


Complete Mitochondrial Genome of the Backswimmer: Notonecta triguttata Motschulsky, 1861 (Hemiptera: Notonectidae): Sequence, Structure, and Phylogenetic Analysis

Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Collection, DNA Extraction, and Sequencing
2.2. Sequence Assembly, Annotation, and Analysis
2.3. Gene Arrangement and Phylogenetic Analysis
3. Results and Discussion
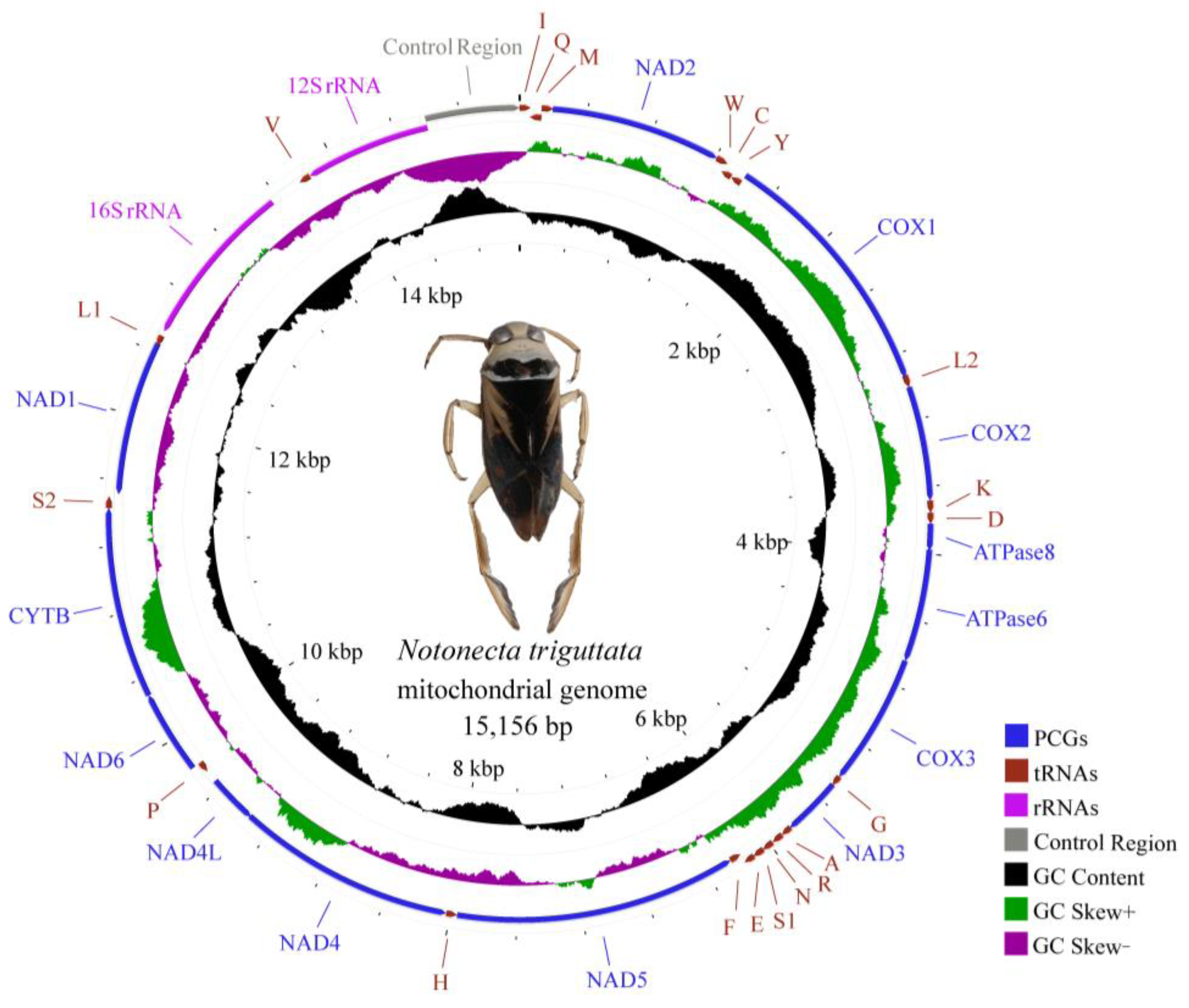
3.1. Genome Organization
3.2. Nucleotide Composition and Codon Usage
3.3. Protein-Coding Genes
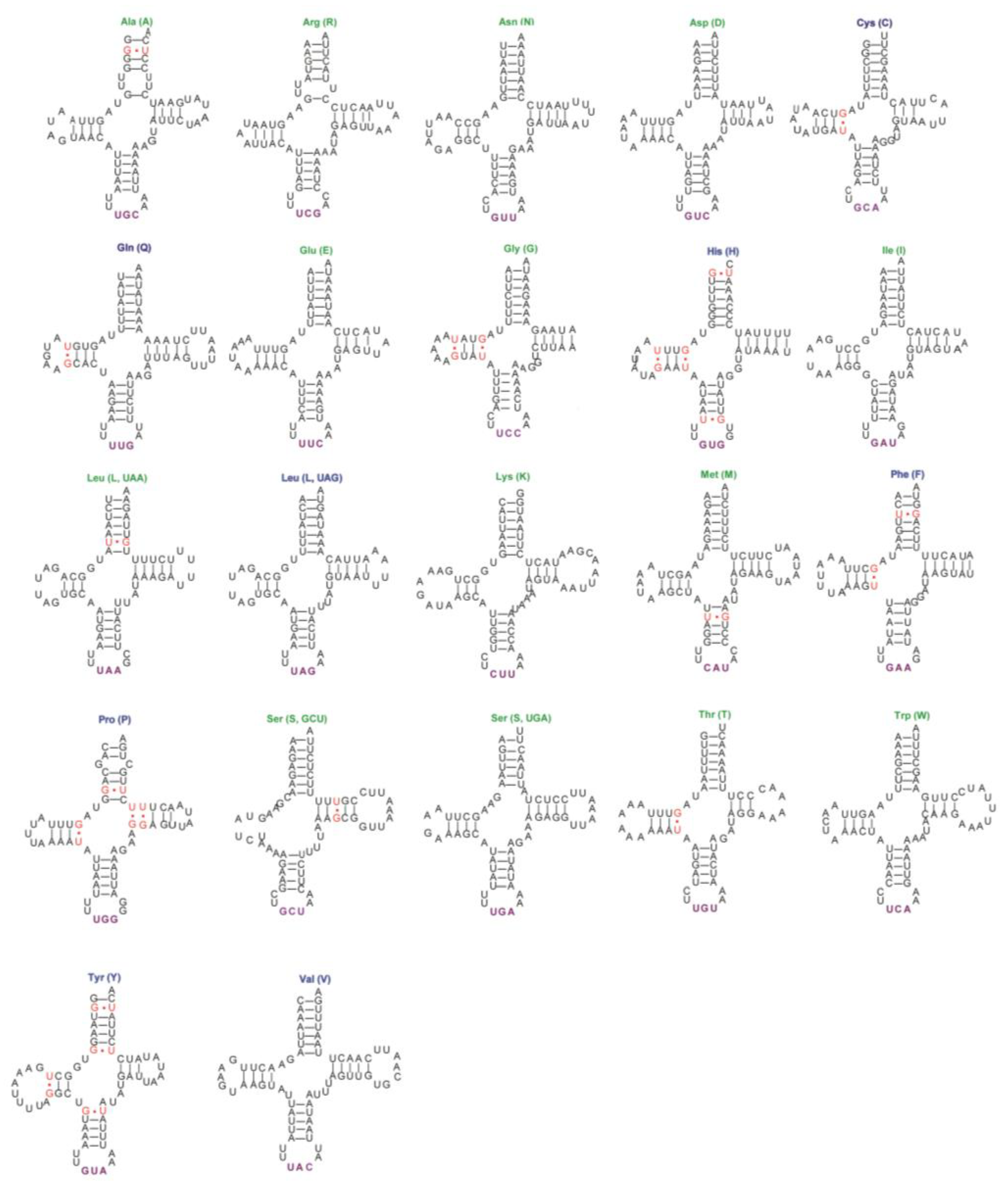
3.4. Transfer, Ribosomal RNA Genes and Control Regions
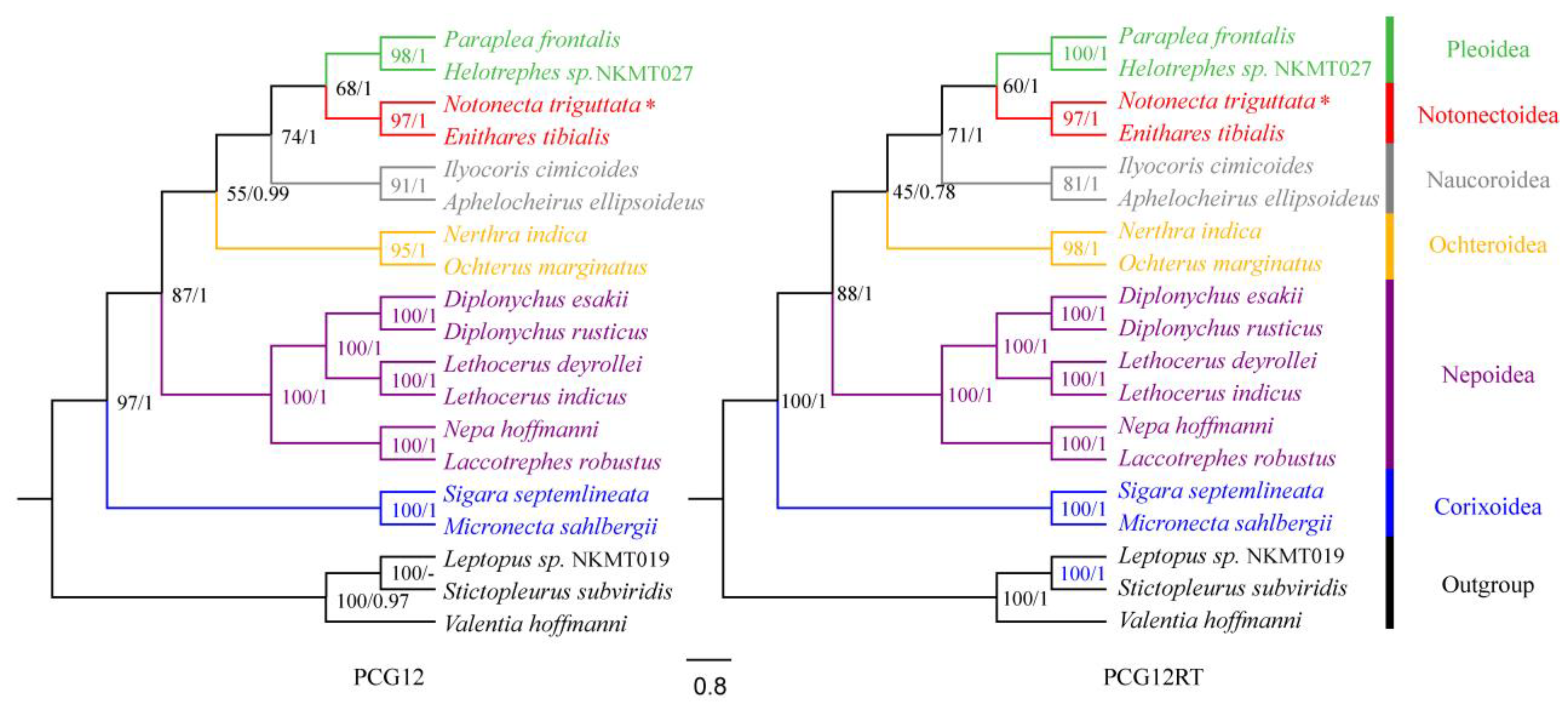
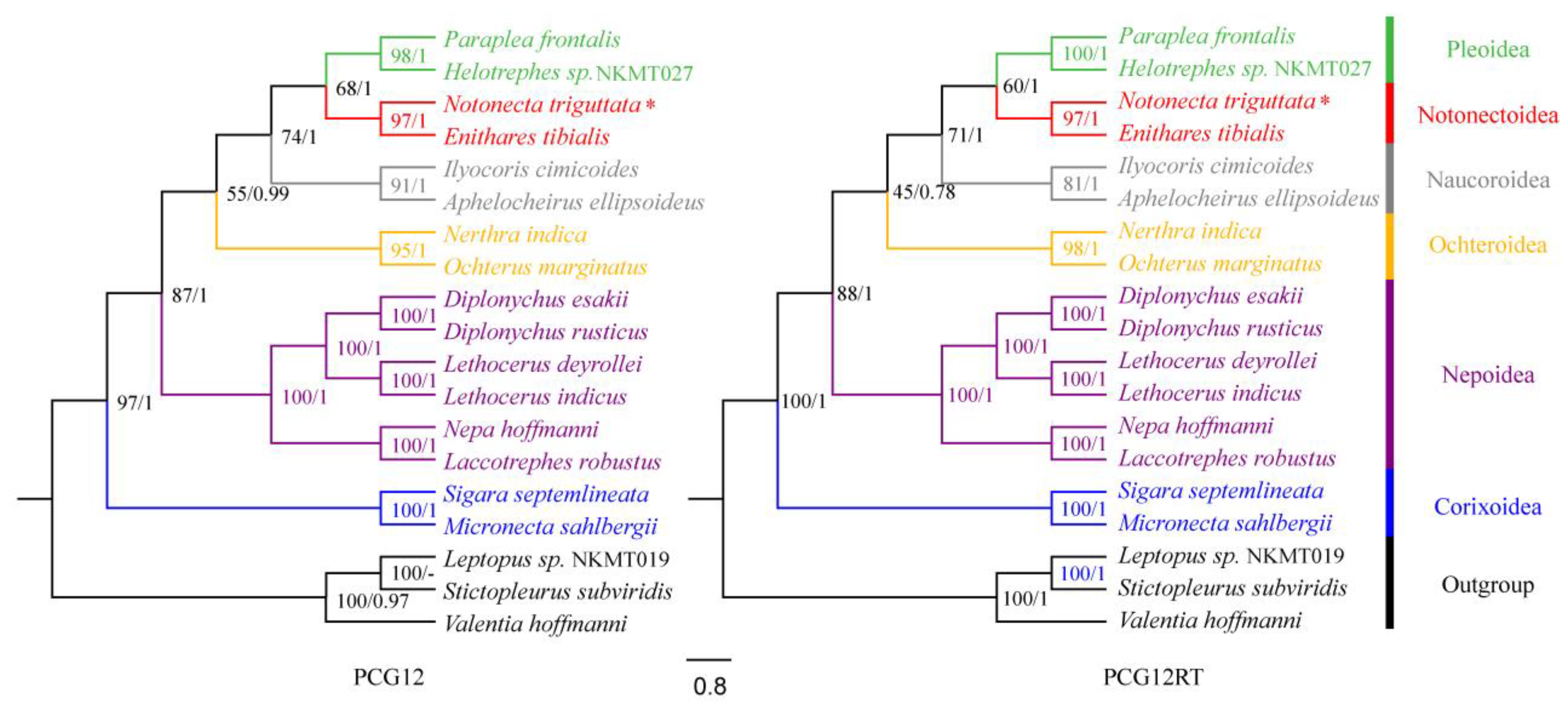
3.5. Gene Arrangement and Phylogenetic Analysis
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Simon, C.; Buckley, T.R.; Frati, F.; Stewart, J.B.; Beckenbach, A.T. Incorporating molecular evolution into phylogenetic analysis, and a new compilation of conserved polymerase chain reaction primers for animal mitochondrial DNA. Annu. Rev. Ecol. Evol. Syst. 2006, 37, 545–579. [Google Scholar] [CrossRef]
- Bourguignon, T.; Lo, N.; Cameron, S.L.; Sobotnik, J.; Hayashi, Y.; Shigenobu, S.; Watanabe, D.; Roisin, Y.; Miura, T.; Evans, T.A. The evolutionary history of termites as inferred from 66 mitochondrial genomes. Mol. Biol. Evol. 2014, 32, 406–421. [Google Scholar] [CrossRef] [PubMed]
- Cameron, S.L. Insect mitochondrial genomics: Implications for evolution and phylogeny. Annu. Rev. Entomol. 2014, 59, 95–117. [Google Scholar] [CrossRef] [PubMed]
- Polhemus, J.T. Hemiptera (True Bugs). In Encyclopedia of Inland Waters; Likens, G.E., Ed.; Academic Press: Cambridge, MA, USA, 2009; pp. 323–334. [Google Scholar]
- Latreille, P.A.; Buffon, G.L.L.; Sonnini, C.S.; Sève, J.E.d. Histoire Naturelle, Générale et Particulière des Crustacés et des Insectes. In Crustacés et des Insectes; Dufart: Paris, France, 1802; Volume 3, p. 468. [Google Scholar]
- Schuh, R.T.; Slater, J.A. True Bugs of the World (Hemiptera: Heteroptera): Classification and Natural History; Cornell University Press: Ithaca, NY, USA; London, UK, 1995; p. xii + 336. [Google Scholar]
- Polhemus, J.T.; Polhemus, D.A. Global diversity of true bugs (Heteroptera; Insecta) in freshwater. Hydrobiologia 2007, 595, 379–391. [Google Scholar] [CrossRef]
- Shaalan, E.A.; Canyon, D.V. Aquatic insect predators and mosquito control. Trop. Biomed. 2009, 26, 223–261. [Google Scholar] [PubMed]
- Fischer, S.; Pereyra, D.; Fernandez, L. Predation ability and non-consumptive effects of Notonecta sellata (Heteroptera: Notonectidae) on immature stages of Culex pipiens (Diptera: Culicidae). J. Vector Ecol. 2012, 37, 245–251. [Google Scholar] [CrossRef] [PubMed]
- Dalal, A.; Cuthbert, R.N.; Dick, J.T.A.; Gupta, S. Water depth-dependent notonectid predatory impacts across larval mosquito ontogeny. Pest Manag. Sci. 2019, 75, 2610–2617. [Google Scholar] [CrossRef]
- Dambach, P. The use of aquatic predators for larval control of mosquito disease vectors: Opportunities and limitations. Biol. Control 2020, 150, 104357. [Google Scholar] [CrossRef]
- Hungerford, H.B. The genus Notonecta of the world (Notonectidae-Hemiptera). Univ. Kans. Sci. Bull. 1933, 21, 5–195. [Google Scholar]
- Lim, J.S.; Lee, B.W.; Park, S.Y.; Jo, D.G. Insect fauna of Maebongsan Mountain, Hongcheon-gun, Gangwon-do. J. Korean Nat. 2011, 4, 293–307. [Google Scholar] [CrossRef]
- Ohba, S. Feeding habits of the diving beetle larvae, Cybister brevis Aubé (Coleoptera: Dytiscidae) in Japanese wetlands. Appl. Entomol. Zool. 2009, 44, 447–453. [Google Scholar] [CrossRef]
- Ohba, S. Ontogenetic dietary shift in the larvae of Cybister japonicus (Coleoptera: Dytiscidae) in Japanese rice fields. Environ. Entomol. 2009, 38, 856–860. [Google Scholar] [CrossRef] [PubMed]
- Hirayama, H.; Kasuya, E. Effect of adult females’ predation risk on oviposition site selection in a water strider. Entomol. Exp. Appl. 2013, 149, 250–255. [Google Scholar] [CrossRef]
- Kobashi, K.; Harada, T.; Adachi, Y.; Mori, M.; Ihara, M.; Hayasaka, D. Comparative ecotoxicity of imidacloprid and dinotefuran to aquatic insects in rice mesocosms. Ecotoxicol. Environ. Saf. 2017, 138, 122–129. [Google Scholar] [CrossRef] [PubMed]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Coil, D.; Jospin, G.; Darling, A.E. A5-miseq: An updated pipeline to assemble microbial genomes from Illumina MiSeq data. Bioinformatics 2015, 31, 587–589. [Google Scholar] [CrossRef]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef]
- Marcais, G.; Delcher, A.L.; Phillippy, A.M.; Coston, R.; Salzberg, S.L.; Zimin, A. MUMmer4: A fast and versatile genome alignment system. PLoS Comput. Biol. 2018, 14, e1005944. [Google Scholar] [CrossRef]
- Walker, B.J.; Abeel, T.; Shea, T.; Priest, M.; Abouelliel, A.; Sakthikumar, S.; Cuomo, C.A.; Zeng, Q.; Wortman, J.; Young, S.K.; et al. Pilon: An integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS ONE 2014, 9, e112963. [Google Scholar] [CrossRef]
- Bernt, M.; Donath, A.; Juhling, F.; Externbrink, F.; Florentz, C.; Fritzsch, G.; Putz, J.; Middendorf, M.; Stadler, P.F. MITOS: Improved de novo metazoan mitochondrial genome annotation. Mol. Phylogenet. Evol. 2013, 69, 313–319. [Google Scholar] [CrossRef]
- Grant, J.R.; Stothard, P. The CGView Server: A comparative genomics tool for circular genomes. Nucleic Acids Res. 2008, 36, W181–W184. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Stecher, G.; Kumar, S. MEGA11: Molecular evolutionary genetics analysis version 11. Mol. Biol. Evol. 2021, 38, 3022–3027. [Google Scholar] [CrossRef] [PubMed]
- Xiang, C.Y.; Gao, F.L.; Jakovlić, I.; Lei, H.P.; Hu, Y.; Zhang, H.; Zou, H.; Wang, G.T.; Zhang, D. Using PhyloSuite for molecular phylogeny and tree-based analyses. iMeta 2023, 2, e87. [Google Scholar] [CrossRef]
- Perna, N.T.; Kocher, T.D. Patterns of nucleotide composition at fourfold degenerate sites of animal mitochondrial genomes. J. Mol. Evol. 1995, 41, 353–358. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.L.; Li, M.; Li, T.; Yuan, J.J.; Bu, W.J. A mitochondrial genome of Micronectidae and implications for its phylogenetic position. Int. J. Biol. Macromol. 2018, 119, 747–757. [Google Scholar] [CrossRef]
- Ronquist, F.; Teslenko, M.; van der Mark, P.; Ayres, D.L.; Darling, A.; Hohna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. MrBayes 3.2: Efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef]
- Lanfear, R.; Frandsen, P.B.; Wright, A.M.; Senfeld, T.; Calcott, B. PartitionFinder 2: New methods for selecting partitioned models of evolution for molecular and morphological mhylogenetic analyses. Mol. Biol. Evol. 2017, 34, 772–773. [Google Scholar] [CrossRef]
- Nguyen, L.T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef]
- Li, H.; Liu, H.; Song, F.; Shi, A.; Zhou, X.; Cai, W. Comparative mitogenomic analysis of damsel bugs representing three tribes in the family Nabidae (Insecta: Hemiptera). PLoS ONE 2012, 7, e45925. [Google Scholar] [CrossRef]
- Li, T.; Yang, J.; Li, Y.W.; Cui, Y.; Xie, Q.; Bu, W.J.; Hillis, D.M. A mitochondrial genome of Rhyparochromidae (Hemiptera: Heteroptera) and a comparative analysis of related mitochondrial genomes. Sci. Rep. 2016, 6, 35175. [Google Scholar] [CrossRef]
- Men, Y.; Kment, P.; Stahlavsky, F.; Ye, F.; Wang, Y.H.; Xie, Q. The mitochondrial genomes of Macrocheraia grandis grandis and Myrmoplasta mira and the unique mitogenome rearrangement in Pyrrhocoroidea. Entomotaxonomia 2019, 41, 96–113. [Google Scholar]
- Zhao, W.Q.; Zhao, Q.; Li, M.; Wei, J.F.; Zhang, X.H.; Zhang, H.F. Comparative mitogenomic analysis of the Eurydema genus in the context of representative Pentatomidae (Hemiptera: Heteroptera) taxa. J. Insect Sci. 2019, 19, 20. [Google Scholar] [CrossRef] [PubMed]
- Xia, X.H. Genomic Features: Content Sensors, Nucleotide Skew Plot, Strand Asymmetry, and DNA Methylation. In Bioinformatics and the Cell; Springer: Berlin/Heidelberg, Germany, 2018; pp. 255–268. [Google Scholar]
- Hassanin, A.; Leger, N.; Deutsch, J. Evidence for multiple reversals of asymmetric mutational constraints during the evolution of the mitochondrial genome of metazoa, and consequences for phylogenetic inferences. Syst. Biol. 2005, 54, 277–298. [Google Scholar] [CrossRef] [PubMed]
- Parvathy, S.T.; Udayasuriyan, V.; Bhadana, V. Codon usage bias. Mol. Biol. Rep. 2022, 49, 539–565. [Google Scholar] [CrossRef]
- Ojala, D.; Montoya, J.; Attardi, G. tRNA punctuation model of RNA processing in human mitochondria. Nature 1981, 290, 470–474. [Google Scholar] [CrossRef]
- Zhang, D.; Xu, L.; Wang, S.; Liang, J.; Li, M.; Zhang, H. The first complete mitochondrial genome of Dufouriellini (Hemiptera: Anthocoridae) and implications for its phylogenetic position. Arch. Insect Biochem. Physiol. 2022, 111, e21885. [Google Scholar] [CrossRef]
- Zhu, W.; Yang, L.; Long, J.; Chang, Z.; Gong, N.; Mu, Y.; Lv, S.; Chen, X. Characterizing the Complete Mitochondrial Genomes of Three Bugs (Hemiptera: Heteroptera) Harming Bamboo. Genes 2023, 14, 342. [Google Scholar] [CrossRef]
- Li, T.; Hua, J.M.; Wright, A.M.; Cui, Y.; Xie, Q.; Bu, W.J.; Hillis, D.M. Long-branch attraction and the phylogeny of true water bugs (Hemiptera: Nepomorpha) as estimated from mitochondrial genomes. BMC Evol. Biol. 2014, 14, 99. [Google Scholar] [CrossRef]
- Hebsgaard, M.B.; Andersen, N.M.; Damgaard, J. Phylogeny of the true water bugs (Nepomorpha: Hemiptera–Heteroptera) based on 16S and 28S rDNA and morphology. Syst. Entomol. 2004, 29, 488–508. [Google Scholar] [CrossRef]
- Li, M.; Tian, Y.; Zhao, Y.; Bu, W. Higher level phylogeny and the first divergence time estimation of Heteroptera (Insecta: Hemiptera) based on multiple genes. PLoS ONE 2012, 7, e32152. [Google Scholar] [CrossRef]
- Ye, Z.; Damgaard, J.; Yang, H.H.; Hebsgaard, M.B.; Weir, T.; Bu, W.J. Phylogeny and diversification of the true water bugs (Insecta: Hemiptera: Heteroptera: Nepomorpha). Cladistics 2020, 36, 72–87. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Infraorder | Superfamily | Family | Species | Accession Number |
|---|---|---|---|---|
| Leptopodomorpha | Leptopodoidea | Leptopodidae | Leptopus sp. NKMT019 | FJ456946 |
| Cimicomorpha | Reduvioidea | Reduviidae | Valentia hoffmanni | FJ456952 |
| Pentatomomorpha | Coreoidea | Rhopalidae | Stictopleurus subviridis | EU826088 |
| Nepomorpha | Nepoidea | Belostomatidae | Diplonychus esakii | MK251109 |
| Diplonychus rusticus | FJ456940 | |||
| Lethocerus deyrollei | KU237288 | |||
| Lethocerus indicus | NC_027194 | |||
| Nepidae | Nepa hoffmanni | NC_028084 | ||
| Laccotrephes robustus | FJ456948 | |||
| Corixoidea | Corixidae | Sigara septemlineata | FJ456941 | |
| Micronectidae | Micronecta sahlbergii | NC_039588 | ||
| Ochteroidea | Gelastocoridae | Nerthra indica | NC_012838 | |
| Ochteridae | Ochterus marginatus | FJ456950 | ||
| Pleoidea | Pleidae | Paraplea frontalis | NC_028629 | |
| Helotrephidae | Helotrephes sp. NKMT027 | NC_012822 | ||
| Notonectoidea | Notonectidae | Enithares tibialis | NC_012819 | |
| Notonecta triguttata | MT043157 | |||
| Naucoroidea | Naucoridae | Ilyocoris cimicoides | NC_012845 | |
| Aphelocheiridae | Aphelocheirus ellipsoideus | FJ456939 |
| Gene | Strand | Position | Anticodon | Site (bp) | Start Codon | Stop Codon | Intergenic Nucleotides |
|---|---|---|---|---|---|---|---|
| tRNA-Ile | J | 1–65 | GAT | 65 | |||
| tRNA-Gln | N | 63–131 | TTG | 69 | −3 | ||
| tRNA-Met | J | 131–199 | CAT | 69 | −1 | ||
| NAD2 | J | 200–1210 | 1011 | GTG | TAA | 0 | |
| tRNA-Trp | J | 1209–1279 | TCA | 71 | −2 | ||
| tRNA-Cys | N | 1272–1335 | GCA | 64 | −8 | ||
| tRNA-Tyr | N | 1340–1405 | GTA | 66 | 4 | ||
| COX1 | J | 1407–2945 | 1539 | TTG | TAA | 1 | |
| tRNA-Leu2 | J | 2941–3005 | TAA | 65 | −5 | ||
| COX2 | J | 3006–3684 | 679 | ATA | T- | 0 | |
| tRNA-Lys | J | 3685–3756 | CTT | 72 | 0 | ||
| tRNA-Asp | J | 3757–3821 | GTC | 65 | 0 | ||
| ATPase8 | J | 3822–3980 | 159 | ATA | TAA | 0 | |
| ATPase6 | J | 3974–4643 | 670 | ATG | T- | −7 | |
| COX3 | J | 4644–5430 | 787 | ATG | T- | 0 | |
| tRNA-Gly | J | 5431–5492 | TCC | 62 | 0 | ||
| NAD3 | J | 5493–5844 | 352 | ATT | T- | 0 | |
| tRNA-Ala | J | 5845–5909 | TGC | 65 | 0 | ||
| tRNA-Arg | J | 5910–5975 | TGC | 66 | 0 | ||
| tRNA-Asn | J | 5982–6050 | GTT | 69 | 6 | ||
| tRNA-Ser1 | J | 6050–6118 | GCT | 69 | −1 | ||
| tRNA-Glu | J | 6118–6181 | TTC | 64 | −1 | ||
| tRNA-Phe | N | 6180–6245 | GAA | 66 | −2 | ||
| NAD5 | N | 6246–7955 | 1710 | ATG | TAA | 1 | |
| tRNA-His | N | 7957–8021 | GTG | 65 | 1 | ||
| NAD4 | N | 8021–9352 | 1332 | ATG | TAG | −1 | |
| NAD4L | N | 9346–9660 | 315 | ATG | TAA | −7 | |
| tRNA-Thr | J | 9663–9726 | TGT | 64 | 2 | ||
| tRNA-Pro | N | 9727–9792 | TGG | 66 | 0 | ||
| NAD6 | J | 9795–10,295 | 501 | ATT | TAA | 2 | |
| CYTB | J | 10,295–11,431 | 1137 | ATG | TAG | −1 | |
| tRNA-Ser2 | J | 11,430–11,497 | TGA | 68 | −2 | ||
| NAD1 | N | 11,515–12,447 | 933 | ATA | TAA | 17 | |
| tRNA-Leu1 | N | 12,442–12,506 | TAG | 65 | −6 | ||
| 16S rRNA | N | 12,539–13,557 | 1019 | 32 | |||
| tRNA-Val | N | 13,761–13,830 | TAC | 70 | 3 | ||
| 12S rRNA | N | 13,833–14,598 | 766 | 2 | |||
| CR | 14,599–15,156 | 558 | 0 |
| Feature | Length (bp) | A% | G% | C% | T% | A + T (%) | AT-Skew | GC-Skew |
|---|---|---|---|---|---|---|---|---|
| Whole genome | 15,156 | 42.93 | 10.40 | 13.64 | 33.03 | 75.96 | 0.13 | −0.14 |
| PCGs | 11,125 | 33.67 | 12.40 | 11.60 | 42.33 | 76.00 | −0.11 | 0.03 |
| PCGs-J | 6835 | 38.22 | 12.07 | 13.36 | 36.36 | 74.57 | 0.02 | −0.05 |
| PCGs-N | 4290 | 26.43 | 12.91 | 8.81 | 51.84 | 78.28 | −0.32 | 0.19 |
| tRNA genes | 1465 | 40.89 | 10.92 | 12.76 | 35.43 | 76.32 | 0.07 | −0.08 |
| tRNA genes-J | 934 | 41.33 | 12.63 | 11.13 | 34.90 | 76.23 | 0.08 | 0.06 |
| tRNA genes-N | 531 | 40.11 | 7.91 | 15.63 | 36.35 | 76.46 | 0.05 | −0.33 |
| rRNA genes | 1785 | 43.47 | 9.41 | 14.29 | 32.83 | 76.30 | 0.14 | −0.20 |
| Control region | 558 | 29.75 | 7.17 | 25.09 | 37.99 | 67.74 | −0.12 | −0.56 |
| Amino Acid | Codon | JN | RSCU | J | RSCU+ | N | RSCU- |
|---|---|---|---|---|---|---|---|
| Phe | UUU(F) | 284 | 1.65 | 123 | 1.46 | 161 | 1.83 |
| UUC(F) | 60 | 0.35 | 45 | 0.54 | 15 | 0.17 | |
| Leu2 | UUA(L) | 402 | 4.66 | 220 | 4.57 | 182 | 4.77 |
| UUG(L) | 24 | 0.28 | 4 | 0.08 | 20 | 0.52 | |
| Leu1 | CUU(L) | 41 | 0.47 | 17 | 0.35 | 24 | 0.63 |
| CUC(L) | 1 | 0.01 | 0 | 0 | 1 | 0.03 | |
| CUA(L) | 49 | 0.57 | 47 | 0.98 | 2 | 0.05 | |
| CUG(L) | 1 | 0.01 | 1 | 0.02 | 0 | 0 | |
| Ile | AUU(I) | 339 | 1.87 | 214 | 1.84 | 125 | 1.92 |
| AUC(I) | 24 | 0.13 | 19 | 0.16 | 5 | 0.08 | |
| Met | AUA(M) | 287 | 1.88 | 199 | 1.9 | 88 | 1.85 |
| AUG(M) | 18 | 0.12 | 11 | 0.1 | 7 | 0.15 | |
| Val | GUU(V) | 76 | 1.55 | 30 | 1 | 46 | 2.42 |
| GUC(V) | 4 | 0.08 | 2 | 0.07 | 2 | 0.11 | |
| GUA(V) | 109 | 2.24 | 82 | 2.77 | 27 | 1.42 | |
| GUG(V) | 6 | 0.12 | 5 | 0.17 | 1 | 0.05 | |
| Ser1 | UCU(S) | 119 | 2.57 | 26 | 1.07 | 93 | 4.23 |
| UCC(S) | 5 | 0.11 | 1 | 0.04 | 4 | 0.18 | |
| UCA(S) | 107 | 2.31 | 91 | 3.73 | 16 | 0.73 | |
| UCG(S) | 3 | 0.06 | 2 | 0.08 | 1 | 0.05 | |
| Pro | CCU(P) | 59 | 1.8 | 34 | 1.39 | 25 | 3.03 |
| CCC(P) | 7 | 0.21 | 6 | 0.24 | 1 | 0.12 | |
| CCA(P) | 63 | 1.92 | 57 | 2.33 | 6 | 0.73 | |
| CCG(P) | 2 | 0.06 | 1 | 0.04 | 1 | 0.12 | |
| Thr | ACU(T) | 75 | 1.6 | 41 | 1.12 | 34 | 3.32 |
| ACC(T) | 5 | 0.11 | 3 | 0.08 | 2 | 0.2 | |
| ACA(T) | 106 | 2.27 | 101 | 2.77 | 5 | 0.49 | |
| ACG(T) | 1 | 0.02 | 1 | 0.03 | 0 | 0 | |
| Ala | GCU(A) | 63 | 1.7 | 25 | 0.97 | 38 | 3.38 |
| GCC(A) | 7 | 0.19 | 7 | 0.27 | 0 | 0 | |
| GCA(A) | 76 | 2.05 | 70 | 2.72 | 6 | 0.53 | |
| GCG(A) | 2 | 0.05 | 1 | 0.04 | 1 | 0.09 | |
| Tyr | UAU(Y) | 145 | 1.81 | 58 | 1.59 | 87 | 2 |
| UAC(Y) | 15 | 0.19 | 15 | 0.41 | 0 | 0 | |
| His | CAU(H) | 50 | 1.39 | 34 | 1.21 | 16 | 2 |
| CAC(H) | 22 | 0.61 | 22 | 0.79 | 0 | 0 | |
| Gin | CAA(Q) | 56 | 1.87 | 44 | 1.96 | 12 | 1.6 |
| CAG(Q) | 4 | 0.13 | 1 | 0.04 | 3 | 0.4 | |
| Asn | AAU(N) | 154 | 1.79 | 96 | 1.7 | 58 | 1.97 |
| AAC(N) | 18 | 0.21 | 17 | 0.3 | 1 | 0.03 | |
| Lys | AAA(K) | 83 | 1.68 | 63 | 1.83 | 20 | 1.33 |
| AAG(K) | 16 | 0.32 | 6 | 0.17 | 10 | 0.67 | |
| Asp | GAU(D) | 59 | 1.71 | 35 | 1.59 | 24 | 1.92 |
| GAC(D) | 10 | 0.29 | 9 | 0.41 | 1 | 0.08 | |
| Glu | GAA(E) | 78 | 1.75 | 60 | 1.97 | 18 | 1.29 |
| GAG(E) | 11 | 0.25 | 1 | 0.03 | 10 | 0.71 | |
| Cys | UGU(C) | 49 | 2 | 13 | 2 | 36 | 2 |
| UGC(C) | 0 | 0 | 0 | 0 | 0 | 0 | |
| Trp | UGA(W) | 91 | 1.84 | 69 | 2 | 22 | 1.47 |
| UGG(W) | 8 | 0.16 | 0 | 0 | 8 | 0.53 | |
| Arg | CGU(R) | 18 | 1.33 | 5 | 0.57 | 13 | 2.74 |
| CGC(R) | 0 | 0 | 0 | 0 | 0 | 0 | |
| CGA(R) | 31 | 2.3 | 29 | 3.31 | 2 | 0.42 | |
| CGG(R) | 5 | 0.37 | 1 | 0.11 | 4 | 0.84 | |
| Ser2 | AGU(S) | 43 | 0.93 | 12 | 0.49 | 31 | 1.41 |
| AGC(S) | 1 | 0.02 | 0 | 0 | 1 | 0.05 | |
| AGA(S) | 91 | 1.96 | 63 | 2.58 | 28 | 1.27 | |
| AGG(S) | 2 | 0.04 | 0 | 0 | 2 | 0.09 | |
| Gly | GGU(G) | 81 | 1.54 | 24 | 0.73 | 57 | 2.89 |
| GGC(G) | 2 | 0.04 | 0 | 0 | 2 | 0.1 | |
| GGA(G) | 114 | 2.17 | 100 | 3.05 | 14 | 0.71 | |
| GGG(G) | 13 | 0.25 | 7 | 0.21 | 6 | 0.3 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, G.; Sun, C.; Hu, H.; Zhang, D.; Li, M. Complete Mitochondrial Genome of the Backswimmer: Notonecta triguttata Motschulsky, 1861 (Hemiptera: Notonectidae): Sequence, Structure, and Phylogenetic Analysis. Diversity 2024, 16, 16. https://doi.org/10.3390/d16010016
Wang G, Sun C, Hu H, Zhang D, Li M. Complete Mitochondrial Genome of the Backswimmer: Notonecta triguttata Motschulsky, 1861 (Hemiptera: Notonectidae): Sequence, Structure, and Phylogenetic Analysis. Diversity. 2024; 16(1):16. https://doi.org/10.3390/d16010016
Chicago/Turabian StyleWang, Guobin, Chengze Sun, Huijun Hu, Danli Zhang, and Min Li. 2024. "Complete Mitochondrial Genome of the Backswimmer: Notonecta triguttata Motschulsky, 1861 (Hemiptera: Notonectidae): Sequence, Structure, and Phylogenetic Analysis" Diversity 16, no. 1: 16. https://doi.org/10.3390/d16010016
APA StyleWang, G., Sun, C., Hu, H., Zhang, D., & Li, M. (2024). Complete Mitochondrial Genome of the Backswimmer: Notonecta triguttata Motschulsky, 1861 (Hemiptera: Notonectidae): Sequence, Structure, and Phylogenetic Analysis. Diversity, 16(1), 16. https://doi.org/10.3390/d16010016