
Barley Developmental Mutants: The High Road to Understand the Cereal Spike Morphology

 , ,
, ,  and
and 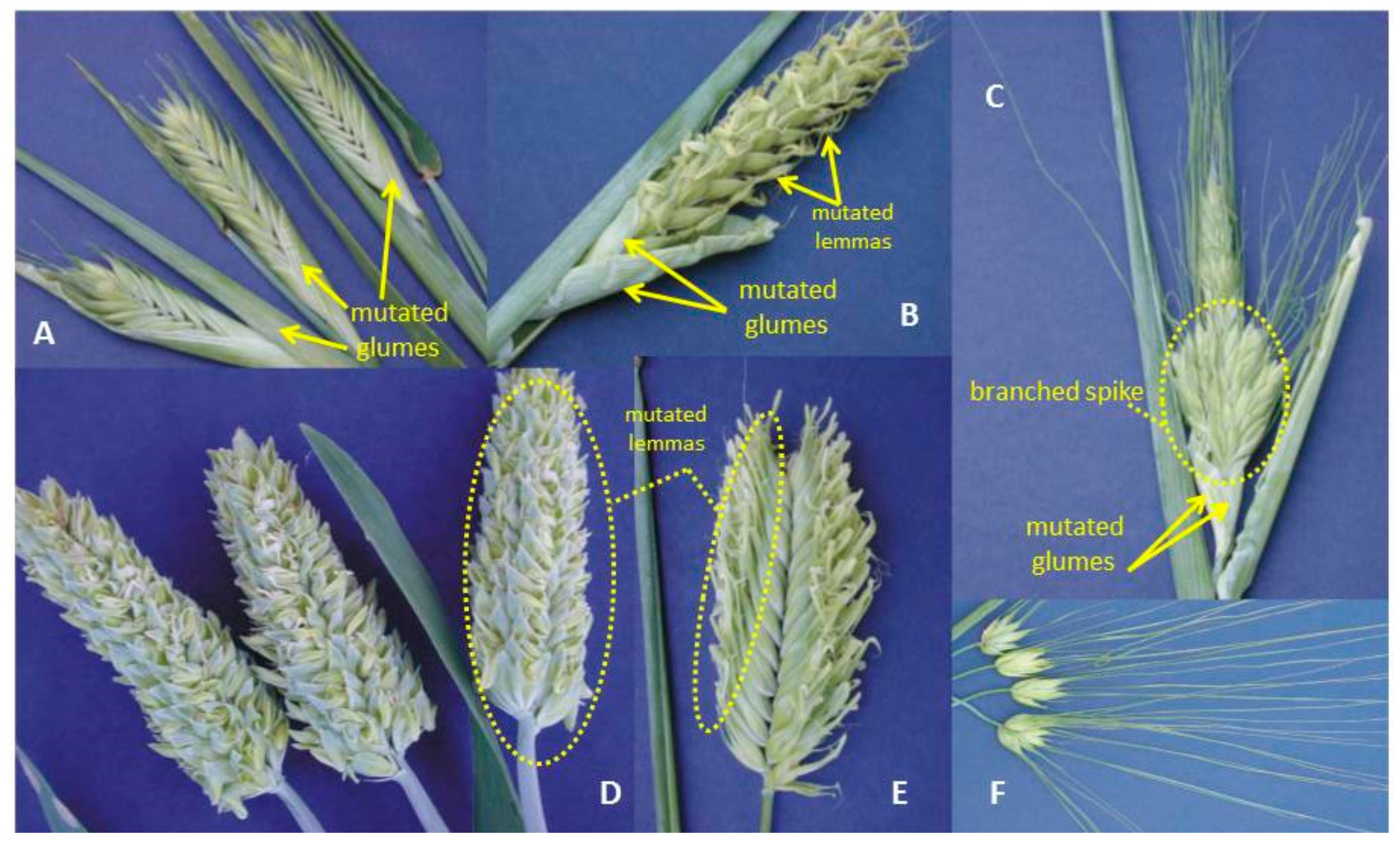{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
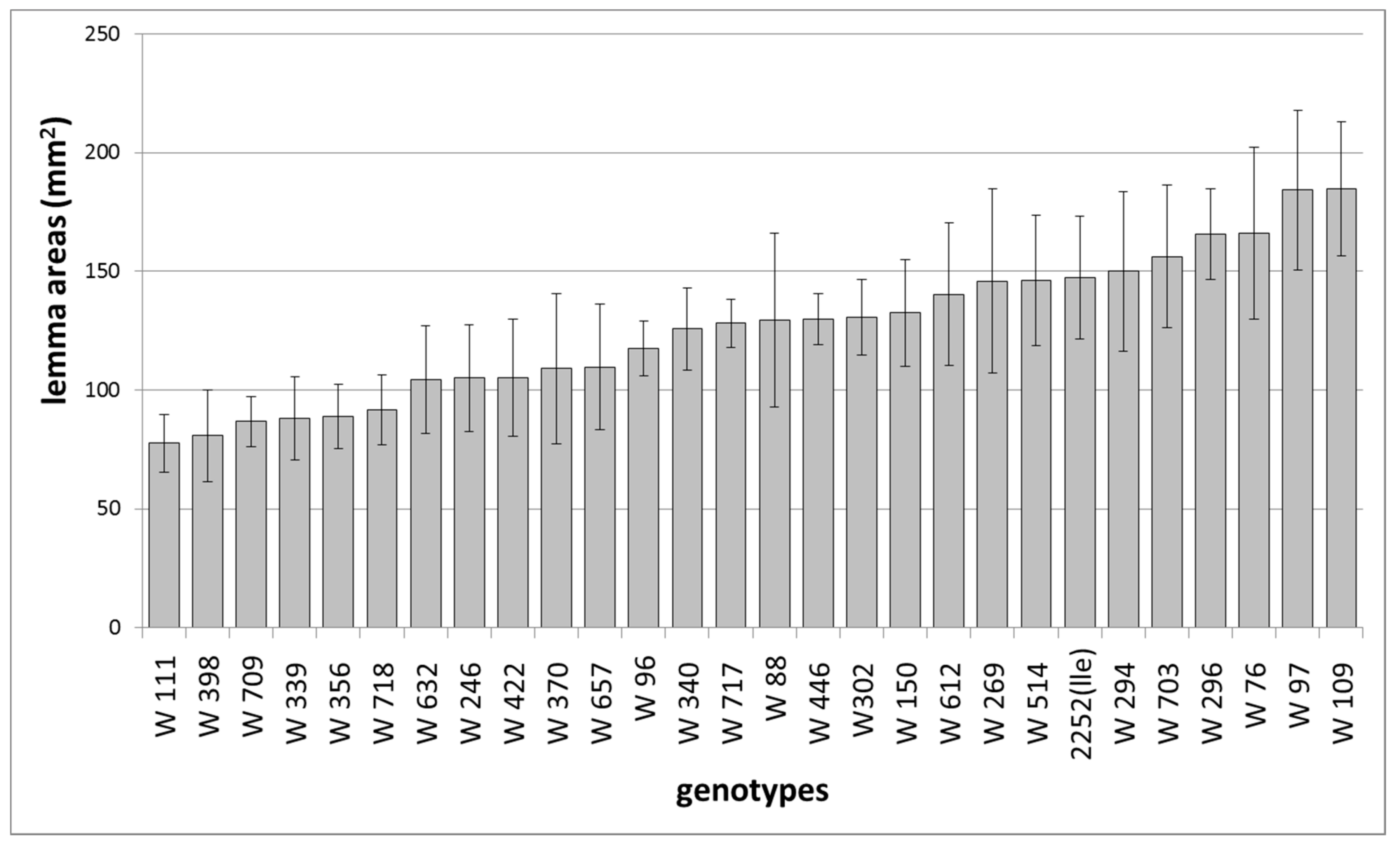{kind=link}
{kind=link}
{kind=link}
Abstract
:1. The Cereal Spike
2. Why Barley?
3. Genetic Dissection of the Key Developmental Trait of Barley Spike
4. The Phytomer Model: Key to Understand the Ontogeny of Barley Inflorescence
5. The Brittle Rachis
6. The Row Number
7. Branched Spike
8. Spike Density
9. Cleistogamy
10. Lemma and Awn
11. Naked Seed
12. The Genetic Background Effect
13. Conclusive Remarks
- -
- from an applied point of view, to identify loci that control traits of agronomic and qualitative relevance for pre-breeding and breeding programs;
- -
- from a speculative point of view, as unique tools to better understand the developmental plan of a crop and the major forces driving its evolution.
Acknowledgments
Author Contributions
Conflicts of Interest
References
- How to Feed the World in 2050. Available online: http://www.fao.org/fileadmin/templates/wsfs/docs/expert_paper/How_to_Feed_the_World_in_2050.pdf (accessed on 9 May 2017).
- Fischer, R.A.; Edmeades, G.O. Breeding and cereal yield progress. Crop Sci. 2010, 50, 85–98. [Google Scholar] [CrossRef]
- Sreenivasulu, N.; Schnurbusch, T. A genetic playground for enhancing grain number in cereals. Trends Plant Sci. 2012, 17, 91–101. [Google Scholar] [CrossRef] [PubMed]
- Mascher, M.; Gundlach, H.; Himmelbach, A.; Beier, S.; Twardziok, S.O.; Wicker, T.; Radchuk, V.; Dockter, C.; Hedley, P.E.; Russell, J.; et al. A chromosome conformation capture ordered sequence of the barley genome. Nature 2017, 544, 427–433. [Google Scholar] [CrossRef] [PubMed]
- International Database for Barley Genes and Barley Genetic Stocks. Available online: www.nordgen.org/bgs/ (accessed on 9 May 2017).
- Druka, A.; Franckowiak, J.; Lundqvist, U.; Bonar, N.; Alexander, J.; Houston, K.; Radovic, S.; Shahinnia, F.; Vendramin, V.; Morgante, M.; et al. Genetic Dissection of Barley Morphology and Development. Plant Physiol. 2011, 155, 617–627. [Google Scholar] [CrossRef] [PubMed]
- Sakuma, S.; Salomon, B.; Komatsuda, T. The Domestication Syndrome Genes Responsible for the Major Changes in Plant Form in the Triticeae Crops. Plant Cell Physiol. 2011, 52, 738–749. [Google Scholar] [CrossRef] [PubMed]
- Mascher, M.; Schuenemann, V.J.; Davidovich, U.; Marom, N.; Himmelbach, A.; Hübner, S.; Korol, A.; David, M.; Reiter, E.; Riehl, S.; et al. Genomic analysis of 6000-year-old cultivated grain illuminates the domestication history of barley. Nat. Genet. 2016, 48, 1089–1093. [Google Scholar] [CrossRef] [PubMed]
- Kirby, E.J.M.; Appleyard, M. Cereal Development Guide, 2nd ed.; Arable Unit, National Agricultural Centre: Warwickshire, UK, 1986; pp. 53–59. [Google Scholar]
- Bossinger, G.; Rohde, W.; Lundqvist, U.; Salamini, F. Genetics of barley development: Mutant phenotypes and molecular aspects. In Barley: Genetics, Biochemistry, Molecular Biology and Biotechnology; Shewry, P.R., Ed.; CAB International: Wallingford, UK, 1992; pp. 231–264. [Google Scholar]
- Forster, B.P.; Franckowiak, J.D.; Lundqvist, U.; Lyon, J.; Pitkethly, I.; Thomas, W.T.B. The Barley Phytomer. Ann. Bot. 2007, 100, 725–733. [Google Scholar] [CrossRef] [PubMed]
- Pourkheirandish, M.; Hensel, G.; Kilian, B.; Kumlehn, J.; Sato, K.; Komatsuda, T. Evolution of the Grain Dispersal System in Barley. Cell 2015, 162, 527–539. [Google Scholar] [CrossRef] [PubMed]
- Civáň, P.; Brown, T.A. A novel mutation conferring the non brittle phenotype of cultivated barley. New Phytol. 2017, 214, 468–472. [Google Scholar]
- Schmalenbach, I.; March, T.J.; Bringezu, T.; Waugh, R.; Pillen, K. High-Resolution Genotyping of Wild Barley Introgression Lines and Fine-Mapping of the Threshability Locus thresh-1 Using the Illumina GoldenGate Assay. G3 Genes Genomes Genet. 2011, 1, 187–196. [Google Scholar] [CrossRef] [PubMed]
- Koppolu, R.; Anwar, N.; Sakuma, S.; Tagiri, A.; Lundqvist, U.; Pourkheirandish, M.; Rutten, T.; Seiler, C.; Himmelbach, A.; Ariyadasa, R.; et al. Six-rowed spike4 (Vrs4) controls spikelet determinacy and row-type in barley. Proc. Natl. Acad. Sci. USA 2013, 110, 13198–13203. [Google Scholar] [CrossRef] [PubMed]
- Komatsuda, T.; Pourkheirandish, M.; He, C.; Azhaguvel, P.; Kanamori, H.; Perovic, D.; Stein, N.; Graner, A.; Wicker, T.; Tagiri, A.; et al. Six-rowed barley originated from a mutation in a homeodomain-leucine zipper I-class homeobox gene. Proc. Natl. Acad. Sci. USA 2007, 104, 1424–1429. [Google Scholar] [CrossRef] [PubMed]
- Ramsay, L.; Comadran, J.; Druka, A.; Marshall, D.F.; Thomas, W.T.W.; Macaulay, M.; MacKenzie, K.; Simpson, C.; Fuller, J.; Bonar, N.; et al. INTERMEDIUM-C, a modifier of lateral spikelet fertility in barley, is an ortholog of the maize domestication gene TEOSINTE BRANCHED 1. Nat. Genet. 2011, 43, 169–173. [Google Scholar] [CrossRef] [PubMed]
- Youssef, H.M.; Koppolu, R.; Rutten, T.; Korzun, V.; Schweizer, P.; Schnurbusch, T. Genetic mapping of the labile (lab) gene: A recessive locus causing irregular spikelet fertility in labile-barley (Hordeum vulgare convar. labile). Theor. Appl. Genet. 2014, 127, 1123–1131. [Google Scholar] [CrossRef] [PubMed]
- Helmy, M.Y.; Eggert, K.; Koppolu, R.; Alqudah, H.M.; Poursarebani, N.; Fazeli, A.; Sakuma, S.; Tagiri, A.; Rutten, T.; Govind, G.; et al. VRS2 regulates hormone-mediated inflorescence patterning in barley. Nat. Genet. 2017, 49, 157–161. [Google Scholar]
- Boden, S.A. How hormones regulate floral architecture in barley. Nat. Gen. 2017, 49, 8–9. [Google Scholar] [CrossRef] [PubMed]
- Shang, Y.; Zhu, J.; Hua, W.; Wang, J.; Jia, Q.; Yang, J. Characterization and mapping of a Prbs gene controlling spike development in Hordeum vulgare L. Genes Genome 2014, 36, 275–282. [Google Scholar] [CrossRef]
- Poursarebani, N.; Seidensticker, T.; Koppolu, R.; Trautewig, C.; Gawroński, P.; Bini, F.; Govind, G.; Rutten, R.; Sakuma, S.; Tagiri, A.; et al. The genetic basis of composite spike form in barley and ‘Miracle-Wheat’. Genetics 2015, 115, 176628. [Google Scholar] [CrossRef] [PubMed]
- Brown, R.H.; Bregitzer, P. A Ds Insertional Mutant of a Barley miR172 Gene results in Indeterminate Spikelet Development. Crop Sci. 2011, 51, 1664–1672. [Google Scholar] [CrossRef]
- Jost, M.; Taketa, S.; Mascher, M.; Himmelbach, A.; You, T.; Shahinnia, F.; Rutten, T.; Druka, A.; Schmutzer, T.; Steuernagel, B.; et al. A homolog of Blade-On-Petiole 1 and 2 (BOP1/2) controls internode length and homeotic changes of the barley inflorescence. Plant Physiol. 2016, 171, 1113–1127. [Google Scholar] [CrossRef] [PubMed]
- Shahinnia, F.; Druka, A.; Franckowiak, J.; Morgante, M.; Waugh, R.; Stein, N. High resolution mapping of Dense spike-ar (dsp.ar) to the genetic centromere of barley chromosome 7H. Theor. Appl. Genet. 2012, 124, 373–384. [Google Scholar] [CrossRef] [PubMed]
- Taketa, S.; Yuo, T.; Sakurai, Y.; Miyake, S.; Ichii, M. Molecular mapping of the short awn 2 (lks2) and dense spike 1 (dsp1) genes on barley chromosome 7H. Breed. Sci. 2011, 61, 80. [Google Scholar] [CrossRef]
- Houston, K.; McKim, S.M.; Comadran, J.; Bonar, N.; Druka, I.; Uzrek, N.; Cirillo, E.; Guzy-Wrobelska, J.; Collins, N.C.; Halpin, C.; et al. Variation in the interaction between alleles of HvAPETALA2 and microRNA172 determines the density of grains on the barley inflorescence. Proc. Natl. Acad. Sci. USA 2013, 110, 16675–16680. [Google Scholar] [CrossRef] [PubMed]
- Nair, S.K.; Wang, N.; Turuspekov, Y.; Pourkheirandish, M.; Sinsuwongwat, S.; Chen, G.; Sameri, M.; Tagiri, A.; Honda, I.; Watanabe, Y.; et al. Cleistogamous flowering in barley arises from the suppression of microRNA-guided HvAP2 mRNA cleavage. Proc. Natl. Acad. Sci. USA 2010, 107, 490–495. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Ning, S.; Wu, J.; Tagiri, A.; Komatsuda, T. An Epiallele at cly1 Affects the Expression of Floret Closing (Cleistogamy) in Barley. Genetics 2015, 199, 95–104. [Google Scholar] [CrossRef] [PubMed]
- Pozzi, C.; Faccioli, P.; Terzi, V.; Stanca, A.M.; Cerioli, S.; Castiglioni, P.; Fink, R.; Capone, R.; Müller, K.J.; Bossinger, G.; et al. Genetics of mutations affecting the development of a barley floral bract. Genetics 2000, 154, 1335–1346. [Google Scholar] [PubMed]
- Roig, C.; Pozzi, C.; Santi, L.; Müller, J.; Wang, Y.; Stile, M.R.; Rossini, L.; Stanca, A.M.; Salamini, F. Genetics of barley hooded suppression. Genetics 2004, 167, 439–448. [Google Scholar] [CrossRef] [PubMed]
- Osnato, M.; Stile, M.R.; Wang, Y.; Meynard, D.; Curiale, S.; Guiderdoni, E.; Liu, Y.; Horner, D.S.; Ouwerkerk, B.F.; Pozzi, C.; et al. Cross Talk between the KNOX and Ethylene Pathways Is Mediated by Intron-Binding Transcription Factors in Barley. Plant Physiol. 2010, 154, 4, 1616–1632. [Google Scholar] [CrossRef] [PubMed]
- Yuo, T.; Yamashita, Y.; Kanamori, H.; Matsumoto, T.; Lundqvist, U.; Sato, K.; Ichii, M.; Jobling, S.A.; Taketa, S. A SHORT INTERNODES (SHI) family transcription factor gene regulates awn elongation and pistil morphology in barley. J. Exp. Bot. 2012, 63, 5223–5232. [Google Scholar] [CrossRef] [PubMed]
- Von Bothmer, R.; Komatsuda, T. Barley origin and related species. In Barley: Production, Improvement and Uses; Ullrich, S.E., Ed.; Wiley Blackwell: Oxford, UK, 2011; pp. 14–62. [Google Scholar]
- Taketa, S.; Amano, S.; Tsujino, Y.; Sato, T.; Saisho, D.; Kakeda, K.; Nomura, M.; Suzuki, T.; Matsumoto, T.; Sato, K.; et al. Barley grain with adhering hulls is controlled by an ERF family transcription factor gene regulating a lipid biosynthesis pathway. Proc. Natl. Acad. Sci. USA 2008, 105, 4062–4067. [Google Scholar] [CrossRef] [PubMed]
- Dworkin, I.; Kennerly, E.; Tack, D.; Hutchinson, J.; Brown, J.; Mahaffey, J.; Gibson, G. Genomic Consequences of Background Effects on scalloped Mutant Expressivity in the Wing of Drosophila melanogaster. Genetics 2009, 181, 1065–1076. [Google Scholar] [CrossRef] [PubMed]
- Šiukšta, R.; Vaitkūnienė, V.; Kaselytė, G.; Okockytė, V.; Žukauskaitė, J.; Žvingila, D.; Rančelis, V. Inherited phenotype instability of inflorescence and floral organ development in homeotic barley double mutants and its specific modification by auxin inhibitors and 2,4-D. Ann. Bot. 2015, 115, 651–663. [Google Scholar] [CrossRef] [PubMed]
- Stanca, A.M.; Tumino, G.; Pagani, D.; Rizza, F.; Alberici, R.; Lundqvist, U.; Morcia, C.; Tondelli, A.; Terzi, V. The “Italian” barley genetics mutant collection: Conservation, development of new mutants and use. In Advance in Barley Sciences; Zhang, G., Li, C., Liu, X., Eds.; Springer Science & Business Media: New York, NY, USA, 2013; pp. 30–35. [Google Scholar]
- Kim, N.S. The genomes and transposable elements in plants: Are they friends or foes? Genes Genom 2017, 39, 359. [Google Scholar] [CrossRef]
- Wicker, T.; Yu, Y.; Haberer, G.; Mayer, K.F.; Marri, P.R.; Rounsley, S.; Roffler, S. DNA transposon activity is associated with increased mutation rates in genes of rice and other grasses. Nat. Commun. 2016, 7. [Google Scholar] [CrossRef] [PubMed]
- Springer, N.M.; Lisch, D.; Li, Q. Creating order from chaos: Epigenome dynamics in plants with complex genomes. Plant Cell 2016, 28, 314–325. [Google Scholar] [CrossRef] [PubMed]
- Mascher, M.; Jost, M.; Kuon, J.-E.; Himmelbach, A.; Aβfalg, A.; Beier, S.; Scholz, U.; Graner, A.; Stein, N. Mapping by sequencing accelerates forward genetics in barley. Genome Biol. 2014, 15, R78. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Bayer, M.; Druka, A.; Russell, J.R.; Hackett, C.A.; Poland, J.; Ramsay, L.; Hedley, P.E.; Waugh, R. An evaluation of genotyping by sequencing (GBS) to map the Breviaristatum-e (ari-e) locus in cultivated barley. BMC Genom. 2014, 15, 104–115. [Google Scholar] [CrossRef] [PubMed]
- Wendt, T.; Holm, P.B.; Starker, C.G.; Christian, M.; Voytas, D.F.; Brinch-Pedersen, H.; Holme, I.B. TAL effector nucleases induce mutations at a pre-selected location in the genome of primary barley transformants. Plant Mol. Boil. 2013, 83, 279–285. [Google Scholar] [CrossRef] [PubMed]
- Gottwald, S.; Bauer, P.; Komatsuda, T.; Lundquist, U.; Stein, N. TILLING in the two-rowed barley cultivar “Barke” reveals preferred sites of functional diversity in the gene HvHoxI. BMC Res. Not. 2009, 2, 258. [Google Scholar]








© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Terzi, V.; Tumino, G.; Pagani, D.; Rizza, F.; Ghizzoni, R.; Morcia, C.; Stanca, A.M. Barley Developmental Mutants: The High Road to Understand the Cereal Spike Morphology. Diversity 2017, 9, 21. https://doi.org/10.3390/d9020021
Terzi V, Tumino G, Pagani D, Rizza F, Ghizzoni R, Morcia C, Stanca AM. Barley Developmental Mutants: The High Road to Understand the Cereal Spike Morphology. Diversity. 2017; 9(2):21. https://doi.org/10.3390/d9020021
Chicago/Turabian StyleTerzi, Valeria, Giorgio Tumino, Donata Pagani, Fulvia Rizza, Roberta Ghizzoni, Caterina Morcia, and Antonio Michele Stanca. 2017. "Barley Developmental Mutants: The High Road to Understand the Cereal Spike Morphology" Diversity 9, no. 2: 21. https://doi.org/10.3390/d9020021
APA StyleTerzi, V., Tumino, G., Pagani, D., Rizza, F., Ghizzoni, R., Morcia, C., & Stanca, A. M. (2017). Barley Developmental Mutants: The High Road to Understand the Cereal Spike Morphology. Diversity, 9(2), 21. https://doi.org/10.3390/d9020021