Evidence of Recent Fine-Scale Population Structuring in South American Puma concolor


Abstract
:1. Introduction
2. Materials and Methods
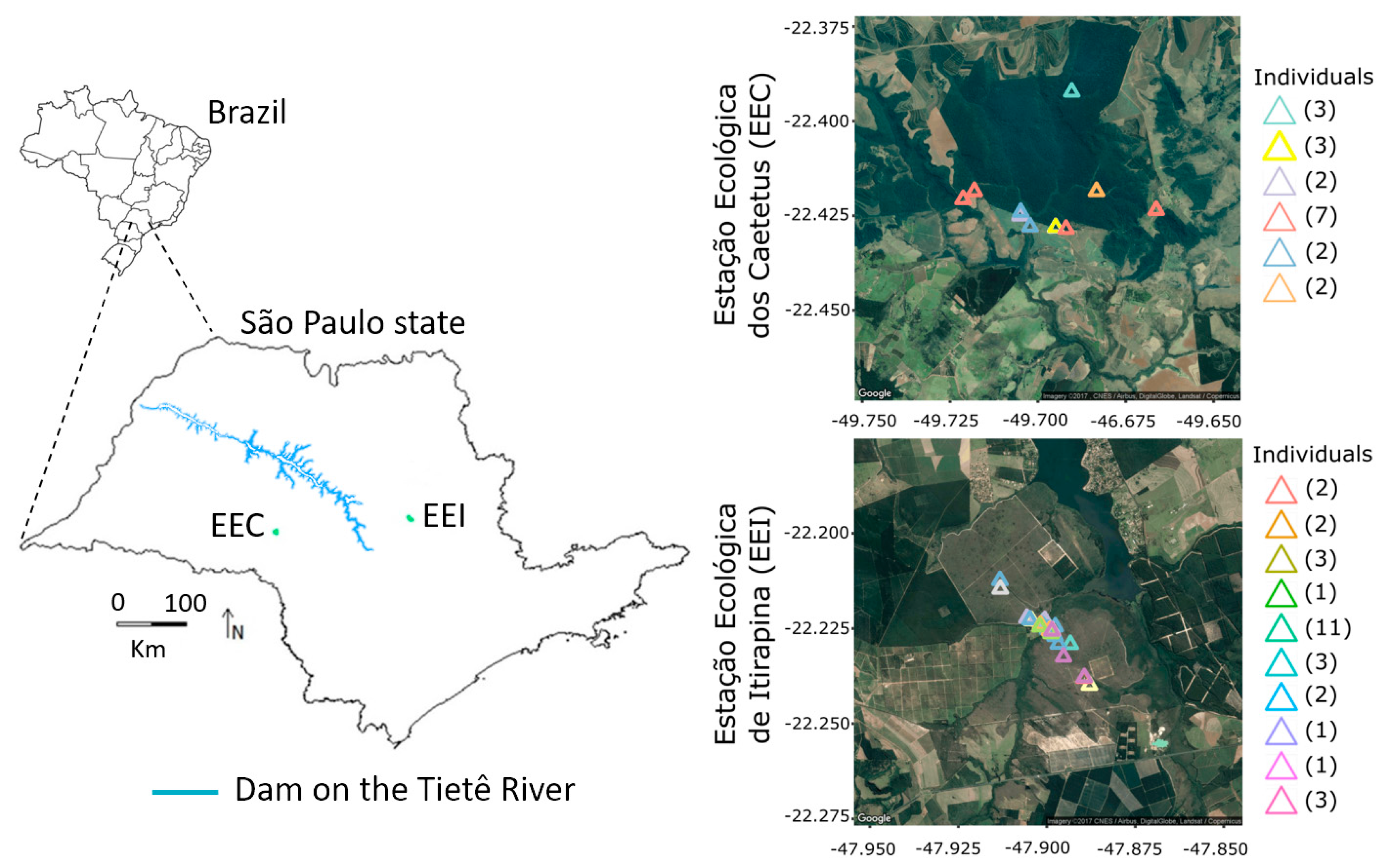
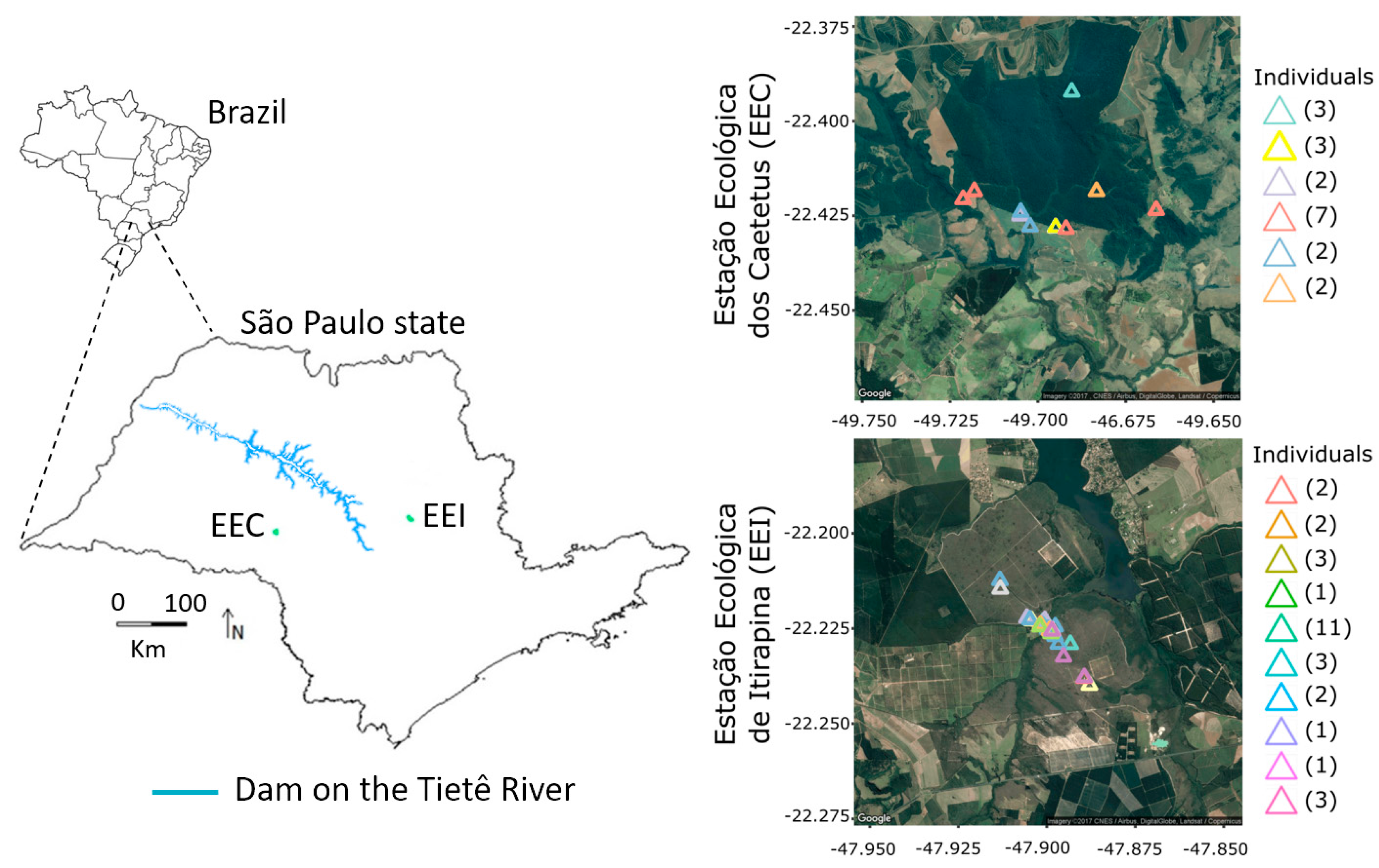
2.1. Study Area
2.2. Sample Collection and DNA Extraction
2.3. Molecular Species Confirmation
2.4. Microsatellite Genotyping, Fecal Samples Individualization, and Relatdeness
2.5. Demographic Analysis
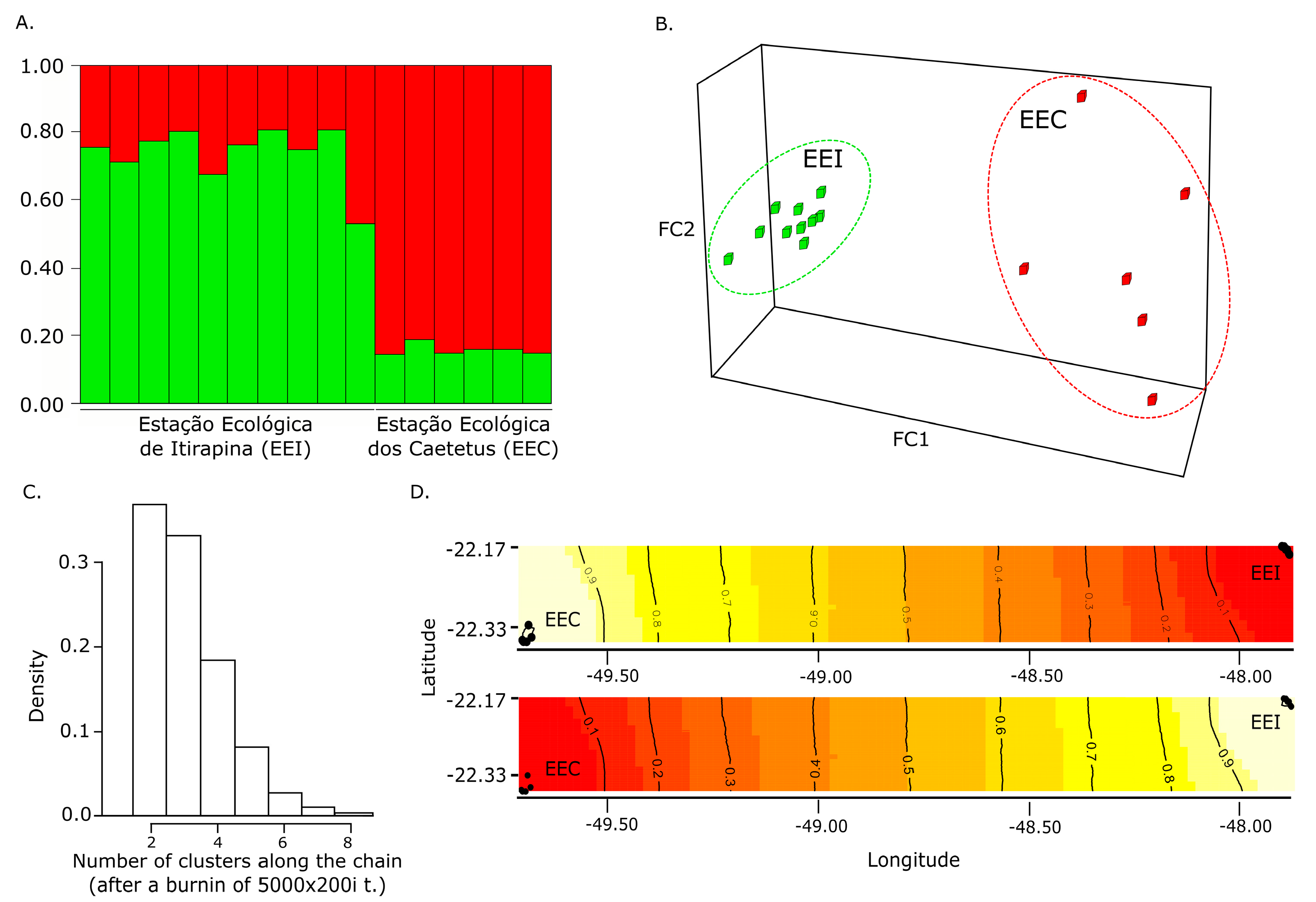
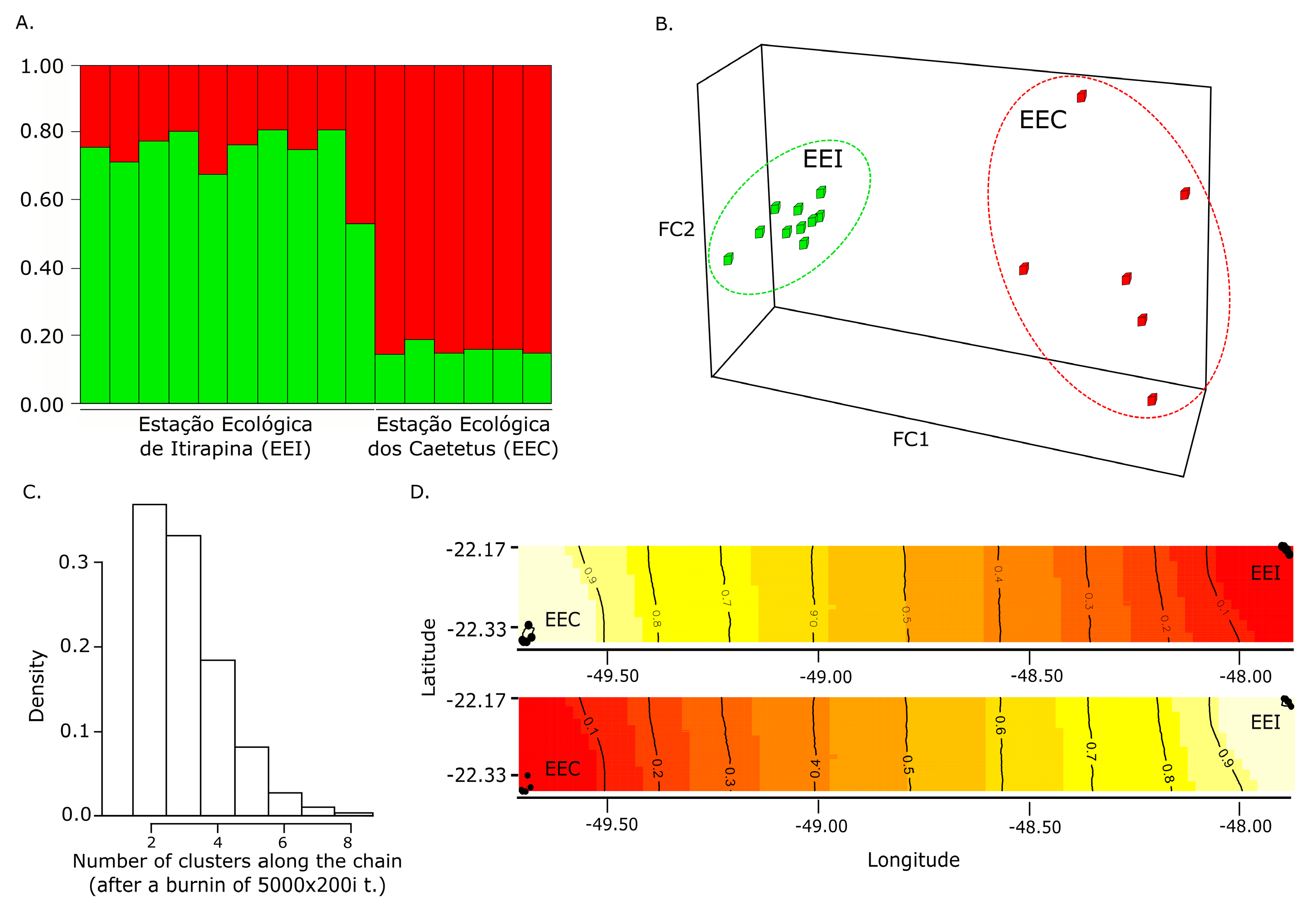
2.6. Genetic Cluster Analyses
2.7. Gene Flow
2.8. Genetic Diversity Analysis
3. Results and Discussion
3.1. Non-Invasive Samples, Discrimination of Individuals, and Demographic Estimatives
3.2. Population Genetic Structuring
3.3. Genetic Diversity
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Haddad, N.M.; Brudvig, L.A.; Clobert, J.; Davies, K.F.; Gonzalez, A.; Holt, R.D.; Lovejoy, T.E.; Sexton, J.O.; Austin, M.P.; Collins, C.D.; et al. Habitat fragmentation and its lasting impact on Earth’s ecosystems. Sci. Adv. 2015, 1. [Google Scholar] [CrossRef] [PubMed]
- Dutta, T.; Sharma, S.; Maldonado, J.E.; Wood, T.C.; Panwar, H.S.; Seidensticker, J. Fine-scale population genetic structure in a wide-ranging carnivore, the leopard (Panthera pardus fusca) in central India. Divers. Distrib. 2013, 19, 760–771. [Google Scholar] [CrossRef]
- Ernest, H.B.; Vickers, T.W.; Morrison, S.A.; Buchalski, M.R.; Boyce, W.M. Fractured genetic connectivity threatens a southern California puma (Puma concolor) population. PLoS ONE 2014, 9, e107985. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.K.; Aspi, J.; Kvist, L.; Sharma, R.; Pandey, P.; Mishra, S.; Singh, R.; Agrawal, M.; Goyal, S.P. Fine-scale population genetic structure of the Bengal tiger (Panthera tigris tigris) in a human-dominated western Terai Arc Landscape, India. PLoS ONE 2017, 12, e0174371. [Google Scholar] [CrossRef] [PubMed]
- Frankham, R.; Ballou, J.D.; Briscoe, D.A. Introduction to Conservation Genetics; Cambridge University Press: Cambridge, UK, 2010; ISBN 0521878470. [Google Scholar]
- Haag, T.; Santos, A.S.; Sana, D.A.; Morato, R.G.; Cullen, L.; Crawshaw, P.G.; De Angelo, C.; Di Bitetti, M.S.; Salzano, F.M.; Eizirik, E. The effect of habitat fragmentation on the genetic structure of a top predator: Loss of diversity and high differentiation among remnant populations of Atlantic Forest jaguars (Panthera onca). Mol. Ecol. 2010, 19, 4906–4921. [Google Scholar] [CrossRef] [PubMed]
- Wultsch, C.; Waits, L.P.; Kelly, M.J. A comparative analysis of genetic diversity and structure in jaguars (Panthera onca), pumas (Puma concolor), and ocelots (Leopardus pardalis) in fragmented landscapes of a critical Mesoamerican linkage zone. PLoS ONE 2016, 11, e0151043. [Google Scholar] [CrossRef] [PubMed]
- Proctor, M.F.; McLellan, B.N.; Strobeck, C.; Barclay, R.M.R. Genetic analysis reveals demographic fragmentation of grizzly bears yielding vulnerably small populations. Proc. R. Soc. Lond. B Biol. Sci. 2005, 272, 2409–2416. [Google Scholar] [CrossRef] [PubMed]
- McManus, J.S.; Dalton, D.L.; Kotzé, A.; Smuts, B.; Dickman, A.; Marshal, J.P.; Keith, M. Gene flow and population structure of a solitary top carnivore in a human-dominated landscape. Ecol. Evol. 2015, 5, 335–344. [Google Scholar] [CrossRef] [PubMed]
- Ripple, W.J.; Estes, J.A.; Beschta, R.L.; Wilmers, C.C.; Ritchie, E.G.; Hebblewhite, M.; Berger, J.; Elmhagen, B.; Letnic, M.; Nelson, M.P. Status and ecological effects of the world’s largest carnivores. Science 2014, 343, 1241484. [Google Scholar] [CrossRef] [PubMed]
- Loxterman, J.L. Fine scale population genetic structure of pumas in the Intermountain West. Conserv. Genet. 2011, 12, 1049–1059. [Google Scholar] [CrossRef]
- Logan, K.A.; Sweanor, L.L. Desert Puma: Evolutionary Ecology and Conservation of an Enduring Carnivore; Island Press: Washington, DC, USA, 2001; ISBN 1559638672. [Google Scholar]
- Hawley, J.E.; Rego, P.W.; Wydeven, A.P.; Schwartz, M.K.; Viner, T.C.; Kays, R.; Pilgrim, K.L.; Jenks, J.A. Long-distance dispersal of a subadult male cougar from South Dakota to Connecticut documented with DNA evidence. J. Mammal. 2016, 97, 1435–1440. [Google Scholar] [CrossRef]
- Morrison, C.D.; Boyce, M.S.; Nielsen, S.E. Space-use, movement and dispersal of sub-adult cougars in a geographically isolated population. PeerJ 2015, 3, e1118. [Google Scholar] [CrossRef] [PubMed]
- Culver, M.; Johnson, W.E.; Pecon-Slattery, J.; O’Brien, S.J. Genomic ancestry of the American puma (Puma concolor). J. Hered. 2000, 91, 186–197. [Google Scholar] [CrossRef] [PubMed]
- Caragiulo, A.; Dias-freedman, I.; Clark, J.A.; Rabinowitz, S.; Amato, G. Neotropic pumas (Puma concolor). 2013, 1736, 1–9. [Google Scholar] [CrossRef]
- Miotto, R.A.; Cervini, M.; Figueiredo, M.G.; Begotti, R.A.; Galetti, P.M. Genetic diversity and population structure of pumas (Puma concolor) in southeastern Brazil: Implications for conservation in a human-dominated landscape. Conserv. Genet. 2011, 12, 1447–1455. [Google Scholar] [CrossRef]
- Castilho, C.S.; Marins-Sá, L.G.; Benedet, R.C.; Freitas, T.R.O. Genetic structure and conservation of Mountain Lions in the South-Brazilian Atlantic Rain Forest. Genet. Mol. Biol. 2012, 35, 65–73. [Google Scholar] [CrossRef]
- Ribeiro, M.C.; Metzger, J.P.; Martensen, A.C.; Ponzoni, F.J.; Hirota, M.M. The Brazilian Atlantic Forest: How much is left, and how is the remaining forest distributed? Implications for conservation. Biol. Conserv. 2009, 142, 1141–1153. [Google Scholar] [CrossRef]
- Dean, W. A Ferro e Fogo: A História e a Devastação da Mata Atlântica Brasileira; Companhia das Letras: São Paulo, Brazil, 1996; ISBN 8571645906. [Google Scholar]
- Martinelli, L.A.; Filoso, S. Expansion of sugarcane ethanol production in Brazil: Environmental and social challenges. Ecol. Appl. 2008, 18, 885–898. [Google Scholar] [CrossRef] [PubMed]
- Victorino, V.I.P. O Acaso Das Águas na Privatização Dos Rios. Estamos Todos a Juzante; Novos Estudos; Cebrap: São Paulo, Brazil, 1999; pp. 176–194. [Google Scholar]
- Massei, R. A Construção da Usina Hidrelétrica Barra Bonita e a Relação Homem-Natureza: Vozes Dissonantes, Interesses Contraditórios (1940–1970). In Proceedings of the XXIV Simpósio Nacional de História, São Leopoldo, Brazil, 15–20 July 2007. [Google Scholar]
- Farrell, L.E.; Roman, J.; Sunquist, M.E. Dietary separation of sympatric carnivores identified by molecular analysis of scats. Mol. Ecol. 2001, 9, 1583–1590. [Google Scholar] [CrossRef]
- Miotto, R.A.; Rodrigues, F.P.; Ciocheti, G.; Manoel, P.G., Jr.; Lu, W. Determination of the Minimum Population Size of Pumas (Puma concolor) Through Fecal DNA Analysis in Two Protected Cerrado Areas in the Brazilian Southeast. Biotropica 2007, 39, 647–654. [Google Scholar] [CrossRef]
- Miotto, R.A.; Cervini, M.; Kajin, M.; Begotti, R.A.; Galetti, P.M. Estimating puma Puma concolor population size in a human-disturbed landscape in Brazil, using DNA mark–recapture data. Oryx 2014, 48, 250–257. [Google Scholar] [CrossRef]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. MEGA6: Molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef] [PubMed]
- Kimura, M. A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. J. Mol. Evol. 1980, 16, 111–120. [Google Scholar] [CrossRef] [PubMed]
- Saitou, N.; Nei, M. The neighbor-joining method: A new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 1987, 4, 406–425. [Google Scholar] [PubMed]
- Kurushima, J.D.; Collins, J.A.; Well, J.A.; Ernest, H.B. Development of 21 microsatellite loci for puma (Puma concolor) ecology and forensics. Mol. Ecol. Notes 2006, 6, 1260–1262. [Google Scholar] [CrossRef]
- Waits, L.P.; Luikart, G.; Taberlet, P. Estimating the probability of identity among genotypes in natural populations: Cautions and guidelines. Mol. Ecol. 2001, 10, 249–256. [Google Scholar] [CrossRef] [PubMed]
- PE, P.R.S. GenAlEx 6.5: Genetic analysis in Excel. Population genetic software for teaching and research—An update. Bioinformatics 2012, 28, 2537–2539. [Google Scholar]
- Kalinowski, S.T.; Wagner, A.P.; Taper, M.L. ML-Relate: A computer program for maximum likelihood estimation of relatedness and relationship. Mol. Ecol. Res. 2006, 6, 576–579. [Google Scholar] [CrossRef]
- Blouin, M. DNA-based methods for pedigree reconstruction and kinship analysis in natural populations. TREE 2003, 18, 503–511. [Google Scholar] [CrossRef]
- Miller, C.R.; Joyce, P.; Waits, L.P. A new method for estimating the size of small populations from genetic mark–recapture data. Mol. Ecol. 2005, 14, 1991–2005. [Google Scholar] [CrossRef] [PubMed]
- Pritchard, J.K.; Stephens, M.; Donnelly, P. Inference of population structure using multilocus genotype data. Genetics 2000, 155, 945–959. [Google Scholar] [PubMed]
- Evanno, G.; Regnaut, S.; Goudet, J. Detecting the number of clusters of individuals using the software STRUCTURE: A simulation study. Mol. Ecol. 2005, 14, 2611–2620. [Google Scholar] [CrossRef] [PubMed]
- Guillot, G.; Mortier, F.; Estoup, A. GENELAND: A computer package for landscape genetics. Mol. Ecol. Resour. 2005, 5, 712–715. [Google Scholar] [CrossRef]
- Belkhir, K.; Borsa, P.; Chikhi, L.; Raufaste, N.; Bonhomme, F. GENETIX, Version 4.05; Windows TM software for population genetics; Lab. Génome, Popul. Interact. CNRS UMR 5000; 2004.
- Wright, S. The genetical structure of populations. Ann. Hum. Genet. 1949, 15, 323–354. [Google Scholar] [CrossRef]
- Jost, L.O.U. GST and its relatives do not measure differentiation. Mol. Ecol. 2008, 17, 4015–4026. [Google Scholar] [CrossRef] [PubMed]
- Goudet, J. FSTAT, a Program to Estimate and Test Gene Diversities and Fixation Indices (Version 2.9.3). Available online: https://www2.unil.ch/popgen/softwares/fstat.htm (accessed on 30 May 2017).
- Team, R.C. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2017. [Google Scholar]
- Jueterbock, A.; Kraemer, P.; Gerlach, G.; Deppermann, J.; Jueterbock, M.A. Package “DEMEtics”. Mol. Ecol. 2012, 19, 3845–3852. [Google Scholar]
- Beerli, P.; Palczewski, M. Unified framework to evaluate panmixia and migration direction among multiple sampling locations. Genetics 2010, 185, 313–326. [Google Scholar] [CrossRef] [PubMed]
- Oosterhout, C.V.; Hutchinson, W.F.; Wills, D.P.M.; Shipley, P. Micro-checker: Software for identifying and correcting genotyping errors in microsatellite data. Mol. Ecol. Notes 2004, 4, 535–538. [Google Scholar] [CrossRef]
- Rousset, F. Genepop’007: A complete re-implementation of the genepop software for Windows and Linux. Mol. Ecol. Resour. 2008, 8, 103–106. [Google Scholar] [CrossRef] [PubMed]
- Rice, W.R. Analyzing tables of statistical tests. Evolution 1989, 43, 223–225. [Google Scholar] [CrossRef] [PubMed]
- Kalinowski, S.T. Hp-Rare 1.0: A Computer Program for Performing Rarefaction on Measures of Allelic Richness. Mol. Ecol. Notes 2005, 5, 187–189. [Google Scholar] [CrossRef]
- Nielsen, E.E.; Bach, L.A.; Kotlicki, P. HYBRIDLAB (version 1.0): A program for generating simulated hybrids from population samples. Mol. Ecol. Resour. 2006, 6, 971–973. [Google Scholar] [CrossRef]
- Anderson, C.R., Jr.; Lindzey, F.G.; McDonald, D.B. Genetic structure of cougar populations across the Wyoming Basin: Metapopulation or megapopulation. J. Mammal. 2004, 85, 1207–1214. [Google Scholar] [CrossRef]
- Miotto, R.A.; Cervini, M.; Begotti, R.A.; Galetti, P.M., Jr. Monitoring a Puma (Puma concolor) Population in a Fragmented Landscape in Southeast Brazil. Biotropica 2012, 44, 98–104. [Google Scholar] [CrossRef]
- McRae, B.H.; Beier, P.; Dewald, L.E.; Huynh, L.Y.; Keim, P. Habitat barriers limit gene flow and illuminate historical events in a wide-ranging carnivore, the American puma. Mol. Ecol. 2005, 14, 1965–1977. [Google Scholar] [CrossRef] [PubMed]
- Fattebert, J.; Robinson, H.S.; Balme, G.; Slotow, R.; Hunter, L. Structural habitat predicts functional dispersal habitat of a large carnivore: How leopards change spots. Ecol. Appl. 2015, 25, 1911–1921. [Google Scholar] [CrossRef] [PubMed]
- Wheeler, D.C.; Waller, L.A. Spatial analysis linking landscape features and genetic population structure in cougars (Puma concolor) in the northern Rocky Mountains. Stat. Interface 2012, 5, 183–193. [Google Scholar]
- Dickson, B.G.; Roemer, G.W.; McRae, B.H.; Rundall, J.M. Models of regional habitat quality and connectivity for pumas (Puma concolor) in the southwestern United States. PLoS ONE 2013, 8, e81898. [Google Scholar] [CrossRef] [PubMed]
- Rabinowitz, A. Cougar: Ecology and Conservation; University of Chicago Press: Chicago, IL, USA, 2009; ISBN 0226353478. [Google Scholar]
- Ernest, H.B.; Boyce, W.M.; Bleich, V.C.; May, B.; Stiver, S.J.; Torres, S.G. Genetic structure of mountain lion (Puma concolor) populations in California. Conserv. Genet. 2003, 4, 353–366. [Google Scholar] [CrossRef]
- Taberlet, P.; Griffin, S.; Goossens, B.; Questiau, S.; Manceau, V.; Escaravage, N.; Waits, L.P.; Bouvet, J. Reliable genotyping of samples with very low DNA quantities using PCR. Nucleic Acids Res. 1996, 24, 3189–3194. [Google Scholar] [CrossRef] [PubMed]
- Gustafson, K.D.; Vickers, T.W.; Boyce, W.M.; Ernest, H.B. A single migrant enhances the genetic diversity of an inbred puma population. R. Soc. Open Sci. 2017, 4, 170115. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
| Locus | A | AR | Ho | He | Hexcess | Fis | |
|---|---|---|---|---|---|---|---|
| Estação Ecológica dos Caetetus (EEC) N = 6 | PcoA208 | 7.00 | 4.521 | 0.60 | 0.82 | 0.98 | 0.434 |
| PcoC108 | 5.00 | 5.059 | 0.70 | 0.64 | 0.67 | 0.444 | |
| PcoB003 | 6.00 | 6.133 | 0.50 | 0.65 | 0.99 | 0.158 | |
| PcoB010 | 9.00 | 3.871 | 0.80 | 0.87 | 0.91 | 0.184 | |
| PcoA216 | 9.00 | 6.133 | 0.90 | 0.88 | 0.71 | 0.600 | |
| PcoB210 | 7.00 | 6.303 | 0.67 | 0.77 | 0.92 | 0.123 | |
| PcoA339 | 6.00 | 3.000 | 0.60 | 0.76 | 0.94 | 1.000 | |
| Mean | 7.00 | 5.003 | 0.68 | 0.77 | - | 0.416 | |
| Estação Ecológica de Itirapina (EEI) N = 10 | PcoA208 | 5.00 | 6.795 | 0.50 | 0.78 | 0.98 | 0.316 |
| PcoC108 | 6.00 | 4.889 | 0.50 | 0.79 | 0.98 | −0.050 | |
| PcoB003 | 7.00 | 5.789 | 0.80 | 0.84 | 0.96 | 0.280 | |
| PcoB010 | 4.00 | 8.689 | 0.67 | 0.74 | 0.91 | 0.127 | |
| PcoA216 | 7.00 | 8.784 | 0.40 | 0.84 | 1.00 | 0.024 | |
| PcoB210 | 8.00 | 7.000 | 0.83 | 0.86 | 0.96 | 0.193 | |
| PcoA339 | 3.00 | 5.795 | 0.20 | 0.63 | 1.00 | 0.260 | |
| Mean | 5.71 | 6.820 | 0.56 | 0.78 | - | 0.166 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Saranholi, B.H.; Chávez-Congrains, K.; Galetti, P.M. Evidence of Recent Fine-Scale Population Structuring in South American Puma concolor. Diversity 2017, 9, 44. https://doi.org/10.3390/d9040044
Saranholi BH, Chávez-Congrains K, Galetti PM. Evidence of Recent Fine-Scale Population Structuring in South American Puma concolor. Diversity. 2017; 9(4):44. https://doi.org/10.3390/d9040044
Chicago/Turabian StyleSaranholi, Bruno H., Karla Chávez-Congrains, and Pedro M. Galetti. 2017. "Evidence of Recent Fine-Scale Population Structuring in South American Puma concolor" Diversity 9, no. 4: 44. https://doi.org/10.3390/d9040044
APA StyleSaranholi, B. H., Chávez-Congrains, K., & Galetti, P. M. (2017). Evidence of Recent Fine-Scale Population Structuring in South American Puma concolor. Diversity, 9(4), 44. https://doi.org/10.3390/d9040044