
Phylogeography of the Endangered Endemic Perkinsiodendron macgregorii Based on Chloroplast Genome Data

Abstract
:1. Introduction
2. Materials and Methods
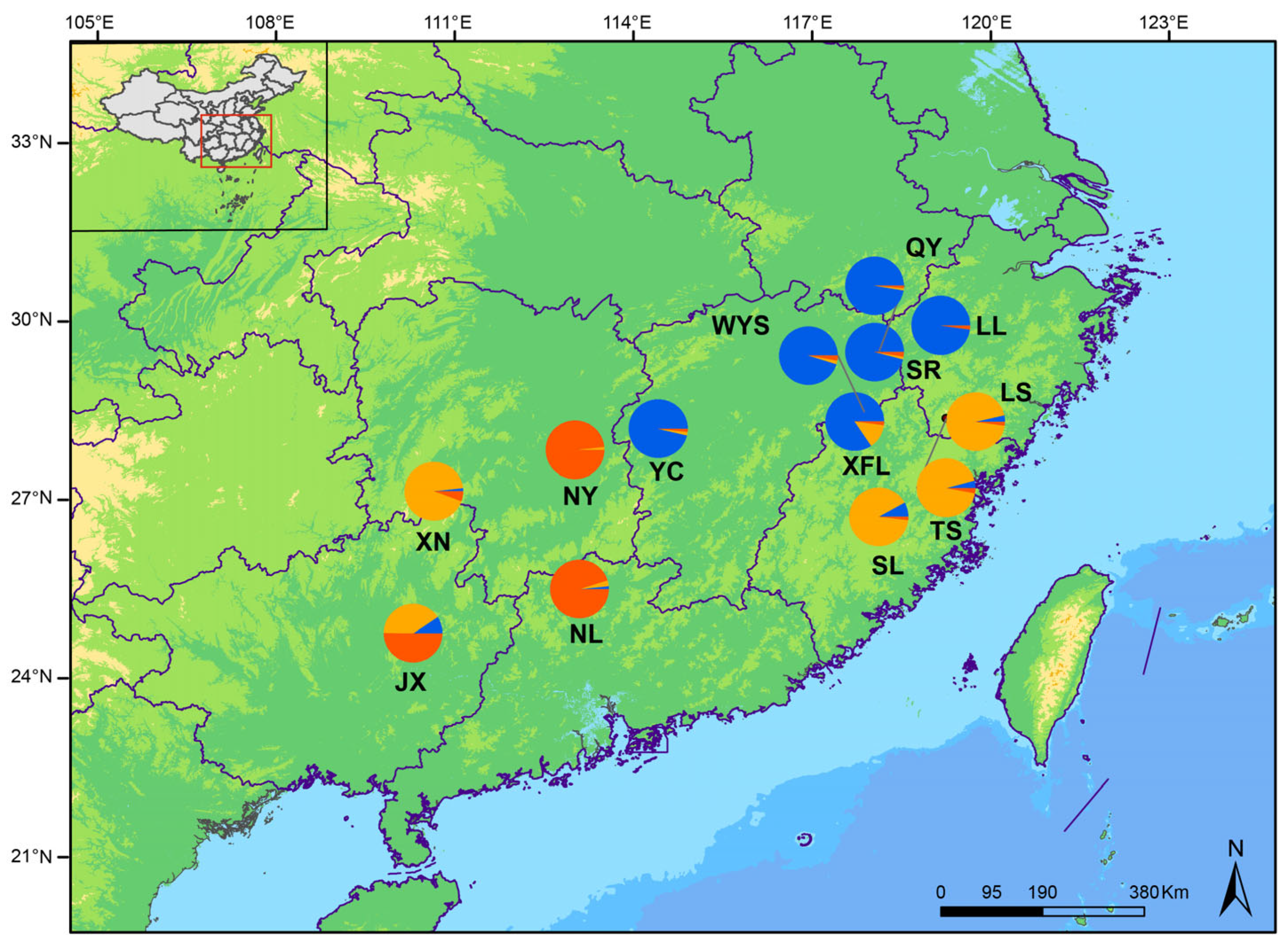
2.1. Sample Collection
2.2. Chloroplast Genome Assembly and Physical Map Construction
2.3. Variant Identification and Statistical Analysis
2.4. Codon Usage Bias and Evolutionary Rate Analysis
2.5. Comparative Analysis of Chloroplast Genome Structure in P. macgregorii Populations
2.6. Phylogenetic and Population Structure Analysis
3. Results
3.1. Structural Features of 134 Chloroplast Genomes in P. macgregorii
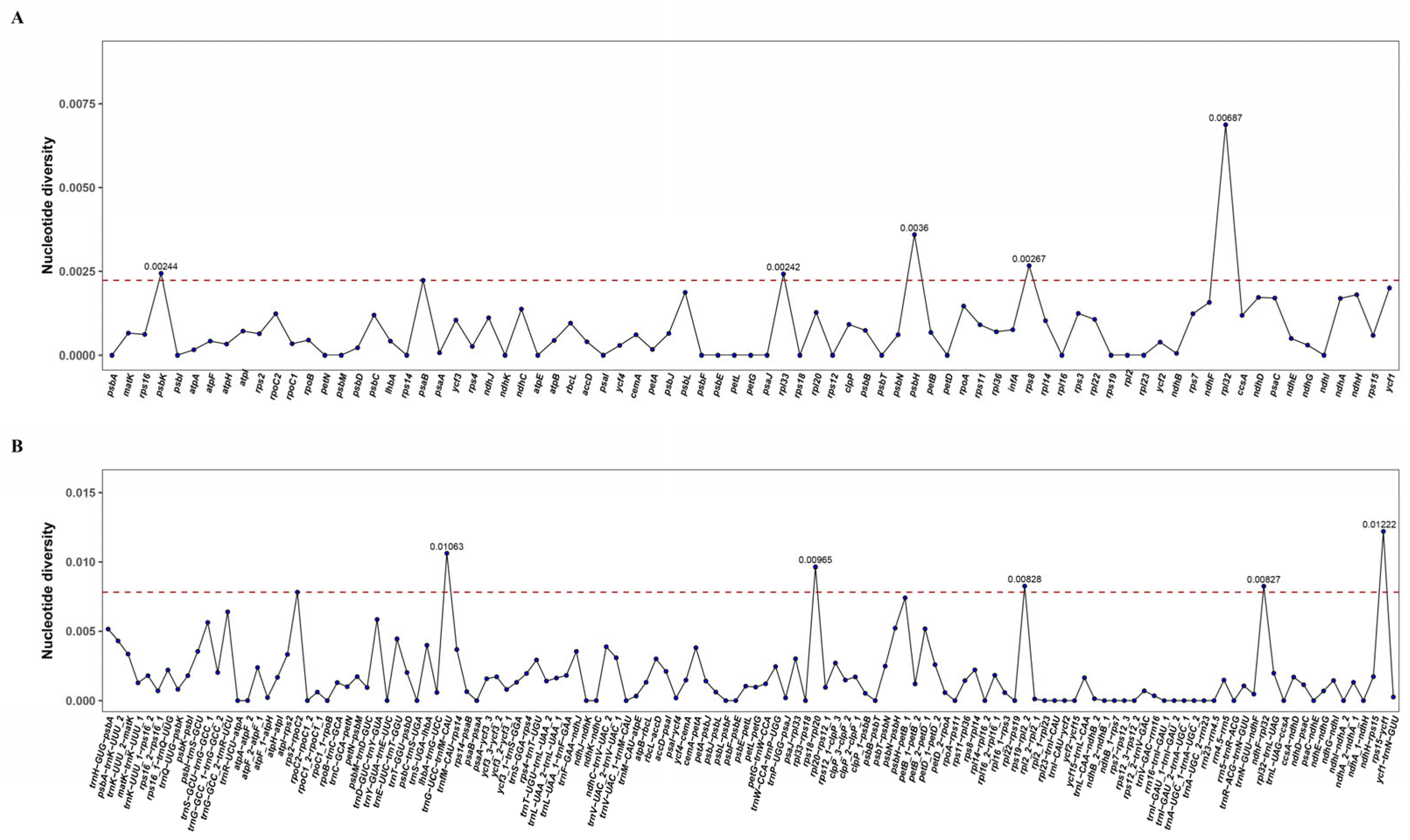
3.2. Chloroplast Genome Variation in P. macgregorii
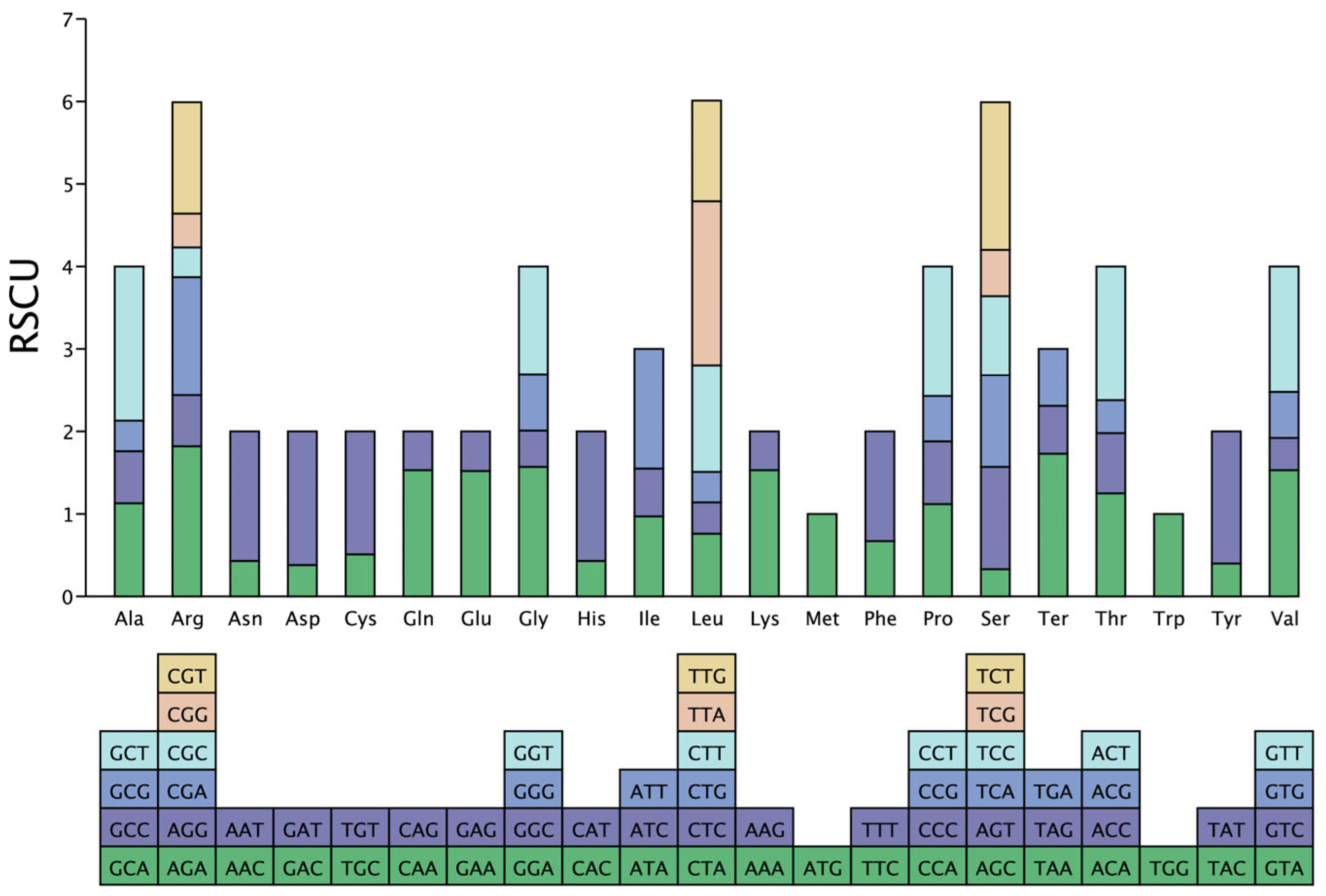
3.3. Codon Usage Bias in P. macgregorii Chloroplast Genomes
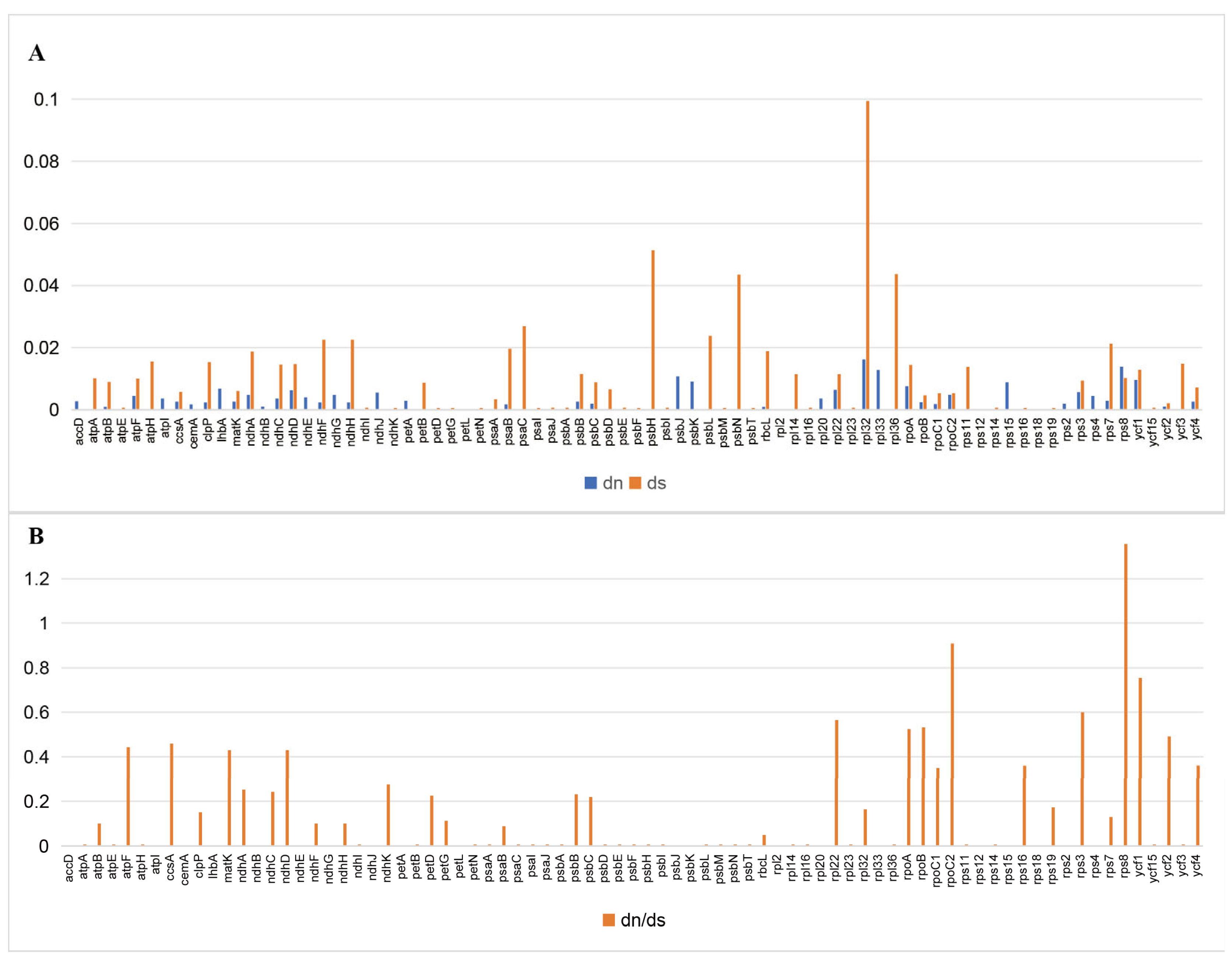
3.4. Adaptive Evolution of Chloroplast Protein-Coding Genes
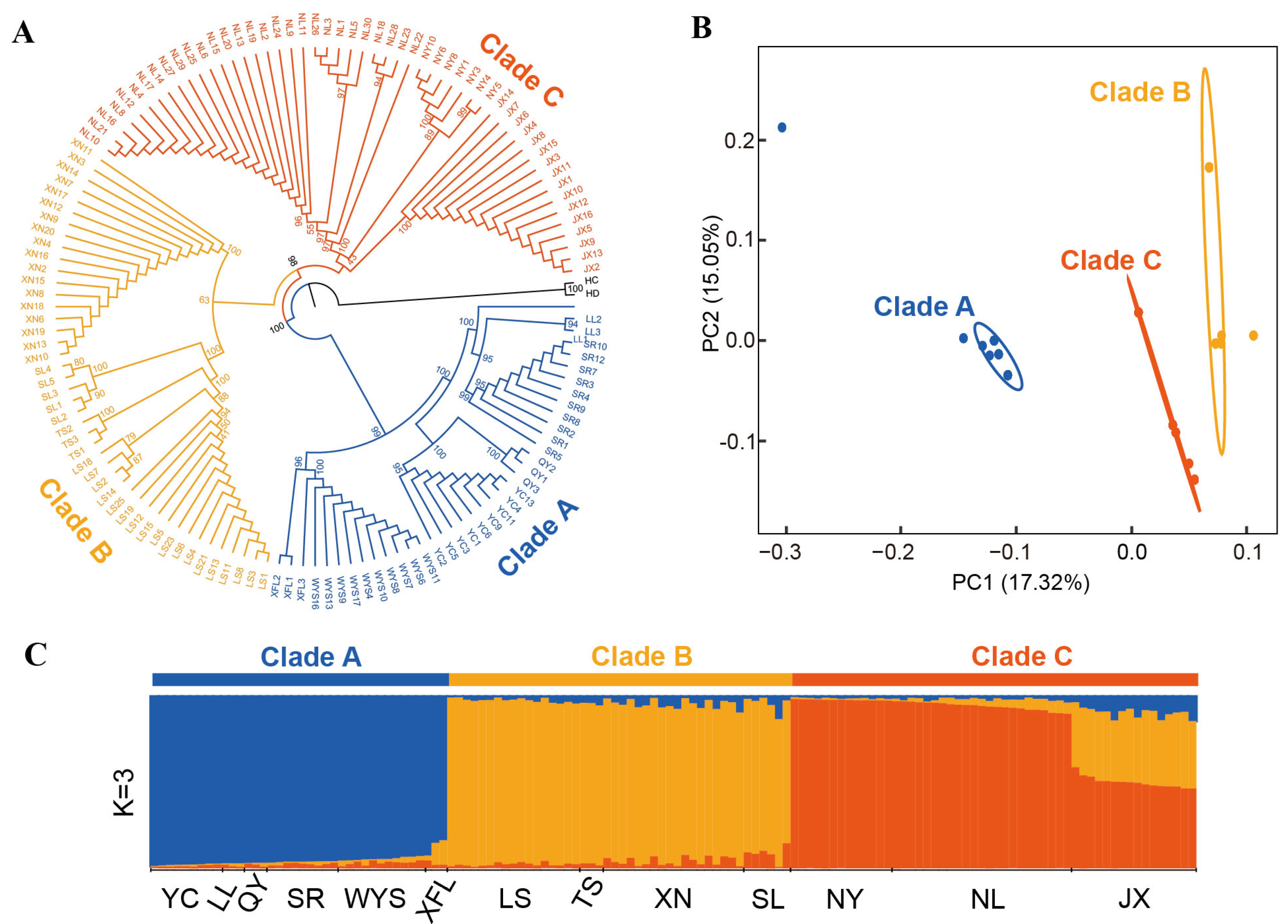
3.5. Phylogenetic Relationships and Population Structure of P. macgregorii Populations
3.6. Genetic Diversity and Differentiation Among Clades
4. Discussion
4.1. Genetic Diversity in P. macgregorii Chloroplast Genome
4.2. P. macgregorii Genetic Divergence and Diversity
4.3. Genetic Conservation Strategies
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Institutional Review Board Statement
Conflicts of Interest
References
- Fritsch, P. Styracaceae. In Flowering Plants·Dicotyledons: Celastrales, Oxalidales, Rosales, Cornales, Ericales; Springer: Berlin/Heidelberg, Germany, 2004; pp. 434–442. [Google Scholar]
- Chase, M.W.; Christenhusz, M.J.M.; Fay, M.F.; Byng, J.W.; Judd, W.S.; Soltis, D.E.; Mabberley, D.J.; Sennikov, A.N.; Soltis, P.S.; Stevens, P.F. An update of the Angiosperm Phylogeny Group classification for the orders and families of flowering plants: APG IV. Bot. J. Linn. Soc. 2016, 181, 1–20. [Google Scholar] [CrossRef]
- Fritsch, P.W.; Morton, C.M.; Chen, T.; Meldrum, C. Phylogeny and biogeography of the Styracaceae. Int. J. Plant Sci. 2001, 162, S95–S116. [Google Scholar] [CrossRef]
- Fritsch, P.W.; Yao, X.; Simison, W.B.; Cruz, B.C.; Chen, T. Perkinsiodendron, a new genus in the Styracaceae based on morphology and DNA sequences. J. Bot. Res. Inst. Tex. 2016, 10, 109–117. [Google Scholar]
- Yan, M.; Fritsch, P.W.; Moore, M.J.; Feng, T.; Meng, A.; Yang, J.; Deng, T.; Zhao, C.; Yao, X.; Sun, H.; et al. Plastid phylogenomics resolves infrafamilial relationships of the Styracaceae and sheds light on the backbone relationships of the Ericales. Mol. Phylogenet. Evol. 2018, 121, 198–211. [Google Scholar] [CrossRef]
- Fu, L. Plant China Plant Red Data Book: Rare and Endangered Plants; Science Publishing House Press: Beijing, China, 1992; p. 741. [Google Scholar]
- Wang, L.; Wu, X.; Li, Y.; Xu, X. Prediction of suitable cultivation area for Halesia carolina L. in China. J. Nanjing For. Univ. 2018, 61, 10–16. [Google Scholar] [CrossRef]
- Guo, Y.; Yang, J.; Bai, M.; Zhang, G.; Liu, Z. The chloroplast genome evolution of Venus slipper (Paphiopedilum): IR expansion, SSC contraction, and highly rearranged SSC regions. BMC Plant Biol. 2021, 21, 248. [Google Scholar] [CrossRef]
- Cauz-Santos, L.A.; da Costa, Z.P.; Sader, M.A.; van den Berg, C.; Vieira, M.L.C. Chloroplast genomic insights into adaptive evolution and rapid radiation in the genus Passiflora (Passifloraceae). BMC Plant Biol. 2025, 25, 192. [Google Scholar] [CrossRef]
- Wang, J.; He, W.; Xiang, K.; Wu, Z.; Gu, C. Advances in plant phylogeny in the genome era. J. Zhejiang A&F Univ. 2023, 40, 227–236. [Google Scholar] [CrossRef]
- Zhu, S.; Liu, Q.; Qiu, S.; Dai, J.; Gao, X. DNA barcoding: An efficient technology to authenticate plant species of traditional Chinese medicine and recent advances. Chin. Med. 2022, 17, 112. [Google Scholar] [CrossRef]
- Gao, J.; Tomlinson, K.W.; Zhao, W.; Wang, B.; Lapuz, R.S.; Liu, J.X.; Pasion, B.O.; Hai, B.T.; Chanthayod, S.; Chen, J.; et al. Phylogeography and introgression between Pinus kesiya and Pinus yunnanensis in Southeast Asia. J. Syst. Evol. 2024, 62, 120–134. [Google Scholar] [CrossRef]
- Jiang, X.L.; Xu, G.; Deng, M. Spatial genetic patterns and distribution dynamics of the rare oak Quercus chungii: Implications for biodiversity conservation in Southeast China. Forests 2019, 10, 821. [Google Scholar] [CrossRef]
- Moner, A.M.; Furtado, A.; Henry, R.J. Two divergent chloroplast genome sequence clades captured in the domesticated rice gene pool may have significance for rice production. BMC Plant Biol. 2020, 20, 472. [Google Scholar] [CrossRef] [PubMed]
- Drouin, G.; Daoud, H.; Xia, J. Relative rates of synonymous substitutions in the mitochondrial, chloroplast and nuclear genomes of seed plants. Mol. Phylogenet. Evol. 2008, 49, 827–831. [Google Scholar] [CrossRef] [PubMed]
- Dong, W.; Xu, C.; Wu, P.; Cheng, T.; Yu, J.; Zhou, S.; Hong, D.Y. Resolving the systematic positions of enigmatic taxa: Manipulating the chloroplast genome data of Saxifragales. Mol. Phylogenet. Evol. 2018, 126, 321–330. [Google Scholar] [CrossRef]
- Dong, W.; Xu, C.; Wen, J.; Zhou, S. Evolutionary directions of single nucleotide substitutions and structural mutations in the chloroplast genomes of the family Calycanthaceae. BMC Evol. Biol. 2020, 20, 96. [Google Scholar] [CrossRef]
- Dong, W.; Liu, Y.; Li, E.; Xu, C.; Sun, J.; Li, W.; Zhou, S.; Zhang, Z.; Suo, Z. Phylogenomics and biogeography of Catalpa (Bignoniaceae) reveal incomplete lineage sorting and three dispersal events. Mol. Phylogenet. Evol. 2022, 166, 107330. [Google Scholar] [CrossRef]
- Shang, C.; Li, E.; Yu, Z.; Lian, M.; Chen, Z.; Liu, K.; Xu, L.; Tong, Z.; Wang, M.; Dong, W. Chloroplast genomic resources and genetic divergence of endangered species Bretschneidera sinensis (Bretschneideraceae). Front. Ecol. Evol. 2022, 10, 873100. [Google Scholar] [CrossRef]
- Torre, S.; Sebastiani, F.; Burbui, G.; Pecori, F.; Pepori, A.L.; Passeri, I.; Ghelardini, L.; Selvaggi, A.; Santini, A. Novel insights into refugia at the southern margin of the distribution range of the endangered species Ulmus laevis. Front. Plant Sci. 2022, 13, 826158. [Google Scholar] [CrossRef]
- Sun, J.; Wang, S.; Wang, Y.; Wang, R.; Liu, K.; Li, E.; Qiao, P.; Shi, L.; Dong, W.; Huang, L.; et al. Phylogenomics and genetic diversity of Arnebiae radix and its allies (Arnebia, Boraginaceae) in China. Front. Plant Sci. 2022, 13, 920826. [Google Scholar] [CrossRef]
- Xiao, S.; Xu, P.; Deng, Y.; Dai, X.; Zhao, L.; Heider, B.; Zhang, A.; Zhou, Z.; Cao, Q. Comparative analysis of chloroplast genomes of cultivars and wild species of sweetpotato (Ipomoea batatas [L.] Lam). BMC Genom. 2021, 22, 1–12. [Google Scholar] [CrossRef]
- Guo, Q.; Xue, X.; Wang, D.; Zhang, L.; Liu, W.; Wang, E.; Cui, X.; Hou, X. Genetic diversity and population genetic structure of Paeonia suffruticosa by chloroplast DNA simple sequence repeats (cpSSRs). Hort. Plant J. 2025, 11, 367–376. [Google Scholar] [CrossRef]
- Yang, R.; Feng, X.; Gong, X. Genetic structure and demographic history of Cycas chenii (Cycadaceae), an endangered species with extremely small populations. Plant Divers. 2017, 39, 44–51. [Google Scholar] [CrossRef] [PubMed]
- Jin, J.; Yu, W.; Yang, J.; Song, Y.; DePamphilis, C.W.; Yi, T.; Li, D. GetOrganelle: A fast and versatile toolkit for accurate de novo assembly of organelle genomes. Genome Biol. 2020, 21, 1–31. [Google Scholar] [CrossRef] [PubMed]
- Danecek, P.; Bonfield, J.K.; Liddle, J.; Marshall, J.; Ohan, V.; Pollard, M.O.; Whitwham, A.; Keane, T.; McCarthy, S.A.; Davies, R.M.; et al. Twelve years of SAMtools and BCFtools. Gigascience 2021, 10, giab008. [Google Scholar] [CrossRef]
- Huang, L.; Yu, H.; Wang, Z.; Xu, W. CPStools: A package for analyzing chloroplast genome sequences. iMetaOmics 2024, 1, e25. [Google Scholar] [CrossRef]
- Shi, L.; Chen, H.; Jiang, M.; Wang, L.; Wu, X.; Huang, L.; Liu, C. CPGAVAS2, an integrated plastome sequence annotator and analyzer. Nucleic Acids Res. 2019, 47, W65–W73. [Google Scholar] [CrossRef]
- Lowe, T.M.; Eddy, S.R. tRNAscan-SE: A program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997, 25, 955–964. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Rambaut, A. Se-Al: Sequence Alignment Editor. 1996. Available online: http://tree.bio.ed.ac.uk/software/seal/ (accessed on 6 February 2025).
- Rozas, J.; Ferrer-Mata, A.; Sánchez-DelBarrio, J.C.; Guirao-Rico, S.; Librado, P.; Ramos-Onsins, S.E.; Sánchez-Gracia, A. DnaSP 6: DNA sequence polymorphism analysis of large data sets. Mol. Biol. Evol. 2017, 34, 3299–3302. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef]
- Yang, Z. PAML 4: Phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 2007, 24, 1586–1591. [Google Scholar] [CrossRef]
- Peden, J.F. Analysis of Codon Usage. Ph.D. Thesis, University of Nottingham, Nottingham, UK, 2000. [Google Scholar]
- Amiryousefi, A.; Hyvönen, J.; Poczai, P. IRscope: An online program to visualize the junction sites of chloroplast genomes. Bioinformatics 2018, 34, 3030–3031. [Google Scholar] [CrossRef]
- Frazer, K.A.; Pachter, L.; Poliakov, A.; Rubin, E.M.; Dubchak, I. VISTA: Computational tools for comparative genomics. Nucleic Acids Res. 2004, 32, W273–W279. [Google Scholar] [CrossRef]
- Xia, E.H.; Li, F.D.; Tong, W.; Li, P.H.; Wu, Q.; Zhao, H.J.; Ge, R.H.; Li, R.P.; Li, Y.Y.; Zhang, Z.Z.; et al. Tea plant information archive: A comprehensive genomics and bioinformatics platform for tea plant. Plant Biotechnol. J. 2019, 17, 1938–1953. [Google Scholar] [CrossRef]
- Ronquist, F.; Teslenko, M.; Van Der Mark, P.; Ayres, D.L.; Darling, A.; Höhna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. MrBayes 3.2: Efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef]
- Leigh, J.W.; Bryant, D.; Nakagawa, S. POPART: Full-feature software for haplotype network construction. Methods Ecol. Evol. 2015, 6, 1110–1116. [Google Scholar] [CrossRef]
- Falush, D.; Stephens, M.; Pritchard, J.K. Inference of population structure using multilocus genotype data: Dominant markers and null alleles. Mol. Ecol. Notes 2007, 7, 574–578. [Google Scholar] [CrossRef]
- Chang, C.C.; Chow, C.C.; Tellier, L.C.; Vattikuti, S.; Purcell, S.M.; Lee, J.J. Second-generation PLINK: Rising to the challenge of larger and richer datasets. Gigascience 2015, 4, s13742-015-0047-8. [Google Scholar] [CrossRef]
- Excoffier, L.; Lischer, H.E. Arlequin suite ver 3.5: A new series of programs to perform population genetics analyses under Linux and Windows. Mol. Ecol. Resour. 2010, 10, 564–567. [Google Scholar] [CrossRef]
- Kavaliauskas, D.; Danusevičius, D.; Baliuckas, V. New insight into genetic structure and diversity of Scots pine (Pinus sylvestris L.) populations in Lithuania based on nuclear, chloroplast and mitochondrial DNA markers. Forests 2022, 13, 1179. [Google Scholar] [CrossRef]
- Huang, X.; Coulibaly, D.; Tan, W.; Ni, Z.; Shi, T.; Li, H.; Hayat, F.; Gao, Z. The analysis of genetic structure and characteristics of the chloroplast genome in different Japanese apricot germplasm populations. BMC Plant Biol. 2022, 22, 354. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Wang, Y.; Qiao, P.; Zhang, L.; Li, E.; Dong, W.; Zhao, Y.; Huang, L. Pueraria montana population structure and genetic diversity based on chloroplast genome data. Plants 2023, 12, 2231. [Google Scholar] [CrossRef] [PubMed]
- Yao, R.; Wang, B.; Heinrich, M.; Wang, Q.; Xiao, P. Genetic diversity of food-medicinal Lycium spp. in China: Insights from chloroplast genome. Chin. Herb. Med. 2024, 16, 401–411. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Wang, Y.; Garran, T.A.; Qiao, P.; Wang, M.; Yuan, Q.; Guo, L.; Huang, L. Heterogeneous genetic diversity estimation of a promising domestication medicinal motherwort Leonurus cardiaca based on chloroplast genome resources. Front. Genet. 2021, 12, 721022. [Google Scholar] [CrossRef]
- Xu, J.; Zang, F.; Wu, Q.; Wang, Y.; Wang, B.; Huang, P.; Zang, D.; Ma, Y.; Zheng, Y. Analysis of the genetic diversity and molecular phylogeography of the endangered wild rose (Rosa rugosa) in China based on chloroplast genes. Glob. Ecol. Conserv. 2021, 28, e01653. [Google Scholar] [CrossRef]
- Casazza, G.; Grassi, F.; Zecca, G.; Minuto, L. Phylogeographic insights into a peripheral refugium: The importance of cumulative effect of glaciation on the genetic structure of two endemic plants. PLoS ONE 2016, 11, e0166983. [Google Scholar] [CrossRef]
- Zhang, X.; Liu, Y.H.; Wang, Y.H.; Shen, S.K. Genetic diversity and population structure of Rhododendron rex Subsp. rex inferred from microsatellite markers and chloroplast DNA sequences. Plants 2020, 9, 338. [Google Scholar] [CrossRef]
- Li, Q.; Guo, X.; Niu, J.; Duojie, D.; Li, X.; Opgenoorth, L.; Zou, J. Molecular phylogeography and evolutionary history of the endemic species Corydalis hendersonii (Papaveraceae) on the Tibetan Plateau inferred from chloroplast DNA and ITS sequence variation. Front. Plant Sci. 2020, 11, 436. [Google Scholar] [CrossRef]
- Ma, X.; Tang, M.; Bi, Y.; Yang, J. Genetic diversity and population structure analysis of wild Cymbidium tortisepalum based on chloroplast DNA in Yunnan Province of China. J. Amer. Soc. Hort. Sci. 2021, 146, 424–434. [Google Scholar] [CrossRef]
- Xiao, X.; Chen, J.; Ran, Z.; Huang, L.; Li, Z. Comparative Analysis of Complete Chloroplast Genomes and Phylogenetic Relationships of 21 Sect. Camellia (Camellia, L.) Plants. Genes 2025, 16, 49. [Google Scholar] [CrossRef]
- Stronen, A.V.; Norman, A.J.; Vander Wal, E.; Paquet, P.C. The relevance of genetic structure in ecotype designation and conservation management. Evol. Appl. 2022, 15, 185–202. [Google Scholar] [CrossRef]
- Teske, D.; Peters, A.; Mollers, A.; Fischer, M. Genomic profiling: The strengths and limitations of chloroplast genome-based plant variety authentication. J. Agric. Food Chem. 2020, 68, 14323–14333. [Google Scholar] [CrossRef]
- Robles, P.; Quesada, V. Unveiling the functions of plastid ribosomal proteins in plant development and abiotic stress tolerance. Plant Physiol. Biochem. 2022, 189, 35–45. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, A.; Li, X.; Lu, C. The role of chloroplast gene expression in plant responses to environmental stress. Int. J. Mol. Sci. 2020, 21, 6082. [Google Scholar] [CrossRef]
- Jia, X.; Feng, S.; Zhang, H.; Liu, X. Plastome phylogenomics provide insight into the evolution of Taxus. Forests 2022, 13, 1590. [Google Scholar] [CrossRef]
- Kim, K.-R.; Park, S.Y.; Kim, H.; Hong, J.M.; Kim, S.-Y.; Yu, J.-N. Complete chloroplast genome determination of Ranunculus sceleratus from Republic of Korea (Ranunculaceae) and comparative chloroplast genomes of the members of the Ranunculus Genus. Genes 2023, 14, 1149. [Google Scholar] [CrossRef]
- Ehlers, J.; Gibbard, P.; Hughes, P. Quaternary glaciations and chronology. In Past Glacial Environments, 2nd ed.; Elsevier: Amsterdam, The Netherlands, 2018; pp. 77–101. [Google Scholar] [CrossRef]
- Fu, J.; Wen, L. Impacts of Quaternary glaciation, geological history and geography on animal species history in continental East Asia: A phylogeographic review. Mol. Ecol. 2023, 32, 4497–4514. [Google Scholar] [CrossRef]
- Tang, C.Q.; Matsui, T.; Ohashi, H.; Dong, Y.F.; Momohara, A.; Herrando-Moraira, S.; Qian, S.; Yang, Y.; Ohsawa, M.; Luu, H.T.; et al. Identifying long-term stable refugia for relict plant species in East Asia. Nat. Commun. 2018, 9, 4488. [Google Scholar] [CrossRef]
- Fan, D.M.; Yue, J.P.; Nie, Z.L.; Li, Z.M.; Comes, H.P.; Sun, H. Phylogeography of Sophora davidii (Leguminosae) across the ‘Tanaka-Kaiyong Line’, an important phytogeographic boundary in Southwest China. Mol. Ecol. 2013, 22, 4270–4288. [Google Scholar] [CrossRef]
- Provan, J.; Bennett, K.D. Phylogeographic insights into cryptic glacial refugia. Trends. Ecol. Evol. 2008, 23, 564–571. [Google Scholar] [CrossRef]
- Luo, D.; Xiao, Y.; Zhan, L.; Zhang, L. The tectogenesis control of Danxia landform in southeast China. J. East China Inst. Technol. 2010, 33, 147–153. [Google Scholar] [CrossRef]
- Jiang, C.; Wu, M.; He, S.; Lu, Y.; Zhao, C. Phylogeography of a herbal Pinellia ternata reveals repeated range expansions and inter/postglacial recolonization routes on the fragmented distribution pattern in China. Ecol. Evol. 2024, 14, e70206. [Google Scholar] [CrossRef]
- Qiu, Y.X.; Fu, C.X.; Comes, H.P. Plant molecular phylogeography in China and adjacent regions: Tracing the genetic imprints of Quaternary climate and environmental change in the world’s most diverse temperate flora. Mol. Phylogenet. Evol. 2011, 59, 225–244. [Google Scholar] [CrossRef]
- Liu, W.; Xie, J.; Zhou, H.; Kong, H.; Hao, G.; Fritsch, P.W.; Gong, W. Population dynamics linked to glacial cycles in Cercis chuniana FP Metcalf (Fabaceae) endemic to the montane regions of subtropical China. Evol. Appl. 2021, 14, 2647–2663. [Google Scholar] [CrossRef]
- Li, N.; Wang, Z.; Deng, Q.; Wang, T.; Su, Y.J. Effects of climatic fluctuations on the fragmented distribution pattern of a Tertiary relict plant, Pseudotaxus chienii (Taxaceae), in subtropical China. Bot. J. Linn. Soc. 2024, 205, 55–74. [Google Scholar] [CrossRef]
- Du, S.; Wang, Z.; Ingvarsson, P.K.; Wang, D.; Wang, J.; Wu, Z.; Tembrock, L.R.; Zhang, J. Multilocus analysis of nucleotide variation and speciation in three closely related Populus (S alicaceae) species. Mol. Ecol. 2015, 24, 4994–5005. [Google Scholar] [CrossRef]
- Li, Y.; Liu, C.; Wang, R.; Luo, S.; Nong, S.; Wang, J.; Chen, X. Applications of molecular markers in conserving endangered species. Biodivers. Sci. 2020, 28, 367. [Google Scholar] [CrossRef]
- Zhao, Y.P.; Fan, G.; Yin, P.P.; Sun, S.; Li, N.; Hong, X.; Hu, G.; Zhang, H.; Zhang, F.M.; Han, J.D.; et al. Resequencing 545 ginkgo genomes across the world reveals the evolutionary history of the living fossil. Nat. Commun. 2019, 10, 4201. [Google Scholar] [CrossRef]
- Wang, C.; Wang, M.; Yang, S.; Wu, X.; Zhu, S.; Yan, Y.; Xu, J.; Wen, Y. Taxonomic status and spatial genetic pattern of Taxus in northern and central China: Insights from integrative taxonomy, ecology and phylogeography. BMC Plant Biol. 2025, 25, 181. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Location | Polymorphic Sites | Singleton Variable Sites | Parsimony Informative Sites | Indels | Nucleotide Diversity | SNP Density/kb |
|---|---|---|---|---|---|---|
| IR | 38 | 0 | 38 | 12 | 0.00031 | 1.93 |
| LSC | 508 | 2 | 506 | 497 | 0.00137 | 11.36 |
| SSC | 157 | 0 | 157 | 131 | 0.00201 | 15.7 |
| Total | 741 | 2 | 739 | 678 | 0.00109 | 8.45 |
| Gene | Protein | Length bp | Nn | Sn | SNP | Density of SNPs/kb |
|---|---|---|---|---|---|---|
| rpl32 | ribosomal protein L32 | 159 | 2 | 3 | 5 | 31.45 |
| rps8 | ribosomal protein S8 | 402 | 4 | 1 | 5 | 12.44 |
| rpl33 | ribosomal protein L33 | 204 | 0 | 2 | 2 | 9.80 |
| ycf1 | hypothetical chloroplast RF19 | 5541 | 12 | 40 | 52 | 9.38 |
| psbH | photosystem II phosphoprotein | 219 | 0 | 2 | 2 | 9.13 |
| rpoA | RNA polymerase alpha subunit | 1023 | 6 | 3 | 9 | 8.80 |
| ndhD | NADH-plastoquinone oxidoreductase subunit 4 | 1500 | 6 | 5 | 11 | 7.33 |
| ndhH | NADH-plastoquinone oxidoreductase subunit 7 | 1179 | 2 | 6 | 8 | 6.79 |
| rps7 | ribosomal protein S7 | 465 | 1 | 2 | 3 | 6.45 |
| ndhA | NADH-plastoquinone oxidoreductase subunit 1 | 1089 | 4 | 3 | 7 | 6.43 |
| Group | N | S | H | Hd | Pi | Tajima’s D | Fu’s Fs |
|---|---|---|---|---|---|---|---|
| Clade A | 38 | 292 | 7 | 0.79943 | 0.00062 | 1.53146 | 2.56614 |
| Clade B | 44 | 241 | 7 | 0.73150 | 0.00068 | 3.43261 * | 2.77411 |
| Clade C | 52 | 229 | 7 | 0.74811 | 0.00061 | 3.25077 * | 7.84627 |
| Group | Accessions | All Variations | |||
|---|---|---|---|---|---|
| SNPs | Indels | Total | Density/kb | ||
| Clade A | 38 | 292 | 375 | 667 | 4.19 |
| Clade B | 44 | 241 | 192 | 433 | 2.72 |
| Clade C | 52 | 229 | 242 | 471 | 2.96 |
| Total | 134 | 741 | 678 | 1571 | 9.88 |
| Comparison | DA | FST |
|---|---|---|
| Clade A vs. Clade B | 0.00067 | 0.16234 |
| Clade A vs. Clade C | 0.00072 | 0.19900 |
| Clade B vs. Clade C | 0.00065 | 0.18308 |
| Source of Variation | Percentage of Variation | |
|---|---|---|
| AMOVA | Among clades | 51.19 |
| Within clades | 48.81 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yan, M.-H.; Du, Y.-R.; Zhao, J.-Y.; Xu, K.-X.; Zhao, L.; Su, J.-M.; Yan, L.-M. Phylogeography of the Endangered Endemic Perkinsiodendron macgregorii Based on Chloroplast Genome Data. Diversity 2025, 17, 439. https://doi.org/10.3390/d17070439
Yan M-H, Du Y-R, Zhao J-Y, Xu K-X, Zhao L, Su J-M, Yan L-M. Phylogeography of the Endangered Endemic Perkinsiodendron macgregorii Based on Chloroplast Genome Data. Diversity. 2025; 17(7):439. https://doi.org/10.3390/d17070439
Chicago/Turabian StyleYan, Ming-Hui, Yan-Rong Du, Jia-Yi Zhao, Ke-Xin Xu, Lu Zhao, Jia-Meng Su, and Lu-Miao Yan. 2025. "Phylogeography of the Endangered Endemic Perkinsiodendron macgregorii Based on Chloroplast Genome Data" Diversity 17, no. 7: 439. https://doi.org/10.3390/d17070439
APA StyleYan, M.-H., Du, Y.-R., Zhao, J.-Y., Xu, K.-X., Zhao, L., Su, J.-M., & Yan, L.-M. (2025). Phylogeography of the Endangered Endemic Perkinsiodendron macgregorii Based on Chloroplast Genome Data. Diversity, 17(7), 439. https://doi.org/10.3390/d17070439