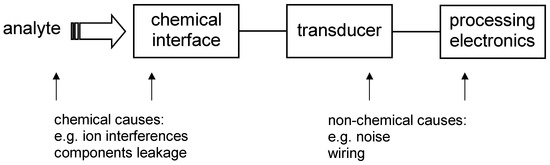
Figure 1.
Chemical sensor construction and possible sources of errors.
Chemical causes
Ion interferences
An ideal chemical sensor exhibits the changes in the analytical signal caused only by the analyte. In laboratory practice, this is not true and the immunity of the sensor to other ions present in the sample under the test is characterised by the selectivity coefficient. Different methods of the selectivity determination can be found in the literature [14,15,16]. The IUPAC suggests two methods: separate solution method (SSM) and fixed interference method (FIM). They differ in the methodology. In SSM method, two separate solutions are measured, each of them contains only a salt of the determined ion. The selectivity coefficient is then calculated based on the different signals obtained. FIM method is based on the determination of the calibration curve for the primary ion in a constant interfering ion background. The selectivity coefficient is calculated with the help of the lower detection limit and the background. Alternative method of the selectivity determination is called matched potential method (MPM). An initial step in this method involves the addition of a given amount of a primary ion to a reference solution. The new potential obtained is noted and then in a separate experiment a solution of interfering ion is added up to a point, when the potential matches this obtained in the initial step of the method. The ratio of the primary ion and interfering ion activity increments gives in formation on the selectivity.
The selectivity determination methods have been compared in several papers [17,18,19]. Each of the methods has got vice and versa, and there are no general rules or guidelines pointing which method gives the best results. The methods proposed by IUPAC with precautions included in ref. [20] will give meaningful data.
Calibration procedure
Several problems may occur during the calibration process of a sensor. In potentiometric sensors, one point calibration can be performed assuming that the slope of the sensor is known. Such a calibration is valid until the sensor follows the theory with an appropriate slope and temperature during the measurements is kept constant. Contrary to such a calibration, a multiple-point calibration was proposed [21] allowing to obtain better accuracy of the measurements. The efficiency of the method was demonstrated on pH determination. The measurement procedure is based on the use of several pH buffers instead of two as usually used in laboratory practice. Having done that, the calibration equation is calculated by linear regression, which results in better accuracy of measurements.
Another problem is connected with the response time of the sensor and its hysteresis, if any. During the calibration process a concentration gradient develops, which influence on diffusion of the analyte through the chemical interface of the sensor until the equilibrium state is achieved. Usually, the diffusion process is quite slow being mainly determined by the thickness of the chemical interface. A proper calibration process requires the user to wait until a steady state is reached. Otherwise, error in the calibration curve is unavoidable. The analyte transport properties of the chemical interfaces can vary in dependence on the sign of the concentration gradient i.e. when the concentration of the analyte is increased or decreased. As a result of this phenomenon there is hysteresis in the sensor calibration curve [22]. From a metrological point of view, such a sensor will have limited applications giving ubiquitous read-outs.
Leakage of the chemical interface components
The process of a chemical sensor design [23] involves a fabrication of an appropriate chemical interface (for example in the form of a membrane) in which the analyte recognition process takes place. Various methods (physical or chemical) can be applied to prepare such an interface. However, the primary requirement, which should be fulfilled is that no chemical interface component will be leaked out to the sample. The best results are obtained if a chemical immobilisation is utilised [8,9]. For example, in optical sensors the chromoionophore is attached to the polymer support or directly to an optical fibre. In the case of potentiometric sensors, usually the ionophore is physically entrapped inside the membrane. So it tends to be washed out from the membrane. This process can be relative slow and to some extends the membrane composition can be optimised in order to achieve durable sensors. The leakage of the membrane components leads to drift in the sensor signal and calibration procedure should be repeated. However if the process of the leakage continuos, the sensor performances vanish and this results in a limited lifetime of the sensor.
Liquid junction potential
Calibration procedure of ISE assumes that the slope of the determined characteristic remains constant during the measurements. However, because the composition of the electrolyte to be measured can differ from the solutions used in the calibration process, this assumption is fulfilled very unlikely, if ever. As a result of such a difference, the slope of the calibration curve varies slightly even if a salt bridge is used. The literature data [12] indicate that the uncertainty in the liquid junction potential causes this phenomenon. The total relative error expected in ISE measurements with respect to the liquid junction potential uncertainty can be evaluated by differentiating the equation describing the work of an ISE obtaining:
where:
relative error of measured activity [%] ≅ 4nΔEj
- n – charge of the determined ion
- ΔEj – uncertainty in liquid junction potential [mV].
The minimal ΔEj uncertainty is in the order of ± 1 mV, which results in a relative error in the measured activity about ± 4% for univalent ion. This error has very strong impact on the whole potentiometric measurements. No matter how precise and sensitive equipment is used, this error cannot be eliminated or compensated for (since generally for unknown composition of a sample the value of liquid junction potential cannot be calculated from theory nor measured directly).
Sample composition
Laboratory measurements of a chemical sensor differ substantially from real sample tests with unknown and unpredictable composition. The sample matrix can influence on the parameters in the sensor response function or even cause its damage or malfunction. For example, in biological samples a deposition of proteins changes physical nature of the ion-selective membrane [24] causing at least delay in the sensor response or in worst case the sensor stops to work.
The influence of the pH of the sample should also be pointed out. In some cases the sensor can work only in a given range of pH and an appropriate buffer or pH correction should be applied.
Instrumental and non-chemical causes
Sensor wiring and electromagnetic fields
Schematic sensor construction showed in Fig. 1 does not show additional parts of a measuring set-up for sensor testing. Among them one can find a peristaltic pump, a stirrer, dosing equipment as well as computerised data acquisition system. In dependence on the type of the sensor this equipment can cause severe noise which could be picked-up either by the sensor either by processing electronics. Especially potentiometric sensors are sensitive to electromagnetic field interferences which are present in the environment or which are generated by other equipment (e.g. a magnetic stirrer). A proper guarding and shielding technique should be applied [25,26,27]. The whole measuring system should be arranged with care with respect to avoid any grounding loops between data acquisition system and processing electronics with the sensor. A temperature probe with a metal case immersed into a beaker with the sensor will act as an antenna leading to the increase of the noise in the signal. The main disadvantage in potentiometric measurements based on ISE and ISFET is that they operate in direct current (DC) mode, which make them very vulnerable to any changes in the electromagnetic and electrostatic fields. A huge contribution in a sensor drift has thermal noise and the noise originated in a transducer [28,29,30].
Ambient light
Typical measurements of a chemical sensor are carried out in a daylight environment. The changes in the optical radiation can influence both optical and potentiometric sensors. If the optical sensor is based on the use of a spectrophotometer (e.g. miniature fibre optic PC2000, Ocean Optics) the environmental light should be kept constant, the sensor should have an optical isolation or the sample under the tests should be covered by a dark box. Optical sensors utilising modulated light and selective detecting system are not sensitive to changes in the ambient light [4]. ISFETs with gates made of SiO2 or Si3N4 are very sensitive to light. The application of Ta2O5 as a gate material solved this problem allowing to commercialise ISFET-based pH-meters [31,32].
Choice of analytical wavelength in optical sensor
Beer’s law generally governs the work of the fibre optic chemical sensor based on the use of an absorbance indicator. The actual description of the output signal generated by a sensor can only be done in a specific construction [4]. There are some fundamental rules, which should be followed in the choice of the analytical wavelength for the sensor. This wavelength should be matched to the maximal changes in the indicator absorbance in order to achieve the highest dynamic range of the sensor. In laboratory based arrangements, this requirement can be easily fulfilled when a monochromator is used or a sensor is based on the use of a commercial spectrophometer. Another guideline for wavelength choice results from general theory of absorption and possibilities of deviations from Beer’s law. Primary assumption of this law is the use of monochromatic radiation. In the case of optical sensors, this is only the truth when a laser source is used. Nevertheless, there is a huge variety of the sensors using chip and stabile light emitting diodes (LED). Then the work of a sensor can be considered as a sum of Beer’s law at different wavelengths [33]. From the point of view of spectrophotometric measurements, the LED radiation should be chosen as close as possible to the analytical wavelength because the changes in the molar absorptivity coefficient versus wavelength are relatively small around it. Otherwise, if the LED emits the radiation quite far from the analytical wavelength, the deviations from the Beer’s law are observed resulting in the smaller dynamic range of the sensor.
Temperature
The sensors used in the measurements are subject to temperature changes [34]. Temperature can change the characteristics of a sensor or a transducer, the potential of a reference electrode (in the case of potentiometric sensors) and the whole measuring system. The latter changes can be easily compensated or constant temperature can be maintained inside the equipment. Whereas in the case of a chemical sensor, it is necessary to investigate the primary function either chemical interface or transducing part of the sensor in dependence on the temperature. Temperature characteristics of three types of chemical sensors i.e. fibre optic pH sensor, pH-selective electrode and pH sensitive field effect transistor were presented in [35]. The data obtained show that the temperature dependence is complicated in nature and the user should beware of using the sensors during variable environmental conditions e.g. field tests. The sensors sometimes do not follow theory of operation and this makes the temperature compensation especially difficult and important. ISFET behaves according to the theory with excellent linearity, which makes the temperature compensation very easy. Additional advantage of these sensors is the possibility of auto-compensation by choice of drain current and drain–source voltage at an isothermal point [36].
Summary
An analytical chemist performing tests of chemical sensors should be very conscious about possible sources of errors influencing the measurements. Some of them have chemical origin and as quite well predicted and recognised can be determined with the help of analytical methods (e.g. determination of selectivity). Such influences can be expected based on the analytical chemistry knowledge. In some cases, differential measurements based on two identical sensors placed in a sample and a reference stream can eliminate instrument errors. However, still there is a possibility of electromagnetic interference and noise pick-up by the measuring electronics. Proper guarding and shielding techniques can only assure low noise, precise measurements. Modern electronic systems can measure chemical sensor signals with high resolution and accuracy. The application of multi-bit analog to digital converters also contributes to this. Nevertheless, there are some inherent errors in chemical sensors (e.g. uncertainty in liquid junction potential), which cannot be eliminated or determined. So the precision of the chemical sensors measurements is determined mainly by the possible instabilities caused by chemical nature of the sensor.
The accuracy of the whole measuring system based on the chemical sensor is still predominantly determined by the performance of the sensor used. This is caused by the fact that the process of transduction of a chemical information into an electric signal is inherently vulnerable to the influences of physical (e.g. temperature) or chemical (e.g. other ions) quantities, which are not the subject of conversion. This seems to be the main obstacle in mass commercialisation of a new developed chemical sensor.
Acknowledgements
The work was financed by a grant of State Committee for Scientific Research – project No. PBZ-015- 19.
References
- Guide to quantifying uncertainty in analytical measurementEURACHEM/CITAC, Second edition; 2000.
- Clarke, P.J.; Hanson, A.R.; Verrill, J.F. Determination of colorimetric uncertainties in the spectrophotometric measurement of colour. Anal. Chim. Acta 1999, 380, 277–284. [Google Scholar] [CrossRef]
- Maroto, A.; Riu, J.; Boque, R.; Rius, F.X. Estimating uncertainties of analytical results using information from the validation process. Anal. Chim. Acta 1999, 391, 173–185. [Google Scholar] [CrossRef]
- Meinrath, G.; Spitzer, P. Uncertainties in determination of pH. Microchim. Acta 2000, 135, 155–168. [Google Scholar] [CrossRef]
- Meinrath, G. Measurement uncertainty of thermodynamic data. Fresenius J. Anal. Chem. 2001, 369, 690–697. [Google Scholar] [CrossRef]
- Guide to the expression of uncertainty in measurement; ISO: Geneva, 1993.
- Cammann, K. Sources of systematic errors in chemical sensor chemometrics. Sens. Actuators B 1995, 24-25, 769–772. [Google Scholar] [CrossRef]
- Janata, J. Principles of chemical sensors; Plenum Press: New York, 1989. [Google Scholar]
- Wolfbeis, O.S. Fiber optic chemical sensors and biosensors; Vol. 1 and 2, CRC: Boca Raton, FL, 1991. [Google Scholar]
- Rabinovich, S.G. Measurement errors: theory and practice; AIP: New York, 1993. [Google Scholar]
- Finkelstein, L.; Grattan, K.T.V. Concise encyclopedia of measurement and instrumentation; Pergamon Press: Oxford, 1994. [Google Scholar]
- Skoog, D.A. Fundamentals of analytical chemistry; Saunders College Publications, 1996. [Google Scholar]
- The measurement, instrumentation and sensors handbook; CRC Press: Heidelberg, Springer, 1999.
- Guibault, G.G.; Durst, R.A.; Frant, M.S.; Freiser, H.; Hansen, E.H.; Light, T.S.; Pungor, E.; Rechnitz, G.; Rice, N.M.; Rohm, T.J.; Simon, W.; Thomas, J.D.R. Recommendations for nomenclature of ion-selective electrodes. Pure & Appl. Chem. 1976, 48, 127–132. [Google Scholar]
- Buck, R.P.; Lindner, E. Recommendations for nomenclature of ion-selective electrodes. Pure & Appl. Chem. 1995, 66, 2527–2536. [Google Scholar]
- Umezawa, Y.; Umezawa, K.; Sato, H. Selectivity coefficient for ion-selective electrodes: recommended methods for reporting KABpot values. Pure & Appl. Chem. 1995, 67, 507–518. [Google Scholar]
- Gadzekpo, V.P.Y.; Christian, G.D. Determination of selectivity coefficients of ion-selective electrodes by a matched-potential method. Anal. Chim. Acta 1984, 164, 279–282. [Google Scholar] [CrossRef]
- Macca, C. Determination of potentiometric selectivity. Anal. Chim. Acta 1996, 321, 1–10. [Google Scholar] [CrossRef]
- Kane, P.; Diamond, D. Determination of ion-selective electrode characteristics by non-linear curve fitting. Talanta 1997, 44, 1847–1858. [Google Scholar] [CrossRef]
- Bakker, E.; Pretsch, E.; Bühlmann, P. Selectivity of potentiometric ion sensors. Anal. Chem. 2000, 72, 1127–1133. [Google Scholar] [CrossRef]
- Baucke, F.G.K.; Naumann, R.; Alexander-Weber, Ch. Multiple-point calibration with linear regression as a proposed standardization procedure for high-precision pH measurements. Anal. Chem. 1993, 65, 3244–3251. [Google Scholar] [CrossRef]
- Allain, L.R.; Xue, Z. Hysteresis in optical sensing and its impact on the analytical error of a calibration-free acid sensor. Anal. Chim. Acta 2001, 433, 97–102. [Google Scholar] [CrossRef]
- Dybko, A.; Wroblewski, W. Analyte recognition and signal conversion in chemical sensor. Chem. Anal. (Warsaw). in press.
- Oesch, U.; Ammann, D.; Simon, W. Ion-selective membrane electrodes for clinical use. Clin. Chem. 1986, 32, 1448–1459. [Google Scholar] [PubMed]
- Ott, H.W. Noise reduction techniques in electronic systems; John Wiley and Sons: New York, 1978. [Google Scholar]
- Carr, J.J. Sensors and circuits; Prentice Hall: New Jersey, 1989. [Google Scholar]
- Cobbold, R.S.C. Transducers for biomedical measurements: principles and applications; John Wiley and Sons: New York, 1982. [Google Scholar]
- Jamasb, S.; Collins, S.; Smith, R.L. A physical model for drift in pH ISFETs. Sens Actuators B 1998, 49, 146–155. [Google Scholar] [CrossRef]
- Jakobson, C.G.; Nemirovsky, Y. 1/f noise in ion sensitive field effect transistors from subthreshold to saturation. IEEE Trans. on Elec. Dev. 1999, 46, 259–261. [Google Scholar] [CrossRef]
- Jakobson, C.G.; Feinsod, M.; Nemirovsky, Y. Low frequency noise and drift in Ion Sensitive Field Effect Transistors. Sens. Actuators B 2000, 68, 134–139. [Google Scholar]
- Ito, Y. Long-term drift mechanism of Ta2O5 gate pH-ISFETs. Sens. Actuators B 2000, 64, 152–155. [Google Scholar] [CrossRef]
- Ito, Y. Stability of ISFET and its new measurement protocol. Sens. Actuators B 2000, 66, 53–55. [Google Scholar]
- Baldini, F. Bile sensor: from the lab to the market. Proc. SPIE 1999, 3860, 144–155. [Google Scholar]
- Zuther, F.; Cammann, K. Temperature compensation for a nitrate sensor. Sens. Actuators B 1994, 18-19, 353–355. [Google Scholar]
- Dybko, A. Influence of temperature on chemical sensors; Chem. Anal. (Warsaw); in press.
- Barabash, P.R.; Cobbold, R.S.C.; Wlodarski, W. Analysis of the threshold voltage and its temperature dependence in electrolyte-insulator-semiconductor field effect transistors (EISFETs). IEEE Trans. on Electron Dev. 1987, ED-34, 1271–1282. [Google Scholar] [CrossRef]
- Sample Availability:
© 2001 by MDPI (http://www.mdpi.net). Reproduction is permitted for noncommercial purposes.