
Lack of Population Genetic Structuring in Ocelots (Leopardus pardalis) in a Fragmented Landscape

Abstract
:1. Introduction
2. Materials and Methods
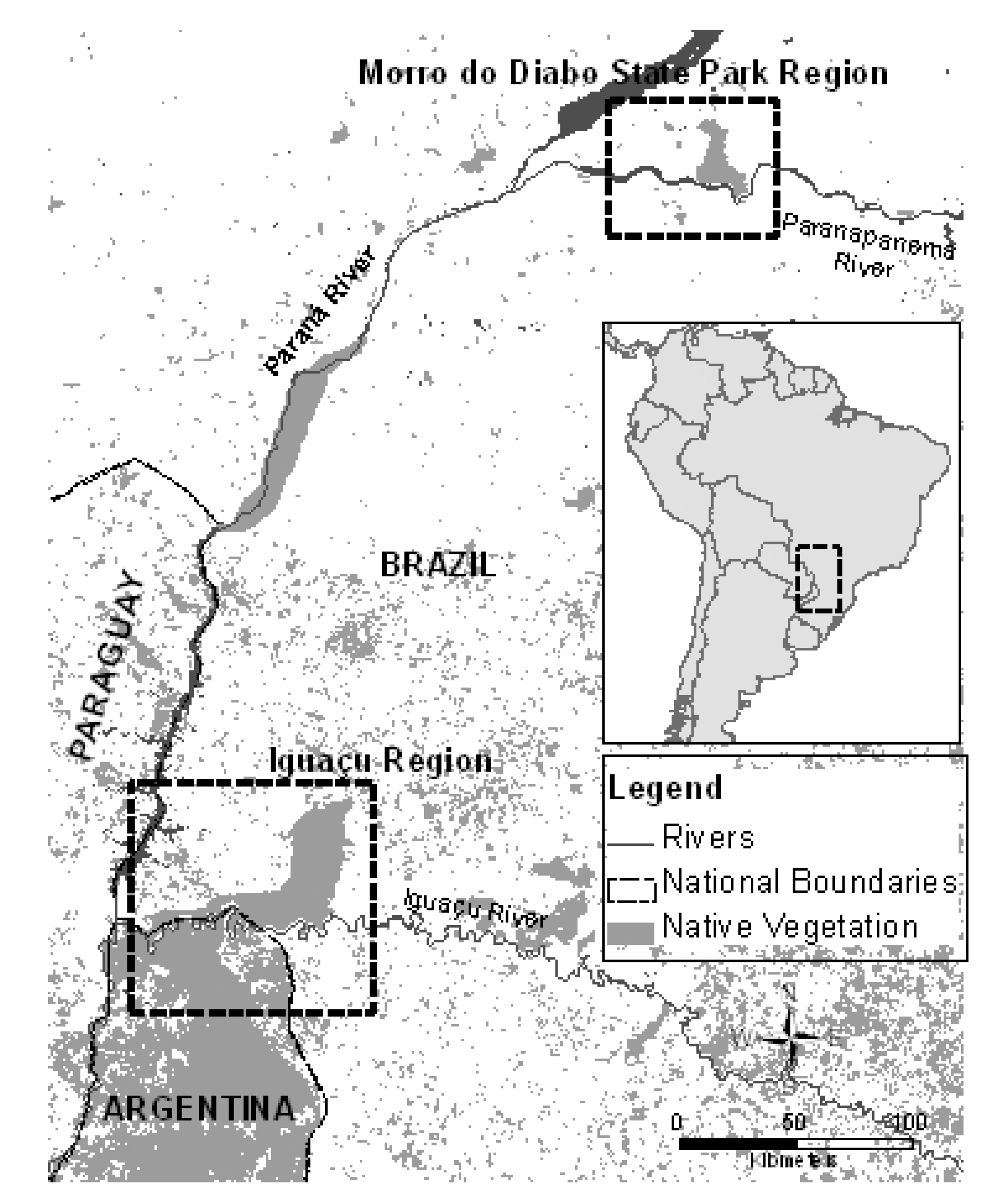
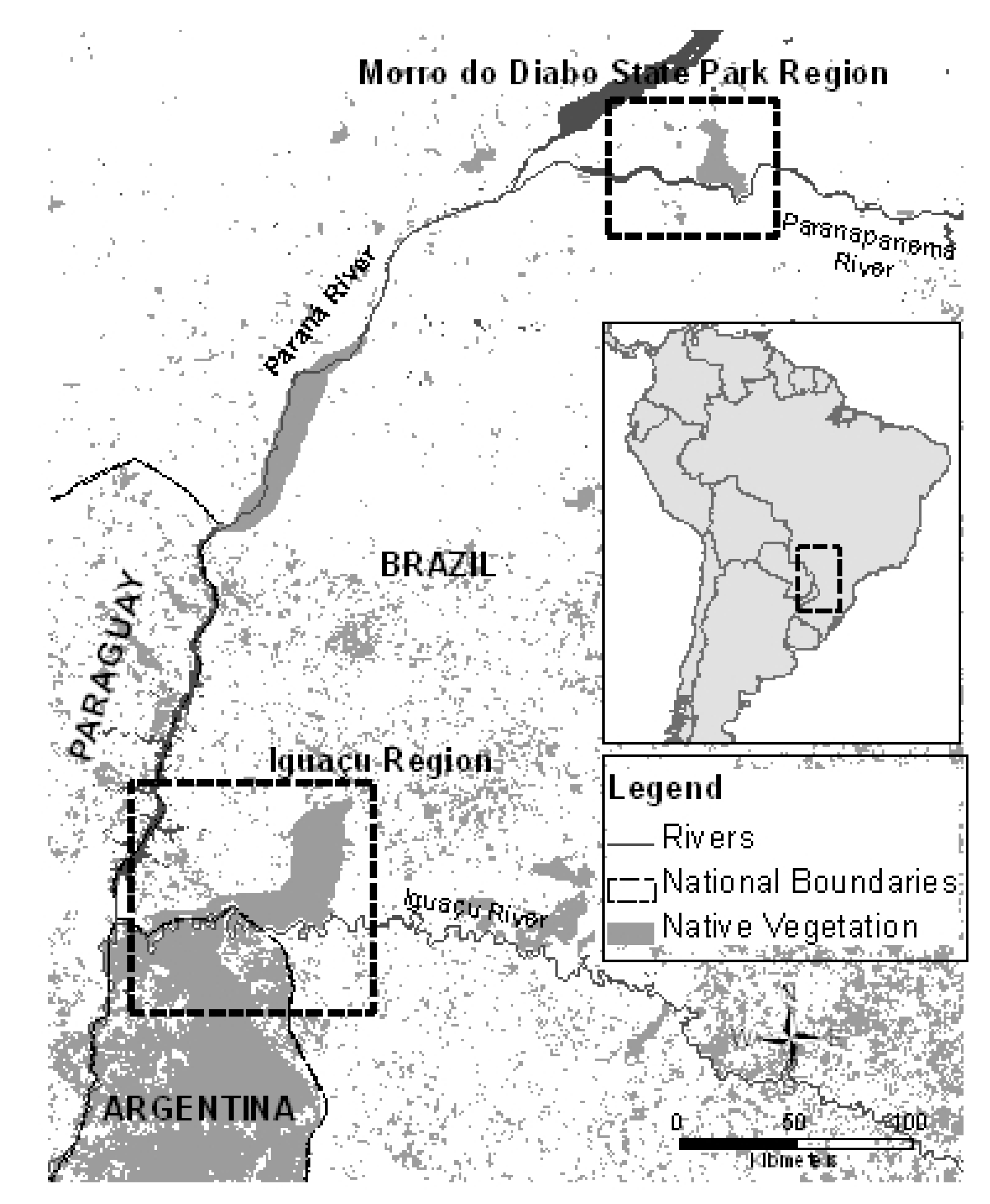
2.1. Study Area and Sampling
2.2. Laboratory Protocols
2.3. Data Analysis
3. Results

{kind=link}
| FCA008 | FCA042 ‡ | FCA053 | FCA077 | FCA124 | FCA391 | FCA424 | FCA441 | FCA453 † | Average | ||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Morro do Diabo (MD) (n = 14) | N | 7 | 9 | 3 | 9 | 2 | 7 | 5 | 3 | 7 | 5.8 |
| R | 6.77 | 8.69 | 3.00 | 8.92 | 2.00 | 6.97 | 5.00 | 3.00 | 6.84 | 5.8 | |
| HE | 0.78 | 0.88 | 0.56 | 0.89 | 0.30 | 0.84 | 0.76 | 0.54 | 0.83 | 0.71 | |
| HO | 0.92 | 1.00 | 0.33 | 0.79 | 0.36 | 0.64 | 0.79 | 0.64 | 0.92 | 0.71 | |
| Fis | −0.190 | −0.147 | 0.417 | 0.123 | −0.182 | 0.243 | −0.036 | −0.200 | −0.099 | 0.001 | |
| Iguaçu Region (IR) (n = 18) | N | 6 | 12 | 5 | 9 | 2 | 8 | 4 | 4 | 7 | 6.3 |
| R | 5.30 | 10.32 | 4.33 | 8.20 | 2.00 | 6.86 | 3.67 | 3.89 | 6.51 | 5.7 | |
| HE | 0.68 | 0.91 | 0.62 | 0.84 | 0.43 | 0.80 | 0.67 | 0.60 | 0.79 | 0.70 | |
| HO | 0.67 | 0.78 | 0.59 | 0.83 | 0.47 | 0.61 | 0.67 | 0.33 | 0.89 | 0.65 | |
| Fis | 0.012 | 0.141 | 0.048 | 0.012 | −0.103 | 0.243 | −0.002 | 0.450 | −0.136 | 0.078 |
4. Discussion
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Marino, J. Threatened Ethiopian wolves persist in small isolated Afroalpine enclaves. Oryx 2003, 37, 62–71. [Google Scholar] [CrossRef]
- Galetti, M.; Eizirik, E.; Beisiegel, B.; Ferraz, K.; Cavalcanti, S.; Srbek-Araujo, A.C.; Crawshaw, P.; Paviolo, A.; Galetti, P.M., Jr.; Jorge, M.L.; et al. Atlantic Rainforest’s Jaguars in Decline. Science 2013, 342, 930. [Google Scholar] [CrossRef] [PubMed]
- Dirzo, R.; Young, H.S.; Galetti, M.; Ceballos, G.; Isaac, N.J.B.; Collen, B. Defaunation in the Anthropocene. Science 2014, 345, 401–406. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, S.J.; Roelke, M.E.; Marker, L.; Newman, A.; Winkler, C.A.; Meltzer, D.; Colly, L.; Evermann, J.F.; Bush, M.; Wildt, D.E. Genetic basis for species vulnerability in the cheetah. Science 1985, 227, 1428–1434. [Google Scholar] [CrossRef] [PubMed]
- Hedrick, P.W.; Kalinowski, S.T. Inbreeding depression in conservation biology. Annu. Rev. Ecol. Evol. Syst. 2000, 31, 139–162. [Google Scholar] [CrossRef]
- Coulon, A.; Guillot, G.; Cosson, J.-F.; Angibault, J.M.A.; Aulagnier, S.; Cargnelutti, B.; Galan, M.; Hewison, A.J.M. Genetic structure is influenced by landscape features: Empirical evidence from a roe deer population. Mol. Ecol. 2006, 15, 1669–1679. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.S.; Ruell, E.W.; Boydston, E.E.; Lyren, L.M.; Alonso, R.S.; Troyer, J.L.; Crooks, K.R.; van de Woude, S. Gene flow and pathogen transmission among bobcats (Lynx rufus) in a fragmented urban landscape. Mol. Ecol. 2012, 21, 1617–1631. [Google Scholar] [CrossRef] [PubMed]
- Ellis, J.S.; Knight, M.E.; Darvill, B.; Goulson, D. Extremely low effective population sizes, genetic structuring and reduced genetic diversity in a threatened bumblebee species, Bombus sylvarum (Hymenoptera: Apidae). Mol. Ecol. 2006, 15, 4375–4386. [Google Scholar] [CrossRef] [PubMed]
- Haag, T.; Santos, A.S.; Sana, D.A.; Morato, R.G.; Cullen, R., Jr.; Crawshaw, P., Jr.; de Angelo, C.; di Bitetti, M.; Salzano, F.M.; Eizirik, E. The effect of habitat fragmentation on the genetic structure of a top predator: Loss of diversity and high differentiation among remnant populations of Atlantic Forest jaguars (Panthera onca). Mol. Ecol. 2010, 19, 4906–4921. [Google Scholar] [CrossRef] [PubMed]
- Galbusera, P.; Githiru, M.; Lens, L.; Matthysen, E. Genetic equilibrium despite habitat fragmentation in an Afrotropical bird. Mol. Ecol. 2004, 13, 1409–1421. [Google Scholar] [CrossRef] [PubMed]
- Purrenhage, J.L.; Niewiarowski, P.H.; Moore, F.B.-G. Population structure of spotted salamanders (Ambystoma maculatum) in a fragmented landscape. Mol. Ecol. 2009, 18, 235–247. [Google Scholar] [CrossRef] [PubMed]
- McCulloch, E.S.; Tello, J.S.; Whitehead, A.; Rolón-Mendoza, C.M.J.; Maldonado-Rodríguez, M.C.D.; Stevens, R.D. Fragmentation of Atlantic Forest has not affected gene flow of a widespread seed-dispersing bat. Mol. Ecol. 2013, 22, 4619–4633. [Google Scholar] [CrossRef] [PubMed]
- Murray, J.L.; Gardner, G.L. Leopardus pardalis. Mammal. Spec. 1997, 548, 1–10. [CrossRef]
- Di Bitetti, M.S.; Placci, G.; Dietz, L. Uma Visão de Biodiversidade Para a Ecorregião Florestas do Alto Paraná—Bioma Mata Atlântica: Planejando a Paisagem de Conservação da Biodiversidade e Estabelecendo Prioridades Para Ações de Conservação; World Wildlife Fund Press: Washington, DC, USA, 2003. (In Portuguese) [Google Scholar]
- Di Bitetti, M.S.; Paviolo, A.; de Angelo, C. Density, habitat use and activity patterns of ocelots (Leopardus pardalis) in the Atlantic Forest of Misiones, Argentina. J. Zool. 2006, 270, 153–163. [Google Scholar] [CrossRef]
- Janečka, J.E.; Tewes, M.E.; Laack, L.L.; Grassman, L.I., Jr.; Haines, A.M.; Honeycutt, R.L. Small effective population sizes of two remnant ocelot populations (Leopardus pardalis albescens) in the United States. Conserv. Genet. 2008, 9, 869–878. [Google Scholar] [CrossRef]
- Janečka, J.E.; Tewes, M.E.; Laack, L.L.; Caso, A.; Grassman, L.I., Jr.; Haines, A.M.; Shindle, D.B.; Davis, B.W.; Murphy, W.J.; Honeycutt, R.L. Reduced genetic diversity and isolation of remnant ocelot populations occupying a severely fragmented landscape in southern Texas. Anim. Conserv. 2011, 14, 608–619. [Google Scholar] [CrossRef]
- Janečka, J.E.; Tewes, M.E.; Laack, L.; Caso, A.; Grassman, L.; Honeycutt, R.L. Loss of Genetic Diversity among Ocelots in the United States during the 20th Century Linked to Human Induced Population Reductions. PLOS 2014, 9, e89384. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, M.C.; Metzger, J.-P.; Martensen, A.C.; Ponzoni, F.J.; Hirota, M.M. The Brazilian Atlantic Forest: How much is left, and how is the remaining forest distributed? Implications for conservation. Biol. Conserv. 2009, 142, 1141–1153. [Google Scholar] [CrossRef]
- Crawshaw, P.G., Jr. Comparative Ecology of Ocelot (Leopardus pardalis) and Jaguar (Panthera onca) in a Protected Subtropical Forest in Brazil and Argentina. Ph.D. Thesis, University of Florida, Gainesville, FL, USA, 1995. [Google Scholar]
- Jacob, A.A. Ecologia e Conservação da Jaguatirica (Leopardus pardalis) no Parque Estadual Morro do Diabo, Pontal do Paranapanema, SP. Master’s Thesis, University of Brasília, Brasília, Brazil, 2002. [Google Scholar]
- Sambrook, J.; Fritsch, E.; Maniatis, T. Molecular Cloning: A Laboratory Manual; Cold Spring Harbor Press: New York, NY, USA, 1989. [Google Scholar]
- Menotti-Raymond, M.; David, V.A.; Lyons, L.A.; Schäffer, A.A.; Tomlin, J.F.; Hutton, M.K.; O’Brien, S.J. A genetic linkage map of microsatellites in the domestic cat (Felis catus). Genomics 1999, 57, 9–23. [Google Scholar] [CrossRef] [PubMed]
- Schuelke, M. An economic method for the fluorescent labeling of PCR fragments. Nat. Biotechnol. 2000, 18, 233–234. [Google Scholar] [CrossRef] [PubMed]
- Rousset, F. Genepop’007: A complete reimplementation of the Genepop software for Windows and Linux. Mol. Ecol. Resour. 2008, 8, 103–106. [Google Scholar] [CrossRef] [PubMed]
- Benjamin, Y.; Yekutieli, D. The control of false discovery rate under dependency. Ann. Stat. 2001, 29, 1165–1188. [Google Scholar]
- Narum, S. Beyond Bonferroni: Less conservative analyses for conservation genetics. Conserv. Genet. 2006, 7, 783–787. [Google Scholar] [CrossRef]
- Van Oosterhout, C.V.; Hutchinson, W.F.; Wills, D.P.M.; Shipley, P. Micro-checker: Software for identifying and correcting genotyping errors in microsatellite data. Mol. Ecol. Notes 2004, 4, 535–538. [Google Scholar] [CrossRef]
- Brookfield, I.F.Y. A simple new method for estimating null allele frequency from heterozygote deficiency. Mol. Ecol. 1996, 5, 453–455. [Google Scholar] [CrossRef] [PubMed]
- Goudet, J. FSTAT, Version 2.9.3.2. Institute of Ecology: Lausanne, 2002; Available online: http://www2.unil.ch/izea/softwares/fstat.html (accessed on 30 October 2009).
- Peakall, R.; Smouse, P.E. GENEALEX 6: Genetic analysis in Excel. Population genetic software for teaching and research. Mol. Ecol. Notes 2006, 6, 288–295. [Google Scholar] [CrossRef]
- Peakall, R.; Smouse, P.E. GenAlEx 6.5: Genetic analysis in Excel. Population genetic software for teaching and research-an update. Bioinformatics 2012, 28, 2537–2539. [Google Scholar] [CrossRef] [PubMed]
- Kalinowski, S.T. HP-RARE 1.0: A computer program for performing rarefaction on measures of allelic richness. Mol. Ecol. Notes 2005, 5, 187–189. [Google Scholar] [CrossRef]
- Cornuet, J.M.; Luikart, G. Description and power analysis of two tests for detecting recent population bottlenecks from allele frequency data. Genetics 1996, 144, 2001–2014. [Google Scholar] [PubMed]
- Garza, J.C.; Williamson, E.G. Detection of reduction in population size using data from microsatellite loci. Mol. Ecol. 2001, 10, 305–318. [Google Scholar] [CrossRef] [PubMed]
- Harley, E.H. AGARST (version 3.3): A Program for Calculating Allele Frequencies, Gst and Rst from Microsatellite Data Plus a Number of Other Population Genetic Estimates and Outputting Files Formatted for Various Other Population Genetic Programs; Department of Chemical Pathology. University of Cape Town: Rondebosch, South Africa, 2003. [Google Scholar]
- Weir, B.S. Genetic Data Analysis II; Sinauer & Associates: Sunderland, MA, USA, 1996. [Google Scholar]
- Chapuis, M.-P.; Estoup, A. Microsatellite null alleles and estimation of population differentiation. Mol. Biol. Evol. 2007, 24, 621–631. [Google Scholar] [CrossRef] [PubMed]
- Pritchard, J.; Stephens, M.; Donnelly, P. Inference of population structure from multilocus genotype data. Genetics 2000, 155, 945–959. [Google Scholar] [PubMed]
- Waples, R.S.; Do, C. LDNE: A program for estimating effective population size from data on linkage disequilibrium. Mol. Ecol. Resour. 2008, 8, 753–756. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-García, M.; Murillo, A.; Corrales, C.; Romero-Aleán, N.; Alvarez-Prada, D. Genética de poblaciones amazónicas: La historia evolutiva del jaguar, ocelote, delfín rosado, mono lanudo y piurí, reconstruida a partir de sus genes. Anim. Biodivers. Conserv. 2007, 30, 115–130. (In Spanish) [Google Scholar]
- Knutsen, H.; Olsen, E.M.; Jorde, P.E.; Espeland, S.H.; Andre’, C.; Stenseth, N.C. Are low but statistically significant levels of genetic differentiation in marine fishes “biologically meaningful”? A case study of coastal Atlantic cod. Mol. Ecol. 2011, 20, 768–783. [Google Scholar] [CrossRef] [PubMed]
- Horne, J.S.; Haines, A.M.; Tewes, M.E.; Laack, L.L. Habitat partitioning by sympatric ocelots and bobcats: Implications for recovery of ocelots in southern Texas. Southwest. Nat. 2009, 54, 119–126. [Google Scholar] [CrossRef]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Figueiredo, M.G.; Cervini, M.; Rodrigues, F.P.; Eizirik, E.; Azevedo, F.C.C.; Cullen, L., Jr.; Crawshaw, P.G., Jr.; Galetti, P.M., Jr. Lack of Population Genetic Structuring in Ocelots (Leopardus pardalis) in a Fragmented Landscape. Diversity 2015, 7, 295-306. https://doi.org/10.3390/d7030295
Figueiredo MG, Cervini M, Rodrigues FP, Eizirik E, Azevedo FCC, Cullen L Jr., Crawshaw PG Jr., Galetti PM Jr. Lack of Population Genetic Structuring in Ocelots (Leopardus pardalis) in a Fragmented Landscape. Diversity. 2015; 7(3):295-306. https://doi.org/10.3390/d7030295
Chicago/Turabian StyleFigueiredo, Marina G., Marcelo Cervini, Fernando P. Rodrigues, Eduardo Eizirik, Fernando C. C. Azevedo, Laury Cullen, Jr., Peter G. Crawshaw, Jr., and Pedro M. Galetti, Jr. 2015. "Lack of Population Genetic Structuring in Ocelots (Leopardus pardalis) in a Fragmented Landscape" Diversity 7, no. 3: 295-306. https://doi.org/10.3390/d7030295
APA StyleFigueiredo, M. G., Cervini, M., Rodrigues, F. P., Eizirik, E., Azevedo, F. C. C., Cullen, L., Jr., Crawshaw, P. G., Jr., & Galetti, P. M., Jr. (2015). Lack of Population Genetic Structuring in Ocelots (Leopardus pardalis) in a Fragmented Landscape. Diversity, 7(3), 295-306. https://doi.org/10.3390/d7030295