rich: An R Package to Analyse Species Richness

Abstract
: The paper describes rich, a new R package to perform species richness estimation and comparison. Species richness is the simplest surrogate for the more complex concept of species biodiversity. It is relatively easy to assess although estimations strongly depend on sampling intensity with the consequence that richness estimations should be standardized to perform valid comparisons. The R package rich allows such corrections as well as the computation of various statistics and implements different randomization tests to compare cumulative and average species richness of two communities. These tests are useful for ranking sites or communities which is a classical goal in restoration ecology and conservation biology.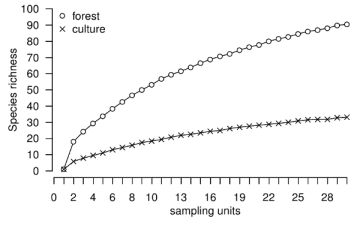
{kind=link}
{kind=link}
{kind=link}
1. Introduction
Species richness is a basic surrogate for the more complex concept of ecological diversity [1]. It is broadly used as a measure of biodiversity with various objectives such as monitoring biodiversity in order to prioritize management or conservation actions [2–4] or design ecological indicators [5,6]. There are many other mathematical indices intended to measure species diversity many of which incorporate species abundance.
However, general agreement among researchers about which index should be used is currently lacking, and this is a major reason for the continued study of species richness in local communities [7,8].
Yet, problems with simple species richness estimation exist [7]. The observed species richness is dependent on sample size and individuals density [9] and this leads to difficulties when comparing communities with, for example, the aim of ranking sites after applying different management options or performing restoration actions. The problem of sample size dependency is broadly acknowledged but the effect of individual density appears perhaps less often considered although it has been very clearly stressed by some authors [9]. The dependence of species richness upon density of sampled individuals is two-fold. First, it requires the use of rarefaction procedures in order to estimate the expected richness of the community with highest density if it was sampled at a density similar to that of the community with lower density [9]. Second, specific statistical tests need to be elaborated in order to compare the former richness estimates while controlling for differences in community densities. The present article focuses on these questions through presenting a new R package entitled rich dedicated to species richness estimation and comparison among communities differing—or not—in terms individuals density.
Comparing measures of species richness is an important task and it is not always straightforward. If the cumulative value (i.e., the overall richness of a set of sampling units) is used to describe a community, only one value is available with no estimate of its dispersion. On the contrary, if one use the average value of the set of sampling units, the resulting mean and standard deviation can be computed. In the latter case, comparing communities can be achieved using standard tests like t-tests or anova tests provided the corresponding assumptions are met. On the other hand, these classical tests cannot be used when the cumulative species richness is considered and other approaches must be used. Evaluating the respective merits of cumulative and average species richness as measures of biodiversity is beyond the scope of this paper. I will here solely mention the statistical tools available in rich to process this type of data.
R is a free statistical software [10] allowing a vast array of data processing and analysis. Various packages are developed by users and constitute a efficient way to spread custom methods and functions. The aim in writing rich was to provide a simple tool for statistical analysis of species richness data. The package provides functions to perform rarefaction [9], bootstrap re-estimation of richness [11] and richness statistical comparison by means of a randomization test [12] with, or without, controlling for differences in individuals density.
2. General Overview of rich
rich comprises different functions that process data in the form of a matrix with species as columns and sampling units as rows. Some of these functions necessitate the R packages boot [13] and vegan [14]. The package also contains different data sets that can be used to illustrate its capacities. For instance the data sets ef and ea describe the abundance of soil macrofauna species in a set of soil samples collected in a secondary forest and a cultivated plot in French Guiana, respectively [15]. These data will be used in the examples examined below.
Rarefaction curves traditionally report the average values of randomized species richness derived from resampling without replacement. A consequence of this is that the variance of the species richness estimation among randomizations decreases with sample size and equals 0 at the right-hand of the curve. As a consequence, such estimations cannot be used to compare different data sets. Sampling with replacement, on the contrary, provides meaningful variance of average species richness and thus allows comparison between different data sets. This corresponds to performing bootstrap on species richness and allows useful additional computations (bias, confidence intervals) [12].
3. Species Richness Computations
The function rich processes a species × sample data matrix and returns various statistics: the bootstrap estimates of species average and cumulative richnesses, their associated bias ([12], p. 36) and confidence interval.
> data(ef)
> test<–rich(matrix=ef, nrandom=499,verbose=TRUE)
> test$cr # observed cumulative species richness [1] 121
> test$mr # observed mean value of species richness over the n samples [1] 10.4
rich returns useful information about rare species: the number of singletons and doubletons (species with at most one or two individual(s), respectively) and the number of uniques and duplicates (species encountered in only one or two sample(s), respectively. rich also returns the total number of zeroes in the raw data table i.e., empty cells, which value is directly related to the difference between the average and the cumulative species richness.
4. Rarefaction
4.1. Rarefaction Curves
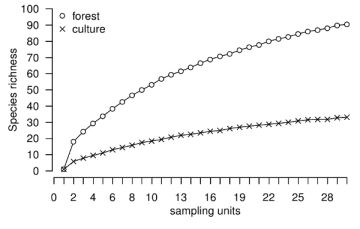
Rarefaction curves are important diagnostic tools that consist of the plot of randomized richness against the sampling intensity. rich operates using resampling with replacement so that the variance among randomizations remains meaningful for large number of sampling units or individuals (i.e., right-hand of rarefaction curves) and thus can be used to compare richnesses. The sampling intensity may be represented by the number of sampling units and may be rescaled to individuals [9]. The function rarc provides such rarefaction data in the form of a data frame which can be used to draw rarefaction curves. Figure 1 illustrates the rarefaction curves based on the number of sampling units (upper panel) and individuals density (lower panel) for species richness of soil macrofauna in two contrasted land-uses, a forest and a cultivated plot in French Guiana (details in [15]). It appears that species richness is higher in the forest. However, because richness depends on the density of the individuals, rarefaction curves may be rescaled to density such as the lower panel in Figure 1. Interestingly, it can be seen that for low densities the cultivated plot might have been considered more diverse than the forest.
4.2. Rarefying Species Richness
The comparison of species richness of 2 communities may be biased if the sampling effort strongly differs. For that reason, some authors (e.g., [9]) recommend that species richness of the community surveyed using the largest number of sampling units (or characterized by the largest individuals density) be rescaled to be comparable to the data set describing the second community. Another strategy implies extrapolation rather than rarefaction [8]. rich comes with a function called raref that interpolates the species richness for a given density on the basis of the rarefaction curve considered above (again, resampling with replacement is used). This is illustrated by analysis the data sets ef and ea to estimate the species richness of soil macrofauna if a forest plot ( ef) for a sampling intensity corresponding to the maximum individuals density recorded in a nearby cultivated plot ( ea) (i.e., 670 individuals). The corresponding richness is estimated as 48 species whereas an overall forest richness of 121 was recorded (see above).
> data (ea) # culture plot
> forest<–raref(matrix=ef, dens=sum(ea), nrandom=500)
> forest$Sinterp[2] [1] 48.09114
A second function, raref2 allows rarefying species richness using bootstrap. For a given density threshold D and a given tolerance t, a subset of the rows (i.e., sampling units) of the community matrix is randomly selected under the constraint that the resulting density is comprised between D − t × D and D + t × D. Using raref2 with the ef data set and a density threshold of d = 670 led to a rarefied richness of 51 species. This method additionally provides a bootstrap standard deviation of the mean richness (10.8 in the example).
> raref2(matrix=ef,dens=sum(ea),tolerance=0.01,nrandom=999)
$mean.boot
[1] 51.13614
$sd.boot
[1] 10.76379
5. Comparing Cumulative Species Richnesses
Comparing cumulative species richnesses is not straightforward because each community is described by only one value and consequently usual statistical tests do not apply. For that reason, published papers often consider average richnesses and this point is discussed below. The test implies evaluating the null hypothesis “there is no difference between observed richnesses but sampling fluctuations” against an alternate hypothesis “richnesses are different”. The nature of the alternate hypothesis will, as usual, determine whether the test is uni or bilateral. rich offers two functions to perform such hypothesis testing.
c2cv is dedicated to simple cases where communities are considered as comparable without rarefaction. Let S1 and S2 be the richness of community 1 and 2. The difference between these values, d = S1 − S2 is computed and compared to n similar differences drand obtained after randomizing samples between communities. The principle of this test is simple and documented in ([12], p. 7). The function c2cv reports a variety of results including the randomized values of d. This test rests on the observed value of d as compared to the quantiles of the corresponding randomized values of a user-fixed probability level. Using the function c2cv with n = 999 randomizations to compare the forest and the cultivated sites examined above leads to an observed difference of 76 species deemed highly significant:
> data(efea)
> c2cv(com1=efea$ef,com2=efea$ea,nrandom=999,verbose=FALSE)
$res
cv1 |
121.000 |
cv2 |
45.000 |
cv1–cv2 |
76.000 |
p |
0.001 |
quantile 0.025 |
−32.025 |
quantile 0.975 |
32.050 |
The function c2rcv operates in a very similar way but incorporates a procedure allowing to control for the effect of differences in density of community 1 and 2. The richness associated to the data matrix with the higher density is first rarefied so as to get an estimate of its richness for a number of individuals comparable to that of the community with lower density. A randomization test similar to the one above is performed on the difference between the observed richness of the community with lower density and the rarefied richness of the community with higher density. Using this function with the example of the forest and the cultivated plots examined above leads to similar conclusion that the forest hosts a higher diversity:
data(efea)
ex<–c2rcv(com1=efea$ef,com2=efea$ea,nrandom=999,tolerance=0.01)
$dmean
[1] 10.35035
$q1 # quantile for p=0.025
[1] −68.05
$q2 #quantile for p=0.975
[1] −4.95
The package rich includes a function c2m which enables the statistical comparison of average species richness over 2 sets of samples. Comparing average species richness may be problematic in cases where standard statistical tests cannot be used safely because some assumptions are violated. This is the case, e.g., for Student t-test when samples have different sample variances. A practical solution is to use randomization tests [12]. The function c2m allows to perform such randomization tests to compare the two sets of sampling units describing the species richness of two communities. If we come back to the example of the species richness in the forest and the cultivated plot, the former appears to have a higher average number of species per sampling unit than the latter:
> data(efea)
> x<–rich(efea$ef,nrandom=50,verbose=TRUE)
> y<–rich(efea$ea,nrandom=50,verbose=TRUE)
> c2m(pop1=x$sumrow,pop2=y$sumrow,
+ nrandom=99,verbose=FALSE)
$res
mv1 |
10.40000000 |
mv2 |
3.46666667 |
mv1–mv2 |
6.93333333 |
p |
0.01000000 |
quantile 0.025 |
−2.06666667 |
quantile 0.975 |
2.27666667 |
7. Discussion and Perspectives
7.1. Rarefaction
There are many indices designed to describe biodiversity but the question of comparing these measures has not received the attention it deserves. The aim in writing rich was to provide a tool to perform simple richness comparisons with randomization procedures. The rarefaction operations were implemented because they are often considered useful when comparing communities sampled with very different intensities. The package provides a function ( raref2) based on a bootstrap procedure to estimate the species richness. Its main interest is that bootstrap additionally provides an estimate of species richness statistical dispersion (i.e., bootstrap standard deviation).
In some cases however, the difference in individuals density is directly reflecting the differences in community structure rather than effects of sampling regimes. In the example of the soil macrofauna richness in the secondary forest and the cultivated plot, strong differences in densities as well as in richness were observed. The first effect of cutting down the forest and settling a cultivated plot is the destruction of many micro-habitats which leads to the decrease of the density of most populations as well as the extinction of numerous species [15]. In this example, it is interesting to consider that for very low densities, the rarefied richness of the forest would appear to be lower than the former which is obviously a wrong conclusion.
Such differences in the rarefaction curves may be explained by very marked differences in species spatial patterning i.e., aggregation and environmental heterogeneity leading to more species being sampled at low sampling intensity in the cultivated plot. The point here is that such different systems as an old secondary tropical forest and a cultivated plot differ by many ecological aspects amongst which the overall organism density and diversity. Controlling for possible confounding density effects may not always be adequate and might even be a confounding operation in itself.
7.2. Average Richness versus Cumulative Richness
rich offers two different ways of comparing the richness associated to two sets of sampling units. These comparisons can be made either on cumulative or on average richnesses. This point is important because differences between communities may depend to a large extent on the the distribution of species among sampling units. In other words, phenomena such as species spatial aggregation or the proportion of rare species may lead to marked differences of our appreciation of the differences in richness of two communities according to the measure considered. The R package rich provides functions to perform such tests and allows to perform rarefaction when using cumulative richness. Future versions of the package may extend the procedure to more than 2 samples comparison and to genetic data so as to allow analyses of allelic richness.
8. Availability
Stable version is available from CRAN: http://cran.r-project.org/mirrors.html. Development version is available from R-Forge https://r-forge.r-project.org/projects/rich/. Both versions can be installed directly from R. rich is distributed under the GNU General Public Licence.

Acknowledgments
The author is grateful to R-Forge for hosting rich and to three anonymous reviewers who provided helpful comments on earlier versions of this note.
References
- Magurran, A. Measuring Biological Diversity; Blackwell Science: Oxford, UK, 2004. [Google Scholar]
- Kerr, J. Species richness, endemism, and the choice of areas for conservation. Conserv. Biol. 1997, 11, 1094–1100. [Google Scholar]
- Caro, T.; O'Doherty, G. On the use of surrogate species in conservation biology. Conserv. Biol. 1999, 13, 805–814. [Google Scholar]
- Mathieu, J.; Rossi, J.P.; Mora, P.; Lavelle, P.; Martins, P.F.D.S.; Rouland, C.; Grimaldi, M. Recovery of soil macrofauna communities after forest clearance in Eastern Amazonia, Brazil. Conserv. Biol. 2005, 19, 1598–1605. [Google Scholar]
- Rossi, J.P.; van Halder, I. Towards indicators of butterfly biodiversity based on a multiscale landscape description. Ecol. Indicat. 2010, 10, 452–458. [Google Scholar]
- Rossi, J.P. Extrapolation and biodiversity indicators: handle with caution! Ecol. Indicat. 2011. [Google Scholar] [CrossRef]
- Hellmann, J.; Fowler, G. Bias, precision, and accuracy of four measures of species richness. Ecol. Appl. 1999, 9, 824–834. [Google Scholar]
- Colwell, R.K.; Coddington, J.A. Estimating Terrestrial Biodiversity through Extrapolation. Phil. Trans.R. Soc. Lond. B 1994, 345, 101–118. [Google Scholar]
- Gotelli, N.; Colwell, R. Quantifying biodiversity: procedures and pitfalls in the measurement and comparison of species richness. Ecol. Lett. 2001, 4, 379–391. [Google Scholar]
- R Development Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2009. [Google Scholar]
- Rossi, J.P.; Mathieu, J.; Cooper, M.; Grimaldi, M. Soil macrofaunal biodiversity in Amazonian pastures: Matching sampling with patterns. Soil Biol. Biochem. 2006, 38, 2178–2187. [Google Scholar]
- Manly, B. Randomization and Monte Carlo Methods in Biology; Chapman & Hall: London, UK, 1997. [Google Scholar]
- Canty, A.; Ripley, B.D. boot: Bootstrap R (S-Plus) Functions; 2009; R package version 1.2-41. [Google Scholar]
- Oksanen, J.; Blanchet, F.G.; Kindt, R.; Legendre, P.; O'Hara, R.B.; Simpson, G.L.; Solymos, P.; Stevens, M.H.H.; Wagner, H. vegan: Community Ecology Package; 2010; R package version 1.17-3. [Google Scholar]
- Rossi, J.P.; Celini, L.; Mora, P.; Mathieu, J.; Lapied, E.; Nahmani, J.; Ponge, J.F.; Lavelle, P. Decreasing fallow duration in tropical slash-and-burn agriculture alters soil macro-invertebrate diversity: A case study in southern French Guiana. Agric. Ecosyst. Environ. 2010, 135, 148–154. [Google Scholar]
© 2011 by the author; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/.)
Share and Cite
Rossi, J.-P. rich: An R Package to Analyse Species Richness. Diversity 2011, 3, 112-120. https://doi.org/10.3390/d3010112
Rossi J-P. rich: An R Package to Analyse Species Richness. Diversity. 2011; 3(1):112-120. https://doi.org/10.3390/d3010112
Chicago/Turabian StyleRossi, Jean-Pierre. 2011. "rich: An R Package to Analyse Species Richness" Diversity 3, no. 1: 112-120. https://doi.org/10.3390/d3010112
APA StyleRossi, J.-P. (2011). rich: An R Package to Analyse Species Richness. Diversity, 3(1), 112-120. https://doi.org/10.3390/d3010112