The Effect of Tillage System and Crop Rotation on Soil Microbial Diversity and Composition in a Subtropical Acrisol


Abstract
:1. Introduction
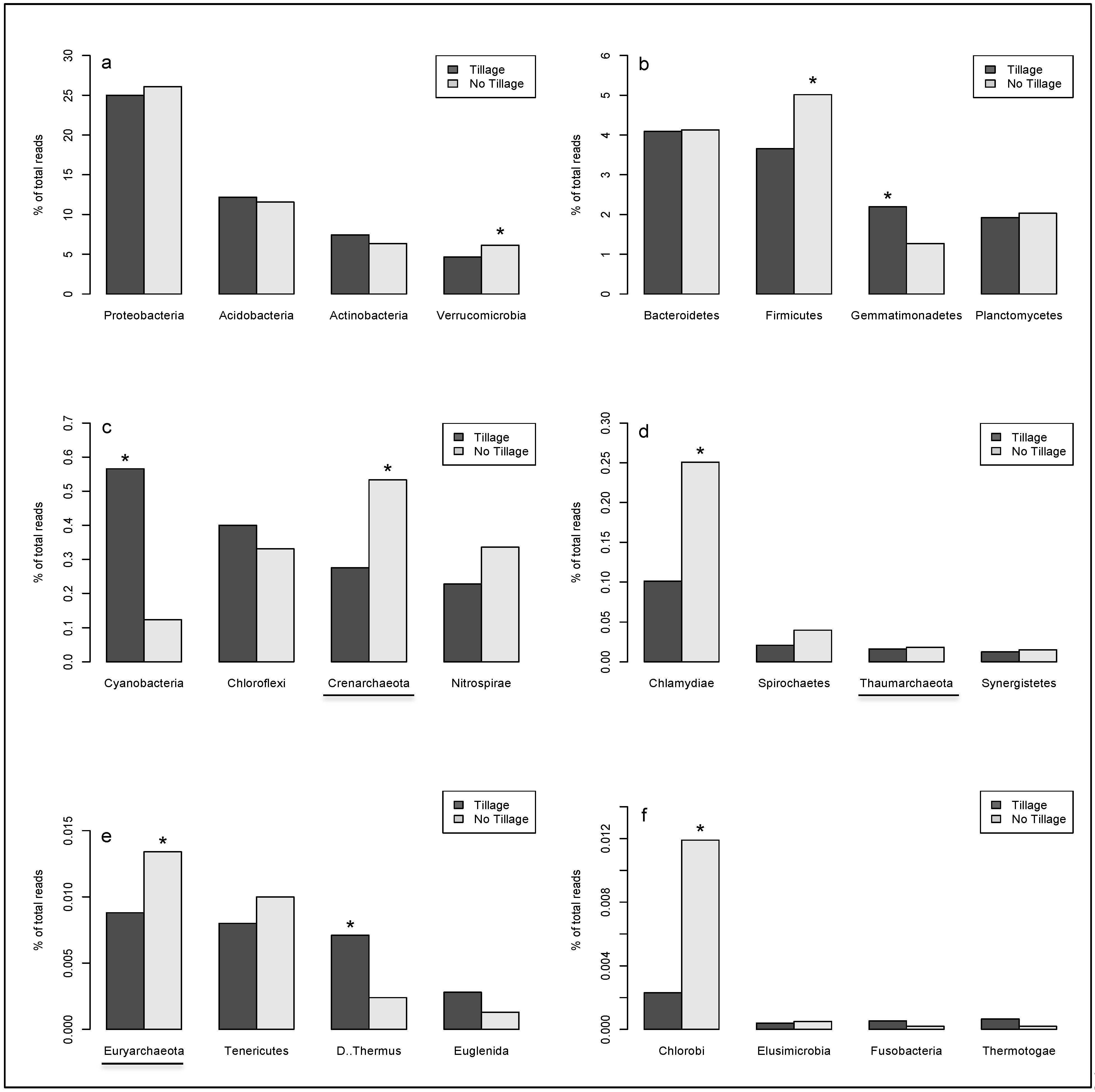
2. Results

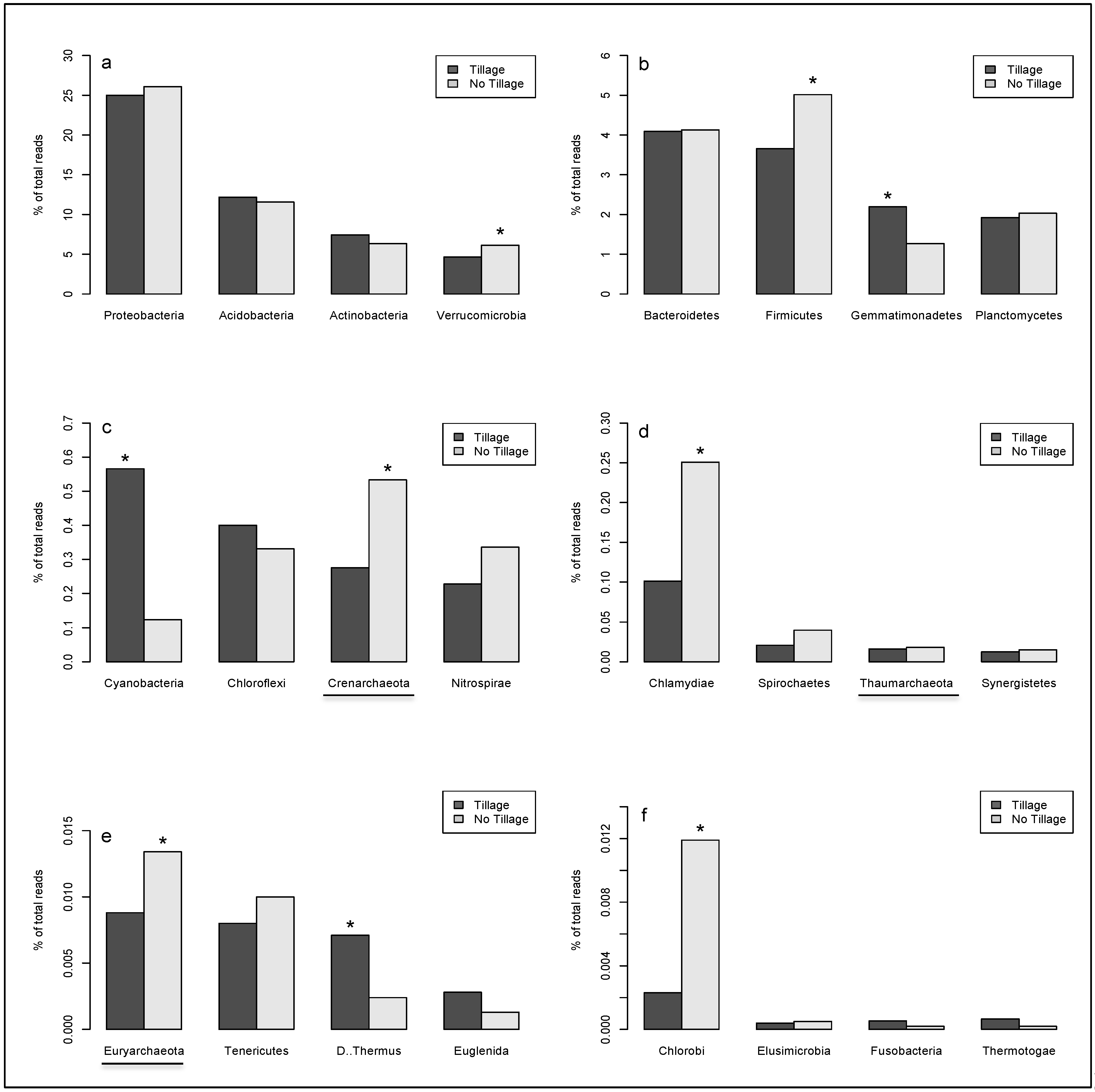{kind=link}
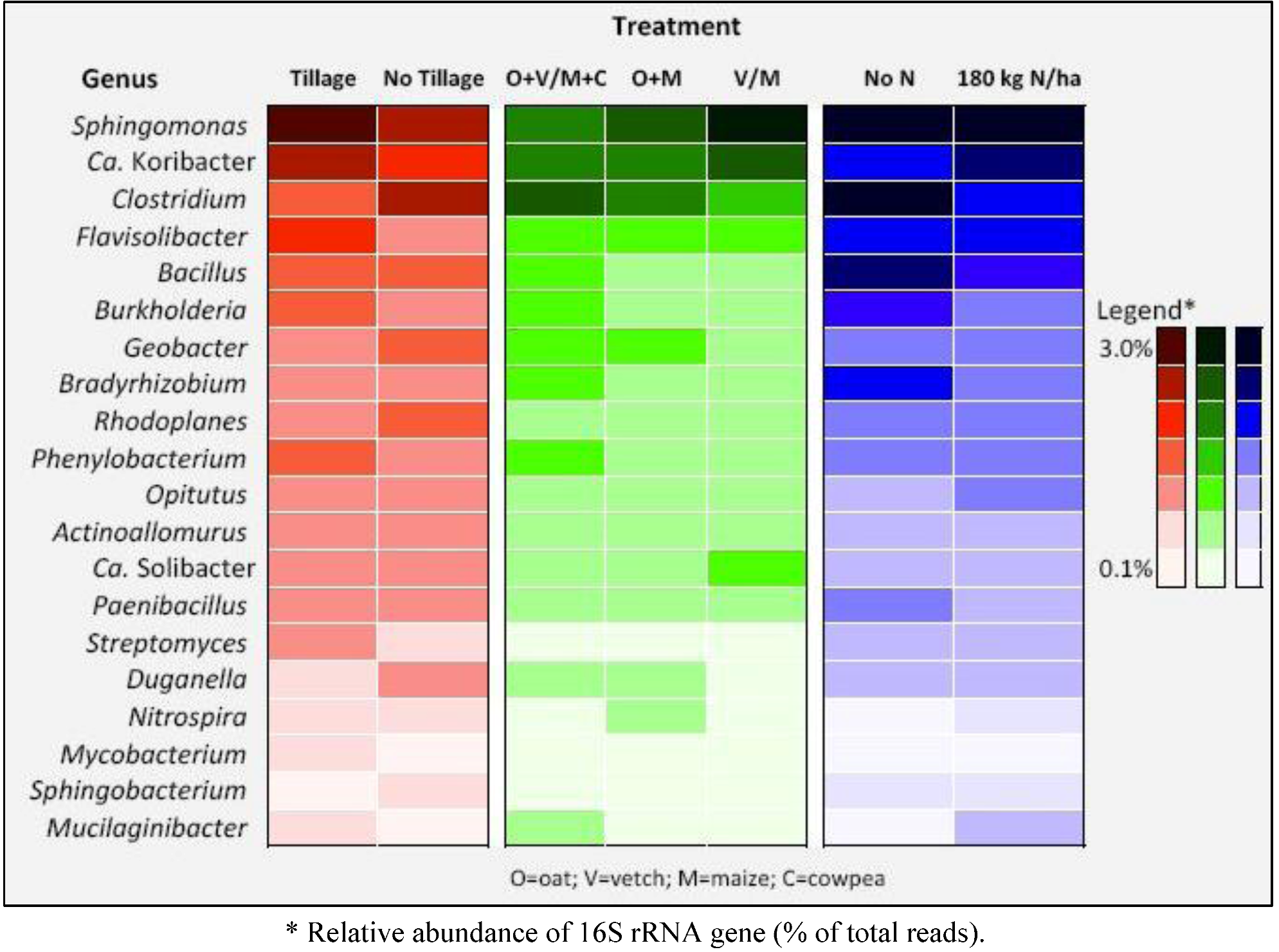{kind=link}
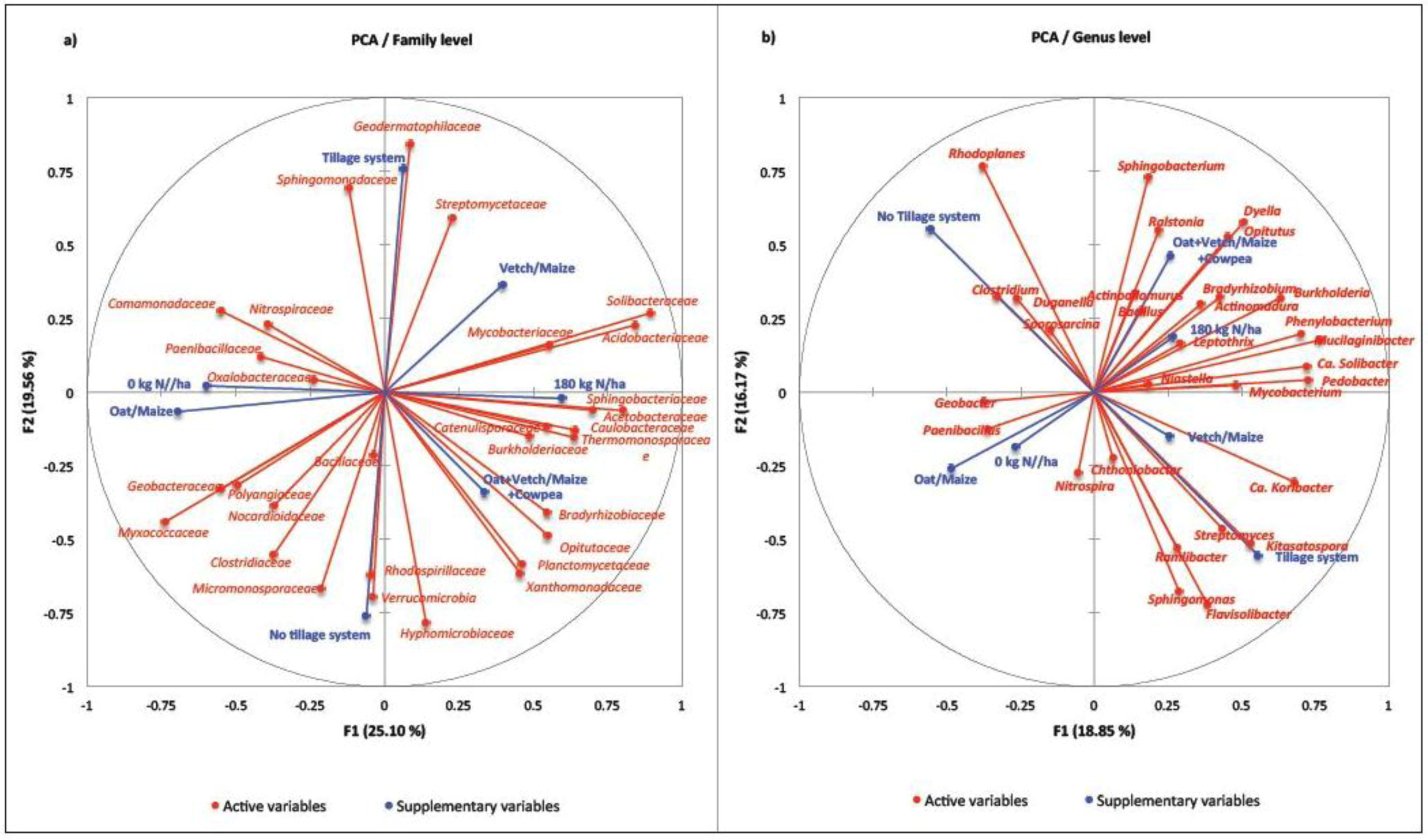{kind=link}
| Plylum | Class | Order | Family | Genus | Species | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Tillage, no N (oat+vetch/maize+cowpea) | 0.16 | abc | 3.48 | b | 3.59 | abc | 3.68 | abc | 5.29 | bcd | 5.29 | bcd |
| Tillage, 180 kg N/ha (oat+vetch/maize+cowpea) | 0.08 | cd | 3.43 | b | 3.45 | c | 3.47 | bc | 5.20 | cd | 5.20 | cd |
| Tillage, no N (oat/maize) | 0.21 | ab | 3.87 | a | 3.93 | ab | 3.93 | ab | 5.73 | ab | 5.73 | ab |
| Tillage, 180 kg N/ha (oat/maize) | 0.23 | a | 3.68 | ab | 3.73 | abc | 3.76 | abc | 5.53 | abc | 5.53 | abc |
| Tillage, no N (vetch/maize) | 0.17 | abc | 3.64 | ab | 3.62 | abc | 3.67 | abc | 5.33 | abcd | 5.33 | abcd |
| Tillage,180 kg N/ha (vetch/maize) | 0.16 | abc | 3.53 | ab | 3.58 | bc | 3.62 | abc | 5.21 | bcd | 5.21 | bcd |
| No tillage, no N (oat+vetch/maize+cowpea) | 0.02 | d | 3.43 | b | 3.42 | c | 3.54 | abc | 4.89 | d | 4.89 | d |
| No tillage, 180 kg N/ha (oat+vetch/maize+cowpea) | 0.14 | abc | 3.54 | ab | 3.57 | bc | 3.64 | abc | 5.29 | bcd | 5.29 | bcd |
| No tillage, no N (oat/maize) | 0.13 | bc | 3.88 | a | 3.96 | a | 3.94 | ab | 5.85 | a | 5.85 | a |
| No tillage, 180 kg N/ha (oat/maize) | 0.18 | abc | 3.77 | ab | 3.89 | ab | 3.98 | a | 5.59 | abc | 5.59 | abc |
| No tillage, no N (vetch/maize) | 0.19 | ab | 3.74 | ab | 3.73 | abc | 3.72 | abc | 5.45 | abc | 5.45 | abc |
| No tillage, 180 kg N/ha (vetch/maize) | 0.16 | abc | 3.43 | b | 3.40 | c | 3.42 | c | 5.11 | cd | 5.11 | cd |
| Soil Tillage System | Crop System | N Fertilization Level | |||||
|---|---|---|---|---|---|---|---|
| Tillage | No Tillage | O+V/M+C | O/M | M/O | 0 kg N ha−1 | 180 kg N ha−1 | |
| pH | 5.2 | 5.2 | 5.0 b | 5.6 a | 4.9 b | 5.4 | 5.0 |
| P (mg dm−3) | 13.7 | 29.1* | 21.1 b | 27.8 a | 17.6 b | 25.7* | 19.4 |
| K(mg dm−3) | 161 | 156 | 157 | 166 | 150 | 162 | 155 |
| Al (cmolc dm−3) | 0.5 | 0.5 | 0.6 b | 0.1 a | 0.9 b | 0.3 | 0.7* |
| Ca (cmolc dm−3) | 2.3 | 2.6 | 2.5 | 2.9 | 2.1 | 2.9* | 2.2 |
| Mg (cmolc dm−3) | 1.2 | 2.6* | 1.4 | 1.5 | 1.2 | 1.5* | 1.2 |
| TOC (%) | 1.17 | 1.52* | 1.54 a | 1.20 b | 1.37 b | 1.32 | 1.38 |
| Total N (%) | 0.096 | 0.127* | 0.127 a | 0.099 c | 0.11 b | 0.110 | 0.114 |
| Mineral N (mg kg−1) | 5.24 | 6.90* | 8.66 a | 2.48 c | 7.29 b | 5.12 | 6.58* |
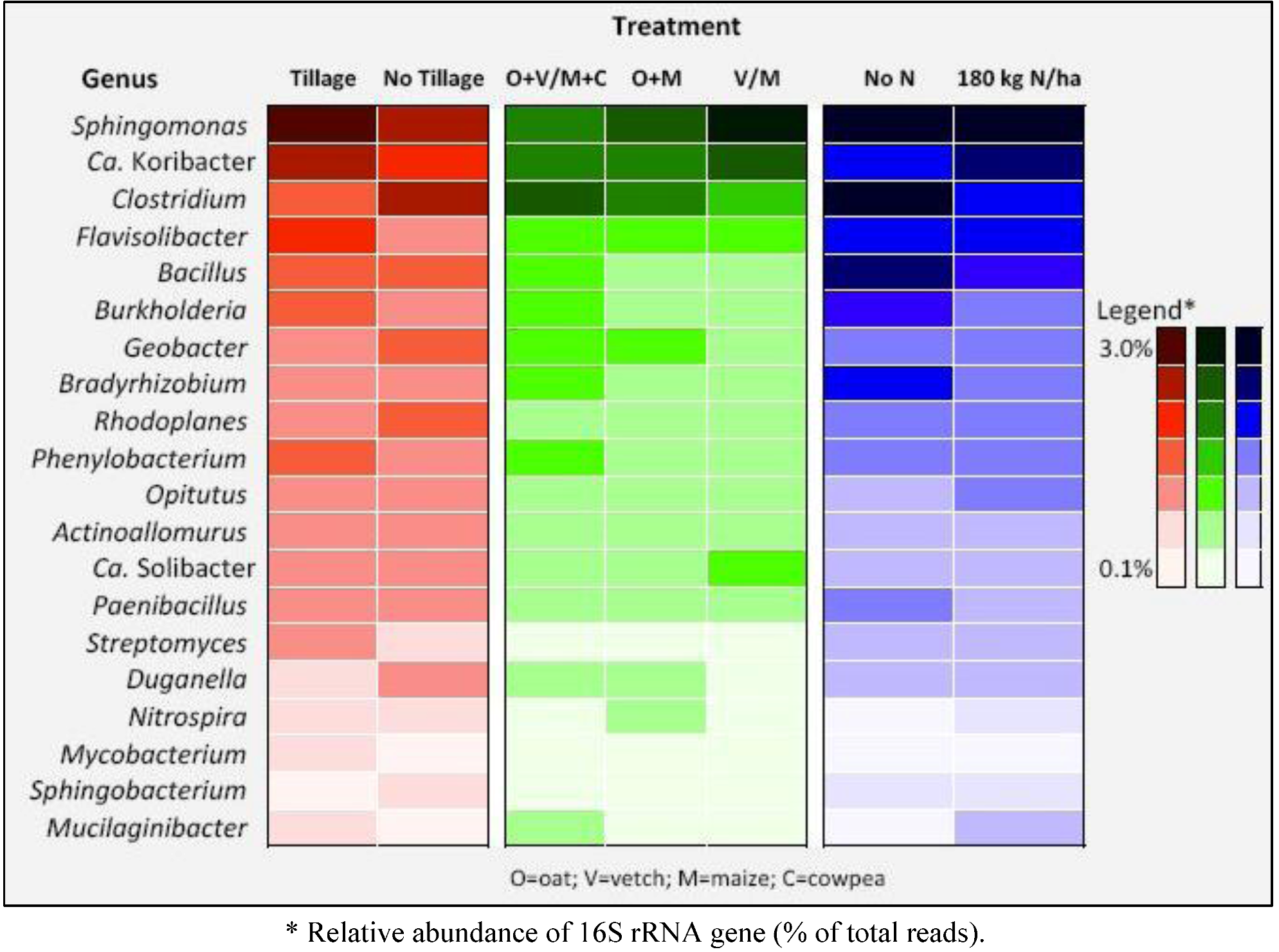
| Phylum | Class | Order | Family | Genus | Species | Tillage | No tillage |
|---|---|---|---|---|---|---|---|
| Chlamydiae | −0.87 | 0.87 | |||||
| Chlamydiae | Chlamidiia | Chlamydiales | −0.82 | 0.82 | |||
| Verrucomicrobia | −0.82 | 0.82 | |||||
| Verrucomicrobia | Verrucomicrobiae | −0.82 | 0.82 | ||||
| Verrucomicrobia | Verrucomicrobiae | Verrucomicrobiales | −0.82 | 0.82 | |||
| Verrucomicrobia | Verrucomicrobiae | Verrucomicrobiales | Verrucomicrobia sub 3 | unclassified | Ellin5121 | −0.72 | 0.72 |
| Verrucomicrobia | Verrucomicrobiae | Verrucomicrobiales | Verrucomicrobia sub 3 | unclassified | Ellin515 | −0.72 | 0.72 |
| Verrucomicrobia | Verrucomicrobiae | Verrucomicrobiales | Verrucomicrobia sub 3 | unclassified | Ellin518 | −0.72 | 0.72 |
| Verrucomicrobia | Verrucomicrobiae | Verrucomicrobiales | Verrucomicrobia sub 3 | unclassified | Ellin5102 | −0.77 | 0.77 |
| Proteobacteria | Gamaproteobacteria | Xanthomonadales | Xanthomonadaceae | −0.82 | 0.82 | ||
| Proteobacteria | Alphaproteobacteria | Rhizobiales | Hyphomicrobiaceae | −0.82 | 0.82 | ||
| Proteobacteria | Alphaproteobacteria | Rhizobiales | Hyphomicrobiaceae | Rhodoplanes | −0.87 | 0.87 | |
| Proteobacteria | Alphaproteobacteria | Rhizobiales | Hyphomicrobiaceae | Rhodoplanes | Rhodoplanes sp. | −0.77 | 0.77 |
| Bacteroidetes | Sphingobacteriia | Sphingobacteriales | Chitinophagaceae | Terrimonas | −0.77 | 0.77 | |
| Actinobacteria | Actinobacteria | Actinomycetales | Streptomycetaceae | 0.82 | −0.82 | ||
| Actinobacteria | Actinobacteria | Actinomycetales | Streptomycetaceae | Streptomyces | 0.87 | −0.87 | |
| Actinobacteria | Actinobacteria | Actinomycetales | Streptomycetaceae | Kitasatospora | 0.72 | −0.72 | |
| Actinobacteria | Actinobacteria | Actinomycetales | Pseudonocardiaceae | Amycolatopsis | 0.87 | −0.87 | |
| Actinobacteria | Actinobacteria | Actinomycetales | Geodermatophilaceae | 0.87 | −0.87 | ||
| Actinobacteria | Actinobacteria | Actinomycetales | Geodermatophilaceae | Modestobacter | Modestobacter sp. | 0.87 | −0.87 |
| Actinobacteria | Actinobacteria | Actinomycetales | Intrasporangiaceae | 0.87 | −0.87 | ||
| Actinobacteria | Actinobacteria | Actinomycetales | Kineosporiaceae | 0.87 | −0.87 | ||
| Actinobacteria | Actinobacteria | Actinomycetales | Micrococcaceae | 0.87 | −0.87 | ||
| Bacteroidetes | Sphingobacteriia | Sphingomonadales | 0.77 | −0.77 | |||
| Bacteroidetes | Sphingobacteriia | Sphingomonadales | Sphingomonadaceae | 0.77 | −0.77 | ||
| Bacteroidetes | Sphingobacteriia | Sphingomonadales | Sphingomonadaceae | Sphingomonas | 0.77 | −0.77 | |
| Bacteroidetes | Sphingobacteriia | Sphingomonadales | Sphingomonadaceae | Sphingomonas | Sphingomonas sp. | 0.77 | −0.77 |
| Bacteroidetes | Sphingobacteriia | Sphingobacteriales | Sphingobacteriaceae | Mucilaginibacter | M. ximonensis | 0.82 | −0.82 |
| Bacteroidetes | Sphingobacteriia | Sphingobacteriales | Sphingobacteriaceae | Pedobacter | 0.72 | −0.72 | |
| Bacteroidetes | Sphingobacteriia | Sphingobacteriales | Chitinophagaceae | Flavisolibacter | 0.87 | −0.87 | |
| Gemmatimonadetes | 0.77 | −0.77 | |||||
| Proteobacteria | Betaproteobacteria | Burkholderiales | Oxalobacteraceae | Herbaspirillum | 0.77 | −0.77 | |
| Proteobacteria | Betaproteobacteria | Burkholderiales | Oxalobacteraceae | Herbaspirillum | Herbaspirillum sp. | 0.87 | −0.87 |
| Proteobacteria | Betaproteobacteria | Burkholderiales | Comamonadaceae | Ramlibacter | 0.72 | −0.72 | |
| Proteobacteria | Betaproteobacteria | Burkholderiales | Comamonadaceae | Ramlibacter | Ramlibacter sp. | 0.72 | −0.72 |
| Phylum | Class | Order | Family | Genus | Species | pH | P | K | Al | Ca | Mg |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Proteobacteria | Deltaproteobacteria | Myxococcales | 0.73 | 0.36 | 0.39 | −0.82 | 0.70 | 0.62 | |||
| Proteobacteria | Deltaproteobacteria | Myxococcales | Myxococcaceae | 0.83 | 0.60 | 0.29 | −0.94 | 0.82 | 0.75 | ||
| Proteobacteria | Deltaproteobacteria | Myxococcales | Myxococcaceae | Anaeromyxobacter | 0.79 | 0.51 | 0.31 | −0.90 | 0.75 | 0.66 | |
| Proteobacteria | Betaproteobacteria | 0.74 | 0.46 | 0.57 | −0.80 | 0.77 | 0.68 | ||||
| Acidobacteria | Acidobacteriia | Acidobacteriales | Acidobacteriaceae | unclassified | Ellin5017 | −0.77 | −0.48 | −0.46 | 0.85 | −0.91 | −0.86 |
| Acidobacteria | Solibacteres | Solibacterales | Solibacteraceae | Ca. Solibacter | −0.75 | −0.53 | −0.19 | 0.85 | −0.75 | −0.70 | |
| Proteobacteria | Betaproteobacteria | 0.74 | 0.46 | 0.57 | −0.80 | 0.77 | 0.68 | ||||
| Proteobacteria | Deltaproteobacteria | Desulfuromonadales | 0.62 | 0.74 | 0.11 | −0.76 | 0.71 | 0.73 | |||
| Proteobacteria | Deltaproteobacteria | Myxococcales | 0.73 | 0.36 | 0.39 | −0.82 | 0.70 | 0.62 | |||
| Proteobacteria | Deltaproteobacteria | Myxococcales | Myxococcaceae | 0.83 | 0.60 | 0.29 | −0.94 | 0.82 | 0.75 | ||
| Proteobacteria | Deltaproteobacteria | Myxococcales | Myxococcaceae | Anaeromyxobacter | 0.79 | 0.51 | 0.31 | −0.90 | 0.75 | 0.66 | |
| Proteobacteria | Alphaproteobacteria | Caulobacterales | Caulobacteraceae | Phenylobacterium | P zucineum | −0.67 | −0.62 | −0.18 | 0.79 | −0.81 | −0.79 |
| Actinobacteria | Actinobacteria | Actinomycetales | Catenulisporaceae | Catenulispora | −0.63 | −0.63 | −0.22 | 0.78 | −0.82 | −0.82 | |
| Acidobacteria | Acidobacteriia | Acidobacteriales | −0.70 | −0.33 | −0.19 | 0.76 | −0.69 | −0.61 | |||
| Acidobacteria | Acidobacteriia | Acidobacteriales | Acidobacteriaceae | −0.70 | −0.33 | −0.19 | 0.76 | −0.69 | −0.61 | ||
| Acidobacteria | Acidobacteriia | Acidobacteriales | Acidobacteriaceae | unclassified | Ellin5017 | −0.77 | −0.48 | −0.46 | 0.85 | −0.91 | −0.86 |
| Acidobacteria | Acidobacteriia | Acidobacteriales | Acidobacteriaceae | unclassified | Ellin5056 | −0.70 | −0.66 | −0.15 | 0.80 | −0.67 | −0.63 |
| Acidobacteria | Acidobacteriia | Acidobacteriales | Acidobacteriaceae | unclassified | Ellin5237 | −0.71 | −0.38 | −0.50 | 0.77 | −0.80 | −0.76 |
| Acidobacteria | Acidobacteriia | Solibacterales | −0.68 | −0.57 | −0.13 | 0.80 | −0.74 | −0.71 | |||
| Acidobacteria | Acidobacteriia | Solibacterales | Solibacteraceae | −0.68 | −0.57 | −0.13 | 0.80 | −0.74 | −0.71 | ||
| Acidobacteria | Acidobacteriia | Solibacterales | Solibacteraceae | Ca. Solibacter | −0.75 | −0.53 | −0.19 | 0.85 | −0.75 | −0.70 | |
| Acidobacteria | unclassified | unclassified | unclassified | Ca. Koribacter | Ca. K. versatilis | −0.58 | −0.69 | −0.23 | 0.73 | −0.77 | −0.83 |
| Firmicutes | 0.50 | 0.73 | 0.31 | −0.56 | 0.77 | 0.85 | |||||
| Firmicutes | Clostridia | Clostridiales | 0.57 | 0.70 | 0.40 | −0.61 | 0.75 | 0.81 | |||
| Firmicutes | Clostridia | Clostridiales | Clostridiaceae | 0.54 | 0.73 | 0.37 | −0.59 | 0.72 | 0.79 | ||
| Firmicutes | Clostridia | Clostridiales | Clostridiaceae | Clostridium | 0.54 | 0.73 | 0.37 | −0.59 | 0.72 | 0.79 | |
| Firmicutes | Clostridia | Clostridiales | Clostridiaceae | Clostridium | C. beijerinckii | 0.59 | 0.53 | 0.42 | −0.61 | 0.77 | 0.78 |
| Firmicutes | Bacilli | Bacillales | Paenibacillaceae | 0.52 | 0.31 | 0.49 | −0.53 | 0.77 | 0.77 | ||
| Proteobacteria | Betaproteobacteria | 0.74 | 0.46 | 0.57 | −0.80 | 0.77 | 0.68 | ||||
| Proteobacteria | Betaproteobacteria | Rhodocyclales | 0.57 | 0.73 | 0.27 | −0.73 | 0.70 | 0.76 | |||
| Proteobacteria | Betaproteobacteria | Rhodocyclales | Rhodocyclaceae | 0.57 | 0.73 | 0.27 | −0.73 | 0.70 | 0.76 | ||
| Proteobacteria | Deltaproteobacteria | Desulfuromonadales | 0.62 | 0.74 | 0.11 | −0.76 | 0.71 | 0.73 | |||
| Proteobacteria | Deltaproteobacteria | Myxococcales | Myxococcaceae | 0.83 | 0.60 | 0.29 | −0.94 | 0.82 | 0.75 | ||
| Proteobacteria | Deltaproteobacteria | Myxococcales | Myxococcaceae | Anaeromyxobacter | 0.79 | 0.51 | 0.31 | −0.90 | 0.75 | 0.66 | |
| Verrucomicrobia | 0.17 | 0.78 | 0.16 | −0.28 | 0.49 | 0.66 | |||||
| Verrucomicrobia | Verrucomicrobiae | −0.38 | 0.81 | 0.01 | 0.54 | −0.56 | −0.67 | ||||
| Proteobacteria | Deltaproteobacteria | Desulfuromonadales | 0.62 | 0.74 | 0.11 | −0.76 | 0.71 | 0.73 | |||
| Proteobacteria | Deltaproteobacteria | Desulfuromonadales | Geobacteraceae | Geobacter | G. bemidjiensis | 0.51 | 0.78 | 0.03 | −0.68 | 0.67 | 0.72 |
| Proteobacteria | Deltaproteobacteria | Desulfuromonadales | Geobacteraceae | Geobacter | Geobacter sp. | 0.43 | 0.76 | −0.10 | −0.61 | 0.59 | 0.64 |
| Proteobacteria | Alphaproteobacteria | Caulobacterales | Caulobacteraceae | Asticcacaulis | −0.57 | −0.81 | −0.18 | 0.70 | −0.81 | −0.88 | |
| Proteobacteria | Alphaproteobacteria | Sphingomonadales | −0.19 | −0.75 | 0.16 | 0.25 | −0.26 | −0.37 | |||
| Proteobacteria | Alphaproteobacteria | Sphingomonadales | Sphingomonadaceae | −0.21 | −0.76 | 0.11 | 0.27 | −0.27 | −0.38 | ||
| Proteobacteria | Alphaproteobacteria | Sphingomonadales | Sphingomonadaceae | Sphingomonas | −0.19 | −0.75 | 0.16 | 0.25 | −0.26 | −0.37 | |
| Proteobacteria | Alphaproteobacteria | Sphingomonadales | Sphingomonadaceae | Sphingomonas | Sphingomonas sp. | −0.19 | −0.75 | 0.16 | 0.25 | −0.26 | −0.37 |
| Proteobacteria | Betaproteobacteria | Rhodocyclales | 0.57 | 0.73 | 0.27 | −0.73 | 0.70 | 0.76 | |||
| Proteobacteria | Betaproteobacteria | Rhodocyclales | Rhodocyclaceae | 0.57 | 0.73 | 0.27 | −0.73 | 0.70 | 0.76 | ||
| Firmicutes | 0.50 | 0.73 | 0.31 | −0.56 | 0.77 | 0.85 | |||||
| Firmicutes | Clostridia | Clostridiales | Clostridiaceae | 0.54 | 0.73 | 0.37 | −0.59 | 0.72 | 0.79 | ||
| Firmicutes | Clostridia | Clostridiales | Clostridiaceae | Clostridium | 0.54 | 0.73 | 0.37 | −0.59 | 0.72 | 0.79 | |
| Firmicutes | Clostridia | Clostridiales | Clostridiaceae | Clostridium | C. magnum | −0.50 | −0.76 | −0.22 | 0.63 | −0.76 | −0.87 |
| Bacteroidetes | Sphingobacteriia | Sphingobacteriales | Sphingobacteriaceae | Mucilaginibacter | M. ximonensis | −0.27 | −0.83 | −0.23 | 0.31 | −0.51 | −0.68 |
| Actinobacteria | −0.36 | −0.75 | −0.11 | 0.51 | −0.62 | −0.72 | |||||
| Actinobacteria | Actinobacteria | Actinomycetales | Intrasporangiaceae | −0.05 | −0.76 | −0.18 | 0.15 | −0.29 | −0.48 | ||
| Actinobacteria | Actinobacteria | Actinomycetales | Micrococcaceae | −0.26 | −0.85 | −0.09 | 0.36 | −0.42 | −0.57 | ||
| Actinobacteria | Actinobacteria | Actinomycetales | Geodermatophilaceae | −0.12 | −0.81 | −0.24 | 0.25 | −0.41 | −0.60 | ||
| Actinobacteria | Actinobacteria | Actinomycetales | Streptomycetaceae | Streptomyces | −0.03 | −0.76 | 0.27 | 0.15 | −0.22 | −0.39 | |
| Actinobacteria | Actinobacteria | Actinomycetales | Pseudonocardiaceae | Amycolatopsis | −0.07 | −0.75 | 0.11 | 0.20 | −0.30 | −0.49 |
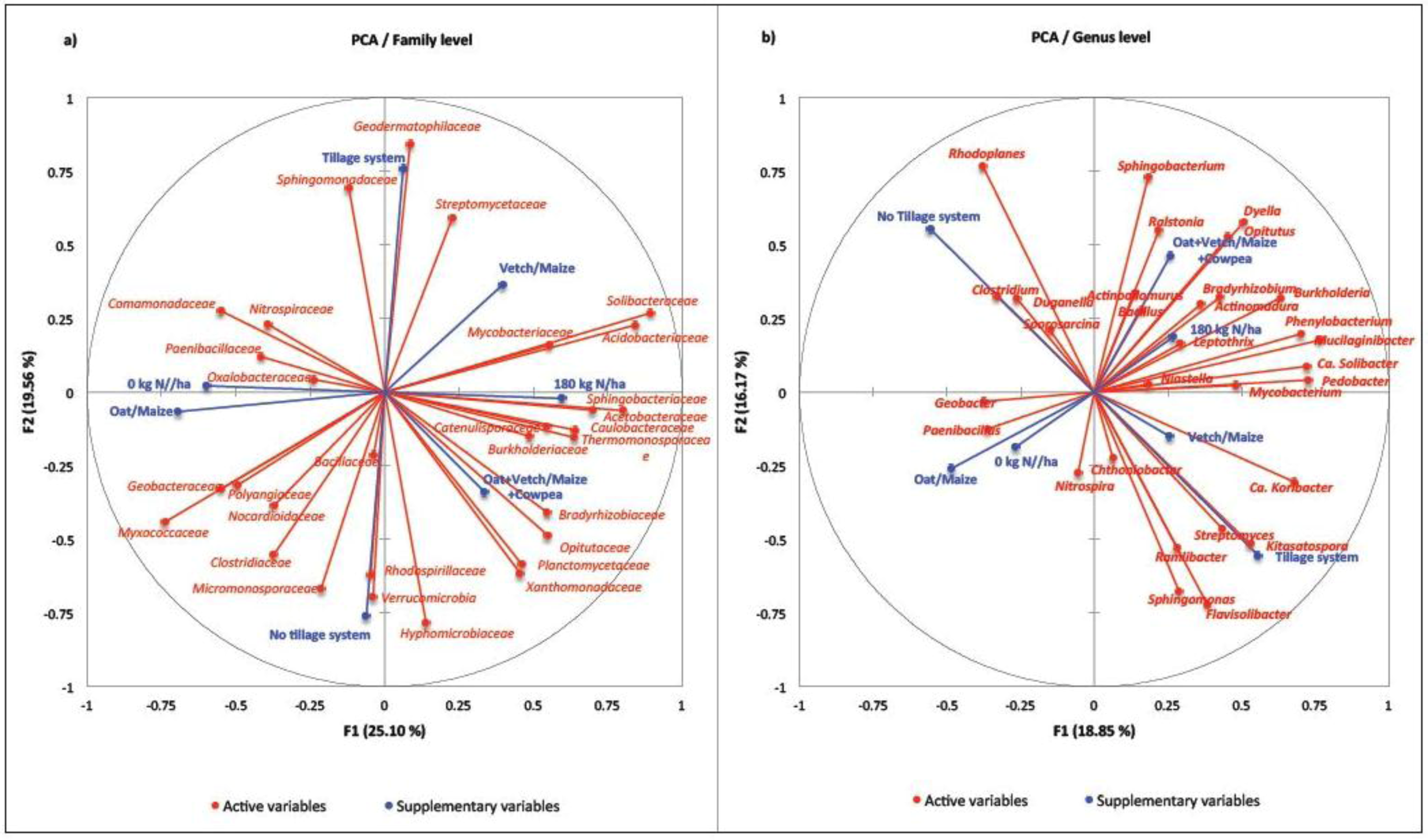
3. Discussion
4. Experimental Section
4.1. Site Description
4.2. Experimental Design and Treatments
4.3. Soil Sampling
4.4. Chemical Analysis
4.5. DNA Extraction and PCR
4.6. Illumina High-Throughput Sequencing of 16S rRNA Genes
4.7. Statistical Analysis
5. Conclusions
Acknowledgments
References
- Lee, K.E. Earthworms: Their Ecology and Relationships with Soil and Land Use; Academic Press: New York, NY, USA, 1985. [Google Scholar]
- Edwards, C.A.; Bohlen, P.J. Biology and ecology of earthworms; Chapman & Hall: London, UK, 1996. [Google Scholar]
- Chan, K.Y. An overview of some tillage impacts on earthworm population abundance and diversity implications for functioning in soils. Soil. Till. Res. 2001, 57, 179–191. [Google Scholar] [CrossRef]
- Fierer, N.; Lauber, C.L.; Ramirez, K.S.; Zaneveld, J.; Bradford, M.A.; Knight, R. Comparative metagenomic, phylogenetic, and physiological analyses of soil microbial communities across nitrogen gradients. ISME J. 2012, 61, 1007–1017. [Google Scholar]
- Hobbs, P.R.; Sayre, K.; Gupta, R. The role of conservation agriculture in sustainable agriculture. Philos. T. Roy. Soc. B 2007, 363, 543–555. [Google Scholar]
- Bayer, C.; Martin-Neto, L.; Mielniczuk, J.; Pavinato, A.; Dieckow, J. Carbon sequestration in two Brazilian Cerrado soils under no-till Soil and Tillage. Soil. Till. Res. 2006, 86, 237–245. [Google Scholar] [CrossRef]
- Omonode, R.A.; Smith, D.R.; Gál, A.; Vyn, T.J. Soil Nitrous Oxide Emissions in Corn following Three Decades of Tillage and Rotation Treatments. Soil. Sci. Soc. Am. J. 2011, 75, 152–163. [Google Scholar]
- Lal, R. Residue Management, Conservation Tillage and Soil Restoration for Mitigating Greenhouse Effect by CO2. Enrichment. Soil. Tillage. Res. 1997, 43, 81–107. [Google Scholar] [CrossRef]
- Campbell, C.A.; McConkey, B.G.; Zentner, R.P.; Dyck, F.B.; Selles, F.; Curtin, D. Tillage and crop rotation effects on soil organic C and N in a coarse- textured Typic Haploboroll in south-western Saskatchewan. Soil. Till. Res. 1996, 37, 3–14. [Google Scholar] [CrossRef]
- Calegari, A.; Hargrove, W.L.; Rheinheimer, D.D.; Ralisch, R.; Tessier, D.; de Tourdonnet, S.; Guimaraes, M.D. Impact of long-term no-tillage and cropping system management on soil organic carbon in an Oxisol: A model for sustainability. Agron. J. 2008, 100, 1013–1019. [Google Scholar] [CrossRef]
- Von Lützow, M.; Leifeld, J.; Kainz, M.; Kögel-Knabner, I.; Munch, J.C. Indications for soil organic matter quality in soils under different management. Geoderma 2002, 105, 243–258. [Google Scholar] [CrossRef]
- Feng, Y.; Motta, A.C.; Reeves, D.W.; Burmester, C.H.; van Santen, E.; Osborne, J.A. Soil microbial communities under conventional-till and no-till continuous cotton systems. Soil. Biol. Biochem. 2003, 35, 1693–1703. [Google Scholar] [CrossRef]
- Derpsch, R.; Friedrich, T.; Kassam, A.; Hongwen, L. Current status of adoption of no-till farming in the world and some of its main benefits. Int. J. Agric. Biol. Eng. 2010, 3, 1–25. [Google Scholar]
- Wang, Y.; Tu, C.; Cheng, L.; Li, C.; Gentry, L.F.; Hoyt, G.D.; Zhang, X.; Hu, S. Long-term impact of farming practices on soil organic carbon and nitrogen pools and microbial biomass and activity. Soil. Till. Res. 2011, 117, 8–16. [Google Scholar] [CrossRef]
- Lu, M.; Yang, Y.; Luo, Y.; Fang, C.; Zhou, X.; Chen, J.; Yang, X.; Li, B. Responses of ecosystem nitrogen cycle to nitrogen addition: a meta-analysis. New. Phytol. 2011, 189, 1040–1050. [Google Scholar] [CrossRef]
- Sarrantonio, M.; Gallandt, E. The role of cover crops in North American cropping systems. J Crop. Prod. 2003, 8, 53–74. [Google Scholar] [CrossRef]
- Fierer, N.; Strickland, M.S.; Liptzin, D.; Bradford, M.A.; Cleveland, C.C. Global patterns in belowground communities. Ecol. Let. 2009, 12, 1–2. [Google Scholar] [CrossRef]
- Ceja-Navarro, J.A.; Rivera, F.N.; Patino-Zuniga, L.; Govaerts, B.; Marsch, R.; Vila-Sanjurjo, A.; Dendooven, L. Molecular characterization of soil bacterial communities in contrasting zero tillage systems. Plant Soil 2010, 329, 127–137. [Google Scholar] [CrossRef]
- Vargas, L.K.; Selbach, P.A.; de Sá, E.L.S. Microbial changes in soil during a maize crop season in no-till and conventional systems. Pesqui. Agropecu. Bras. 2004, 39, 749–755. [Google Scholar]
- Wheatley, R.; Ritz, K.; Griffiths, B. Microbial biomass and mineral N transformations in soil planted with barley, rye cereals, pea or turnip. Plant Soil 1990, 127, 157–167. [Google Scholar] [CrossRef]
- Garcia, F.O.; Rice, C.W. Microbial biomass dynamics in tall cereals prairie. Soil Sci. Soc. Am. Pro. 1994, 58, 816–823. [Google Scholar] [CrossRef]
- Uren, N.C. Types, amounts, and possible function of compounds released into the rizosphere by soil-grown plants. In The rizosphere. Biochemistry and Organic Substances at the Soil-Plant Interface, 2nd; Pinton, R., Varanini, Z., Nannipieri, P., Eds.; CRC Press: New York, NY, USA, 2007. [Google Scholar]
- Badri, D.V.; Vivanco, J.M. Regulation and function of root exudates. Plant Cell Environ. 2009, 32, 666–681. [Google Scholar] [CrossRef]
- Nardi, S.; Concheri, G.; Pizzeghello, D.; Sturaro, A.; Rella, R.; Parvoli, G. Soil organic matter mobilization by root exudates. Chemosphere 2000, 5, 653–658. [Google Scholar]
- Khan, A.R. Influence of Tillage on Soil Aeration. J. Agron. Crop Sci. 1996, 177, 253–259. [Google Scholar] [CrossRef]
- Makarova, K.S.; Omelchenko, M.V.; Gaidamakova, E.K.; Vera, Y.M.; Vasilenko, A.; Zhai, M.; Lapidus, A.; Copeland, A.; Kim, E.; Land, M.; et al. Deinococcus geothermalis: the pool of extreme radiation resistance genes shrinks. PLoS One 2007, 26, e955. [Google Scholar]
- Griffiths, E.; Gupta, R.S. Identification of signature proteins that are distinctive of the Deinococcus-Thermus phylum. Int. Microbiol. 2007, 10, 201–208. [Google Scholar]
- Pukall, R.; Zeytun, A.; Lucas, S.; Lapidus, A.; Hammon, N.; Deshpande, S.; Nolan, M.; Cheng, J.F.; Pitluck, S.; Liolios, K.; et al. Complete genome sequence of Deinococcus maricopensis type strain (LB-34). Stand. Genomic. Sci. 2011, 29, 163–72. [Google Scholar]
- Pagaling, E.; Grant, W.D.; Cowan, D.A.; Jones, B.E.; Ma, Y.; Ventosa, A.; Heaphy, S. Bacterial and archaeal diversity in two hot spring microbial mats from the geothermal region of Tengchong, China. Extremophiles 2012, 16, 617–618. [Google Scholar]
- McLaughlin, R.W.; Chen, M.; Zheng, J.; Zhao, Q.; Wang, D. Analysis of the bacterial diversity in the fecal material of the endangered Yangtze finless porpoise, Neophocaena phocaenoides asiaeorientalis. Mol. Biol. Rev. 2012, 39, 5669–5676. [Google Scholar] [CrossRef]
- Janssen, P.H. Identifying the Dominant Soil Bacterial Taxa in Libraries of 16S rRNA and 16S rRNA Genes. Appl. Environ. Microbiol. 2006, 72, 1719–1728. [Google Scholar] [CrossRef]
- Herrero, A.; Flores, E. The Cyanobacteria: Molecular Biology, Genomics and Evolution; Caister Academic Press: Norfolk, 2008. [Google Scholar]
- Zhalnina, K.; de Quadros, P.D.; Camargo, F.A.O.; Triplett, E.W. Drivers of archaeal ammonia-oxidizing communities in soil. Front. Microb. 2012. [Google Scholar] [CrossRef]
- Ludwig, W.; Klenk, H.P. Overview: A phylogenetic backbone and taxonomic framework for prokaryotic systamatics. In Garrity GM (eds) Bergey’s manual of systematic bacteriology; Brenner, D.J., Krieg, N.R., Staley, J.T., Eds.; Springer-Verlag: Berlin, Germany, 2005. [Google Scholar]
- Bapteste, E.; Brochier, C.; Boucher, Y. Higher-level classification of the Archaea: evolution of methanogenesis and methanogens. Archaea 2005, 1, 353–363. [Google Scholar] [CrossRef]
- Gribaldo, S.; Brochier-Armanet, C. The origin and evolution of Archaea: a state of the art. Philos. Trans. R. Soc. Lond. B. Biol. Sci. 2006, 361, 1007–1022. [Google Scholar] [CrossRef]
- Garrett, R.A.; Klenk, H.P. Archaea: Evolution, Physiology and Molecular Biology; Blackwell Publishing: Oxford, UK, 2006. [Google Scholar]
- Eisen, J.A.; Nelson, K.E.; Paulsen, I.T.; Heidelberg, J.F.; Wu, M.; Dodson, R.J.; Deboy, R.; Gwinn, M.L.; Nelson, W.C.; Haft, D.H.; et al. The complete genome sequence of Chlorobium tepidum TLS, a photosynthetic, anaerobic, green-sulfur bacterium. Proc. Natl. Acad. Sci. USA 2002, 99, 9509–9514. [Google Scholar]
- Li, C.Y.; Hung, L.L. Nitrogen-fixing (acetylene-reducing) bacteria associated with ectomycorrhizae of Douglas-fir. Plant Soil 1987, 98, 425–428. [Google Scholar] [CrossRef]
- Minamisawa, K.; Nishioka, K.; Miyaki, T.; Ye, B.; Miyamoto, T.; You, M.; Saito, A.; Saito, M.; Wilfredo, L.B.; Teaumroong, N.; et al. Anaerobic nitrogen-fixing consortia consisting of Clostridia isolated from gramineous plants. Appl. Environ. Microbiol. 2004, 70, 3096–3102. [Google Scholar]
- Bergmann, G.T.; Bates, S.T.; Eilers, K.G.; Lauber, C.L.; Caporaso, J.G.; Walters, A.W.; Knight, R.; Fierer, N. The under-recognized dominance of Verrucomicrobia in soil bacterial communities. Soil Biol. Biochem. 2011, 43, 1450–1455. [Google Scholar] [CrossRef]
- Haynes, R.J. Effects of lime on phosphate availability in acid soils: a critical review. Plant Soil 1982, 68, 289–308. [Google Scholar] [CrossRef]
- Curtin, D.; Syers, J.K. Lime-Induced Changes in Indices of Soil Phosphate Availability. Soil Sci. Soc. Am. J. 2001, 65, 147–152. [Google Scholar]
- Koch, A.L. Oligotrophs versus copiotrophs. Bioessays 2001, 23, 657–661. [Google Scholar] [CrossRef]
- Kuramae, E.E.; Yergeau, E.; Wong, L.; Pijl, A.; van Veen, J.A.; Kowalchuk, G.A. Soil characteristics more strongly influence soil microbial communities than land management. FEMS Microbiol. Ecol. 2011, 79, 1–271. [Google Scholar]
- Fierer, N.; Bradford, M.; Jackson, R. Toward an ecological classification of soil bacteria. Ecology 2007, 88, 1354–1364. [Google Scholar] [CrossRef]
- Bayer, C.; Martin-Neto, L.; Mielniczuk, J.; Ceretta, C.A. Effect of no-till cropping systems on soil organic matter in a sandy clay loam Acrisol from Southern Brazil monitored by electron spin resonance and nuclear magnetic resonance. Soil. Till. Res. 2000, 53, 95–104. [Google Scholar] [CrossRef]
- Bayer, C.; Martin-Neto, L.; Mielniczuk, J.; Pillon, C.N.; Sangoi, L. Changes in soil organic matter fractions under subtropical No-till Cropping Systems. Soil Sci. Soc. Am. J. 2001, 65, 1473–1478. [Google Scholar]
- Tedesco, M.J.; Gianello, C.; Bissani, C.A.; Bohnen, H.; Volkweiss, S.J. Análises de Solo, Plantas e Outros Materiais; UFRGS: Porto Alegre, Brazil (in Portuguese), 1995. [Google Scholar]
- Page, A.L.; Miller, R.H.; Keeney, D.R. Methods of Soil Analysis, 2nd ed; Soil Science Society of America: Madison, WI, USA, 1982. [Google Scholar]
- Caporaso, J.G.; Lauber, C.L.; Walters, W.A.; Berg-Lyons, D.; Lozupone, C.A.; Turnbaugh, P.J.; Fierer, N.; Knight, R. Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. Proc. Natl. Acad. Sci. USA 2010, 108, 4516–4522. [Google Scholar]
- Giongo, A.; Crabb, D.B.; Davis-Richardson, A.G.; Chauliac, D.; Mobberley, J.M.; Gano, K.; Mukherjee, N.; Casella, G.; Roesch, L.F.; Walts, B.; et al. PANGEA: pipeline for analysis of next generation amplicons. ISME J. 2010, 4, 852–861. [Google Scholar] [CrossRef]
- Fagen, J.R.; Giongo, A.; Brown, C.T.; Davis-Richardson, A.; Gano, K.A.; Triplett, E.W. Characterization of the relative abundance of the citrus pathogen Ca. Liberibacter asiaticus in the microbiome of its insect 525vecor, Diaphorina citri, using high throughput 16S rRNA sequencing. Open Microbiol. J. 2012, 6, 29–33. [Google Scholar] [CrossRef]
- Huang, X.; Wang, J.; Aluru, S.; Yang, S.P.; Hillier, L. PCAP: a whole genome assembly program. Genome. Res. 2003, 13, 2164–2170. [Google Scholar] [CrossRef]
- Trim2_Illumina.pl. Available online: https://gist.github.com/1006830/ (accessed on June 2012).
- Split By Barcode. Available online: https://gist.github.com/1006983/ (accessed on June 2012).
- Cole, J.R.; Wang, O.; Cardenas, E.; Fish, J.; Chai, B.; Farris, R.J.; Kulam-Syed-Mohideen, A.S.; McGarrell, D.M.; Marsh, T.; Garrity, G.M.; et al. The ribosomal database project: improved alignments and new tools for rRNA analysis. Nucleic Acids Res. 2009, 37, 141–145. [Google Scholar]
- National Center for Biotechnology Information–NCBI. Available online: http://www.ncbi.nlm.nih.gov/ (accessed on May 2012).
- Giongo, A.; Davis-Richardson, A.G.; Crabb, D.B.; Triplett, E.W. TaxCollector: tools to modify existing 16S rRNA databases for the rapid classification at six taxonomic levels. Diversity 2010, 2, 1015–1025. [Google Scholar] [CrossRef]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Dorr de Quadros, P.; Zhalnina, K.; Davis-Richardson, A.; Fagen, J.R.; Drew, J.; Bayer, C.; Camargo, F.A.O.; Triplett, E.W. The Effect of Tillage System and Crop Rotation on Soil Microbial Diversity and Composition in a Subtropical Acrisol. Diversity 2012, 4, 375-395. https://doi.org/10.3390/d4040375
Dorr de Quadros P, Zhalnina K, Davis-Richardson A, Fagen JR, Drew J, Bayer C, Camargo FAO, Triplett EW. The Effect of Tillage System and Crop Rotation on Soil Microbial Diversity and Composition in a Subtropical Acrisol. Diversity. 2012; 4(4):375-395. https://doi.org/10.3390/d4040375
Chicago/Turabian StyleDorr de Quadros, Patricia, Kateryna Zhalnina, Austin Davis-Richardson, Jennie R. Fagen, Jennifer Drew, Cimelio Bayer, Flavio A.O. Camargo, and Eric W. Triplett. 2012. "The Effect of Tillage System and Crop Rotation on Soil Microbial Diversity and Composition in a Subtropical Acrisol" Diversity 4, no. 4: 375-395. https://doi.org/10.3390/d4040375
APA StyleDorr de Quadros, P., Zhalnina, K., Davis-Richardson, A., Fagen, J. R., Drew, J., Bayer, C., Camargo, F. A. O., & Triplett, E. W. (2012). The Effect of Tillage System and Crop Rotation on Soil Microbial Diversity and Composition in a Subtropical Acrisol. Diversity, 4(4), 375-395. https://doi.org/10.3390/d4040375