Abstract
This study examines the gut bacterial communities of four necrophagous fly species: Lucilia illustris, L. caesar, Chrysomya megacephala, and C. pinguis. The gut bacterial communities exhibited significant variation across species, showcasing a diverse range of bacterial phyla, classes, and genera. Each species harbored a unique set of bacteria, yet there was considerable overlap in taxa among species. Species richness was comparable across all species. However, measures that account for both richness and evenness, such as the Shannon diversity index and the inverse Simpson’s diversity index, indicated significant differences between species, especially between L. illustris and C. pinguis. The functional profiles of the gut bacterial communities mainly centered on metabolic and environmental information processing functions, with no marked differences between species. While this study had limitations in data collection, it still revealed a significant correlation between the phylogenetic distances of some fly species and the distances of their gut bacterial communities. This supports the hypothesis that the gut microbiota is not random but is influenced by the host’s evolutionary history or seasons. We confirmed that an association between phylogeny and gut bacterial community structure, as determined through entanglement analysis, exists. The study focused on only five individuals from the four fly species sampled during spring and summer, which might affect the generalizability of the results. Future research would benefit from replicating this study with a larger sample size across various seasons to ensure the more widespread applicability of the findings.
1. Introduction
The subject of focus in the present study is insect biodiversity, a complex web of biological and ecological relationships where each species plays a unique role in ecosystem function [,]. Of particular interest in this context are necrophagous insects, specifically dipteran species, which, by virtue of their scavenging behavior, accelerate decomposition processes, facilitate nutrient recycling within trophic networks, and play a significant part in the planet’s biogeochemical cycles. Therefore, the exploration of necrophagous dipteran species becomes pertinent to ecological comprehension and larger ecosystem dynamism.
At the heart of our investigation are four dipteran species exhibiting necrophagous behavior: Lucilia illustris (Meigen, 1826), Lucilia Caesar (Linnaeus, 1758), Chrysomya megacephala (Fabricius, 1794), and Chrysomya pinguis (Walker, 1858). While these species share necrophagous habits, each is a distinct biological entity with its own specific traits and preferences for particular environmental conditions.
L. illustris and L. caesar, members of the Calliphoridae family, demonstrate an affinity for cooler climatic conditions, primarily emerging in the spring season. Their activity is temporally aligned with the availability of carrion—a primary dietary component that is a result of the natural life–death cycles inherent to their habitats. In opposition, C. megacephala and C. pinguis, also Calliphoridae members, exhibit a propensity for warmer conditions, marked by an enhanced presence during summer months. The differential climatic preferences of these species may point towards the existence of adaptive mechanisms facilitating the utilization of diverse niches, thus averting direct interspecies competition [,].
Despite the observed variations in temporal and climatic preferences, all four species converge on the choice of a single dietary source: carrion [,]. This common resource utilization, classifying the species as necrophagous, provides an intriguing platform from which to study survival mechanisms. An investigation into their respective ecological strategies and associated gut microbiota aims to elucidate the role of intestinal microbiota in niche differentiation among closely related species, and the wider ecosystem implications [].
The investigation into the gut microbiota of insects in question is necessitated by its influence over various host–insect biological facets such as nutrition, development, immunity, behavior, and overall fitness []. Furthermore, its correlation with the host’s environmental adaptation capabilities, driven largely by seasonal dietary and temperature fluctuations, cannot be understated [,]. Therefore, a meticulous exploration of these microbial populations may contribute substantially to the understanding of host organism biology and ecological interactions [,,]. This investigation primarily seeks to unravel the interactions between the insects under study and their gut microbiota. Our initial endeavors focus on accurately characterizing the gut microorganisms intrinsic to these necrophagous flies. Such an understanding aims to shed light on the symbiotic relationships between the host insects and their gut microbiota, highlighting implications for survival, reproduction, ecological function, and organic matter decomposition processes [,,].
Our secondary objective assesses whether the gut bacterial communities of these flies correlate with their phylogenetic relationships. The working hypothesis suggests that these factors wield considerable influence over the gut bacterial community compositions of L. illustris, L. caesar, C. megacephala, and C. pinguis, thus unveiling an, up to the present time, unexplored aspect of insect–microbe symbioses.
The study hypothesizes that the formation of gut bacterial communities in these insects is not random but is influenced by their evolutionary lineage. We speculate that phylogenetically close insects will possess more similar gut microbiota. This is based on the notion that closely related species often exhibit comparable physiological and ecological traits, which might extend to their associated microbiota.
2. Materials and Methods
2.1. Sample Collection and Preparation
Sampling for this study took place from March to September 2022 in Gongju City, Chungnam, South Korea (36°28′16″ N, 127°08′24″ E). Lucilia species were collected from March to June during the spring, while Chrysomya species were collected from July to September in the summer. To ensure a comprehensive representation of each necrophagous fly species, we used a trapping method with fresh pig liver as bait. Each sample consisted of five individuals from each species. To reduce variability due to sexual dimorphism, only male specimens were used for subsequent examinations.
2.2. DNA Extraction
The gut was extracted from five samples of each of the four fly species under sterile conditions. Following this, genomic DNA was isolated using the DNeasy PowerSoil Pro Kits from QIAGEN (Qiagen, Hilden, Germany), strictly adhering to the manufacturer’s instructions. The samples were homogenized with a lysis buffer using a TissueLyser II. According to the kit’s protocol, the DNA was subsequently extracted, and the quality and concentration of the extracted genomic DNA were verified using electrophoresis and a DeNovix-QFX fluorometer. This procedure was aimed at investigating the gut bacterial community. For phylogenetic scrutiny, additional genomic DNA was extracted from the remaining exoskeleton following gut extraction using the DNeasy Blood and Tissue Kit (Qiagen).
2.3. PCR Amplification and Sequencing
For the investigation of the gut bacterial community, the V4 region of the 16S rRNA gene was targeted for amplification []. The Polymerase chain reaction (PCR) employed the 515F forward primer and the 806R reverse primer. After the primary amplification, the PCR product was purified utilizing the Agencourt AMPure XP PCR purification system. This was followed by an index PCR process, which incorporated the Nextera XT Index Kit. In a similar manner to the primary PCR, the products from the index PCR were purified. To ensure the integrity, size, and quality of the final PCR product, it underwent evaluation via electrophoresis. Additionally, its concentration was ascertained with the use of a DeNovix-QFX fluorometer. Once these quality controls were complete, the samples were diluted and pooled. Subsequent to these steps, sequencing was carried out on the MiniSeq Illumina platform, specifically targeting the V4 region of the 16S rRNA gene.
For the purpose of phylogenetic investigation, the Cytochrome c oxidase subunit I (COI) gene from the samples was targeted for amplification. A set of universal primers was chosen for the PCR amplification of the COI gene, given its widespread application in molecular taxonomy and the identification of metazoan species. Following the amplification, the PCR products were visualized using gel electrophoresis to ensure the successful amplification of the desired region. After verification, the products underwent purification procedures similar to those described earlier for the 16s rRNA gene region. Post-purification, the quality and concentration of the amplified COI gene were confirmed using a DeNovix-QFX fluorometer. Once the quality checks were complete and deemed satisfactory, the samples were prepped for sequencing. Using established methodologies, the COI gene PCR products were sequenced to generate data for phylogenetic analysis.
2.4. Bioinformatics and Statistical Analysis
Sequencing reads from the 16S rRNA gene underwent quality filtering, sequence alignment, chimera checking, and taxonomic assignment using the QIIME2 pipeline (version 2017.6.0) []. Simultaneously, sequences from the Bacteria domain were extracted for a focused analysis of the gut bacterial community. The COI sequences were aligned, and a phylogenetic tree was constructed using the ggtree package within the R software framework (version 2.4.1). The functional capabilities of the gut bacterial community were inferred using the Tax4Fun package (version 0.3.1). Comprehensive visualization and statistical analysis of the bacterial communities were carried out using the Microeco package for R (version 0.20.0). Mantel tests were utilized to examine the correlation between gut bacterial community distances and species’ phylogenetic distances. This test is typically used to assess the significance of associations between two distance matrices [].
The Sloan’s neutral model was employed to examine the distribution of microbial taxa at the genus level within each fly species. The model posits:
Here, m signifies the neutral migration rate of microbes between individual hosts, representing the balance between the stochastic and deterministic processes governing microbial distributions. For each fly species, the microbial data at the genus level were fitted to the Sloan’s neutral model using R’s non-linear regression functionalities. The initial parameter guess for m was set to 1 and, through iterative optimization techniques, the optimal value of m that provided the best fit to the observed data was derived, along with a 95% confidence interval for the parameter estimate. The adequacy of the model for each species was assessed using the R2 coefficient of determination. This metric, computed in R, quantifies the proportion of variance in the observed occurrence frequencies that is captured by the model, providing an indication of its explanatory power for the microbial distributions at the genus level [].
Occurrence frequency = 1 − (1 + m × Mean relative abundance) exp(−m × Mean relative abundance)
In addition, an entanglement analysis was performed to study the relationship between phylogeny and gut bacterial community structure. Entanglement analysis is a statistical method that investigates how multiple variables are intertwined. In this context, entanglement refers to the interconnectedness and complex dependence between phylogenetic distance and gut bacterial community distance. To further understand the relationship between phylogeny and gut bacterial community structure within each genus, we performed separate entanglement analyses for both Lucilia and Chrysomya genera. This approach allowed us to delve deeper into the intricacies of the relationship within each genus and assess if there were genus-specific patterns that might be masked when analyzing all species together. It allows for the quantification of the degree to which these factors influence each other, providing a multifaceted understanding of the relationship between host evolutionary lineage and microbiota composition. The analysis was carried out using the factoextra, cluster, and dendextend package.
3. Results
3.1. Gut Bacterial Composition of Necrophagous Fly Species
The gut microbiota of the four necrophagous fly species—C. megacephala, L. illustris, C. pinguis, and L. caesar—showed phylum-level variation (Figure 1a). Actinobacteria (58.87%), Bacteroidetes (13.31%), Firmicutes (54.99%), and Proteobacteria (29.59%) were the predominant phyla across all species. C. megacephala (57.10%) and L. illustris (64.57%) were primarily colonized by Firmicutes. In contrast, Actinobacteria were most prevalent in C. pinguis (73.85%) and L. caesar (68.56%). Proteobacteria were more abundant in L. caesar and L. illustris, while Bacteroidetes were notably prevalent in C. megacephala (29.71%) and C. pinguis (18.02%), and less abundant in L. caesar (3.64%) and L. illustris (1.88%).
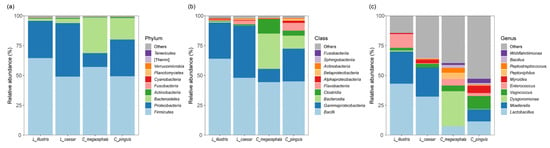
Figure 1.
Taxonomic breakdown of gut bacterial composition in necrophagous fly species at various levels: (a) phylum, (b) class, and (c) genus.
At the class level, Bacilli (50.37%), Bacteroidia (10.45%), and Gammaproteobacteria (28.10%) were dominant across all species (Figure 1b). Bacilli had a consistent presence in all species. L. caesar (43.72%) and L. illustris (30.22%) hosted more Gammaproteobacteria, while Bacteroidia were especially common in C. megacephala (29.57%).
The gut bacterial communities of the four species exhibited distinct compositions at the genus level (Figure 1c). The genera Bacillus (1.33%), Dysgonomonas (7.67%), Enterococcus (5.01%), Lactobacillus (23.53%), Moellerella (15.36%), Myroides (2.44%), Peptoniphilus (1.42%), Peptostreptococcus (1.38%), Vagococcus (5.21%), and Wohlfahrtiimonas (1.31%) were present across the species. Lactobacillus was dominant in L. illustris (32.25%) and L. caesar (43.07%). C. megacephala prominently hosted Dysgonomonas (29.50%). Conversely, C. pinguis exhibited a balanced distribution of Lactobacillus (11.48%), Moellerella (10.05%), and Vagococcus (10.87%).
3.2. Alpha and Beta Diversity of Gut Bacterial Community of Necrophagous Fly Species
Regarding observed species richness, no significant differences were detected among C. pinguis, L. illustris, L. caesar, and C. megacephala (H = 1.080, p = 0.782) (Figure 2a).
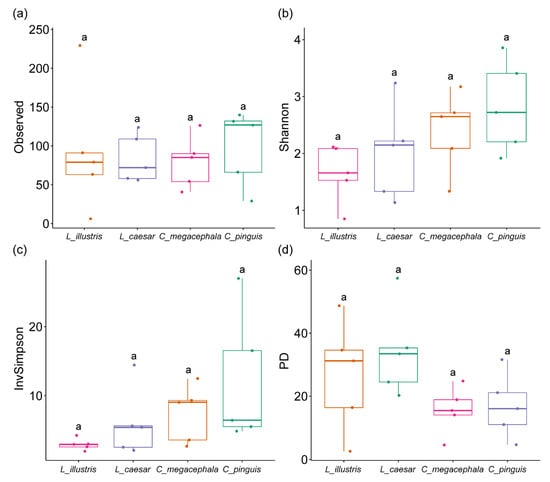
Figure 2.
Measures of alpha diversity in the gut bacterial communities of necrophagous fly species: (a) Observed species richness, (b) Shannon diversity index, (c) Inverse Simpson’s diversity index, and (d) Phylogenetic diversity (PD). Different letters indicate significant differences between groups, as determined by Dunn’s post hoc test following a Kruskal–Wallis test.
Both the Shannon diversity index (H = 2.399, p = 0.106) and the inverse Simpson’s diversity index (H = 7.194, p = 0.066), which account for the richness and evenness of the bacterial communities, showed no significant differences among the four groups (Figure 2b,c).
Regarding phylogenetic diversity (PD), there were no significant differences across the four species (H = 6.040, p = 0.110), suggesting that the gut bacterial communities of these species may have similar evolutionary histories (Figure 2d).
The principal coordinate analysis (PCoA) plot showed no distinct separation of the bacterial communities. The combined PCo1 and PCo2 axes accounted for 36.6% of the total variance in the gut bacterial communities from the four fly species. Clusters mostly overlapped among the groups, indicating a significant similarity in their gut microbial compositions (Figure 3a). Using Bray–Curtis distances in the PCoA, there were no statistically significant differences among the species. This suggests that the overall dissimilarity in the composition of the gut microbial communities is not significant (Figure 3b).
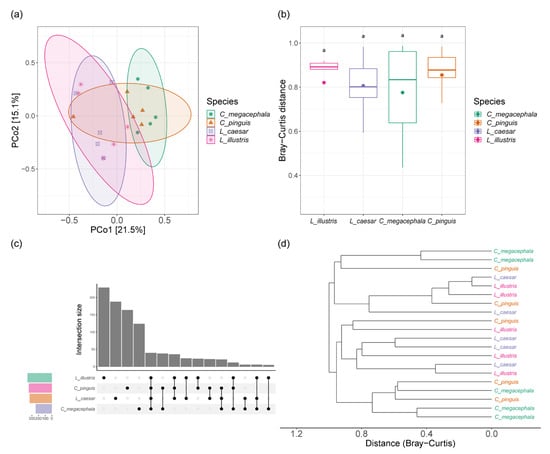
Figure 3.
Assessment of beta diversity and shared taxa among the gut bacterial communities of necrophagous fly species: (a) Principal coordinate analysis (PCoA) visualization based on Bray–Curtis distances, (b) boxplots showing the distribution of Bray–Curtis distances within the gut bacterial communities. Same letters above boxes signify no differences between groups (adjusted p < 0.05), as determined by Dunn’s post hoc test following a Kruskal–Wallis test, (c) an upset plot highlighting both shared and species-specific bacterial taxa among the four fly species, and (d) distance dendrogram based on Bray–Curtis distances.
An upset plot showcased both shared and unique bacterial taxa among the species. While each species harbored a unique set of bacteria (ranging from 123 in C. megacephala to 228 in L. illustris), there was a considerable overlap, with the most extensive shared set comprising 39 taxa across all four species (Figure 3c). The distance dendrogram, utilizing Bray–Curtis distances, showed mixed clustering by genus, with Lucilia and Chrysomya species intermingling (Figure 3d).
3.3. Gut Bacterial Community Functional Profile
The functional profiles of the gut bacterial communities may not differ significantly across the species. However, any differences should be validated using a much larger dataset (Figure 4). The communities we studied predominantly participate in metabolic processes and environmental information processing. The primary functional categories, listed in decreasing order of prevalence, are Carbohydrate Metabolism, Membrane Transport, Amino Acid Metabolism, Metabolism of Cofactors and Vitamins, Nucleotide Metabolism, Translation, Signal Transduction, Energy Metabolism, Replication and Repair, and Lipid Metabolism.
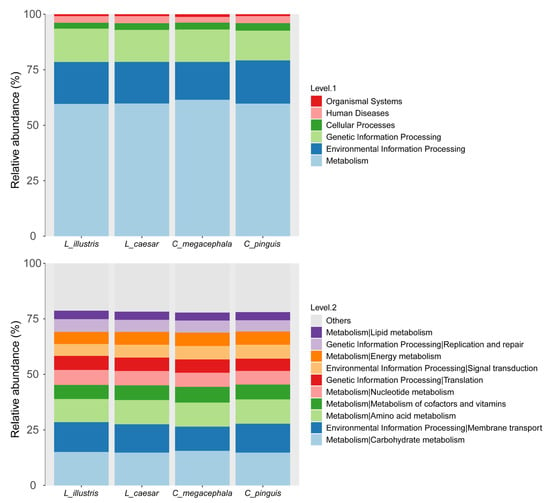
Figure 4.
Functional categorization of gut bacterial communities in four necrophagous fly species as represented by the bar graph, depicting the relative abundance of diverse bacterial functional groups.
3.4. Correlation between Phylogenetic Distances and Gut Bacterial Community Distances
Our analysis revealed a significant correlation between the phylogenetic distances among the four species across two genera of necrophagous flies and their gut bacterial community distances (Mantel test: r = 0.1861, p = 0.0131). This correlation suggests that fly species that are more closely related tend to have gut bacterial communities that are more similar than those of more distantly related species. To delve deeper into this relationship, we conducted an entanglement analysis, which evaluates the interconnectedness between host phylogeny and gut bacterial community structure. This analysis yielded a score of 0.26, indicating a moderate degree of association between the host’s evolutionary lineage and its gut microbiota composition (Figure 5a).
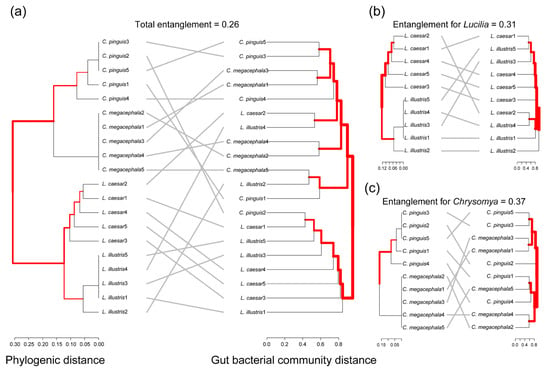
Figure 5.
Examination of correlation between phylogenetic distances among fly species and gut bacterial community distances. (a) Combined analysis for Lucilia and Chrysomya. (b) Analysis specific to Lucilia. (c) Analysis for Chrysomya. The red, bold lines indicate groups of species that are consistently clustered in both dendrograms, suggesting a strong correlation between their phylogenetic relationships and gut bacterial community compositions.
When this entanglement analysis was applied separately to the Lucilia genus, it produced a score of 0.31 (Figure 5b). This score suggests a slightly stronger association between the evolutionary lineage of Lucilia species and their gut microbiota composition compared to the combined analysis of both genera.
Conversely, for the Chrysomya genus, the entanglement score was observed to be 0.37 (Figure 5c). This score denotes an even stronger association between the host’s evolutionary lineage and its gut microbiota composition within this genus. The higher entanglement score for Chrysomya, when compared to Lucilia, implies that evolutionary lineage might exert a more pronounced influence in shaping the gut bacterial community structure in Chrysomya than in Lucilia.
3.5. Sloan Neutral Model Analysis of Gut Bacterial Community
Utilizing Sloan’s neutral model, we evaluated the relationship between the occurrence frequency and the mean relative abundance of gut bacteria in the four necrophagous fly species. The model revealed distinct patterns of microbial occurrence across different species (Figure 6).
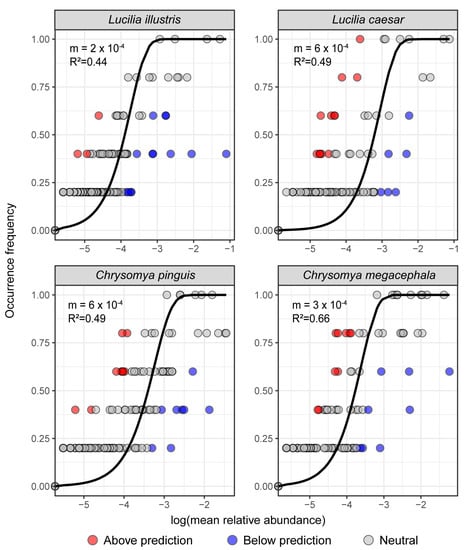
Figure 6.
Sloan neutral model analysis illustrating the relationship between the log-transformed mean relative abundance and occurrence frequency across the four fly species. The best-fitted Sloan curve, along with its 95% confidence interval, is depicted as a solid line. Points representing microbial genera above, below, and in line with the neutral model predictions are color-coded in red, blue, and gray, respectively.
For L. caesar, the fit with Sloan’s neutral model indicated a significant correspondence between the data and the fitted curve with an R2 value of 0.49. Similar patterns were observed for L. illustris, C. megacephala, and C. pinguis with respective R2 values of 0.44, 0.66, and 0.49.
Further insights were obtained from an analysis of deviations from the neutral model predictions (Figure 7). For L. caesar, 9 microbial genera were observed above, and 8 below the model’s prediction, while 221 were consistent with neutral expectations. L. illustris showed 3 genera above and 13 below the predictions, with 222 fitting the neutral model. Both C. megacephala and C. pinguis presented 9 genera above and 9 and 8 genera below the predictions, respectively, with each having 220 and 221 genera aligning with the neutral model.
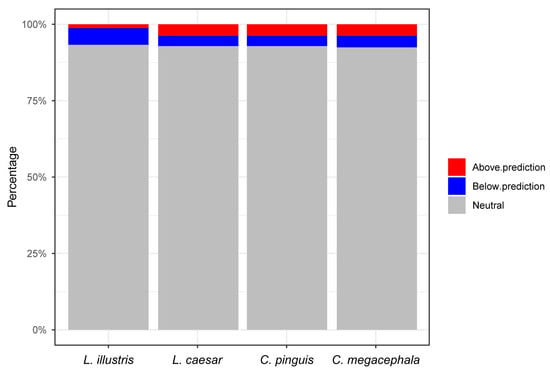
Figure 7.
Deviations from the Sloan neutral model predictions (with a 95% confidence interval) across the four fly species. The number of microbial genera that fall above, below, or align with neutral model expectations are presented for each species. Genera above the predictions are indicated in red, those below in blue, and those aligning with the neutral model in gray. This breakdown offers insights into the specific microbial genera that deviate from or adhere to neutral distribution patterns within each fly species.
4. Discussion
4.1. Gut Bacterial Composition of Necrophagous Fly Species
Actinobacteria, Bacteroidetes, Firmicutes, and Proteobacteria were the predominant phyla in the gut microbiota of the four necrophagous fly species. The relative abundances of these phyla fluctuated among the species: Firmicutes was prevalent in C. megacephala and L. illustris, while Actinobacteria dominated in C. pinguis and L. caesar. Notably, Lactobacillus was consistently present in significant amounts across all species, emphasizing its likely importance in their ecology and physiology [,]. In the fly gut, Lactobacillus potentially influences developmental processes. However, the probiotic attributes of Lactobacillus can vary based on the specific strains []. As a primary metabolic facilitator, Lactobacillus might play a crucial role in decomposing and fermenting the organic matter found in the carrion diet, ensuring nutrient uptake and thereby enhancing fly survival and reproduction []. Its potential immunomodulatory properties could also fortify the flies against pathogenic threats [,,,,,]. Hence, the prevalence of Lactobacillus may signify an adaptive symbiotic relationship that shapes the biology and ecological adaptability of these flies [,]. In light of the Sloan’s neutral model predictions, it is noteworthy that the genus Lactobacillus, despite its prominence in our discussion and its significant representation in the gut bacterial communities, aligns with the model’s neutral expectations. This suggests that the occurrence and distribution of Lactobacillus across the four fly species might be governed by neutral processes rather than specific deterministic factors. While several genera were observed to deviate from the model’s predictions, either being above or below, Lactobacillus was neither overrepresented nor underrepresented in comparison to the model’s predictions []. Despite these foundational findings, we observed significant overlap in bacterial taxa across all species, suggesting a core bacterial community resilient to seasonal variations. Notably, the functional profiles remained consistent, pointing to factors beyond mere seasonality that shape these communities [].
In future research, comparisons with a suitable control group could further validate the hypothesized importance of Lactobacillus []. The diverse composition of the gut microbiota underscores the intricate interplay between the flies and their resident microbes, potentially influencing their biology and ecological roles [,]. These microbial communities might assist in various physiological functions, such as digestion, detoxification, immune modulation, and nutrient synthesis which, in turn, support the flies’ survival and reproductive viability in their habitats [,].
Our investigations revealed a discernible pattern of shared bacterial taxa across the four fly species, pointing towards the presence of a “core” gut microbiota. This core ensemble of microbes is not merely a collection of shared species but is indicative of bacterial populations that collectively perform indispensable biological functions common across the flies. A core gut microbiota might act as the stable base upon which each fly species can accommodate additional, unique bacterial taxa suited to its particular ecological context. While a core microbiota provides a baseline functional profile, the unique sets of bacteria in each fly species could be seen as adaptive components. These specialized microbial sets may confer distinct advantages, allowing each fly species to thrive in their specific ecological niches [,]. The composition of these microbial communities is likely shaped by a combination of factors: the dietary preferences of each fly species, the unique environmental interactions, and the evolutionary histories that guide host–microbe associations over time. It is crucial to distinguish between the concepts of a core microbiota and keystone taxa. Whereas the core refers to the assemblage of microbial taxa commonly found across multiple hosts, keystone taxa are individual or specific groups of microbes that have a disproportionately large impact on the structure and function of the entire microbial community, despite their potentially low abundance. Keystone taxa can anchor the microbial community, and their removal or reduction might lead to significant shifts in community composition and function. In the context of our study, the core microbiota offers insights into the foundational bacterial taxa shared across the necrophagous flies, while any identified keystone taxa would highlight microbes that play a pivotal role in shaping and stabilizing the gut microbial ecosystem of these flies [,].
4.2. Gut Bacterial Community Diversity and Functional Profile
The observed alpha diversity across the gut bacterial communities of the four fly species suggests a consistent number of taxa present across them. This uniformity might indicate that the necrophagous nature of these flies necessitates a particular bacterial diversity level to effectively decompose organic matter and assimilate essential nutrients []. While the gut bacterial communities in adult flies appeared homogenous, some variations in bacterial abundance and diversity were evident. These differences may correlate with the functional roles of the bacteria tailored to specific fly species or individual characteristics.
The congruence in species richness across the communities implies that there might be a common set of fundamental ecological functions performed by the gut bacteria. Yet, it is worth noting that, even with these shared functionalities, the exact roles and adaptations of these bacteria may vary, catering to the specific ecological requirements of each fly species []. To discern these roles conclusively, further in-depth studies involving functional assays and transcriptomic and metabolomic analyses are warranted. These would shed light on the bacteria’s active metabolic pathways and their respective functional contributions to the fly hosts. Notably, the consistent phylogenetic diversity across these species could hint at a parallel evolutionary lineage of their gut bacterial communities. Such nuances or similarities in gut microbiota could be a manifestation of the flies’ distinct evolutionary and ecological journeys [,]. However, it is also possible that the observed patterns stem from inadequate data. Future studies with more extensive sample sizes and appropriate controls are essential to validate these observations.
Regarding beta diversity, the minimal compositional disparities observed suggest that a delicate interplay exists. It is the species-specific determinants that shape the gut microbial makeup and overarching factors contributing to their similarities, encapsulating the flies’ shared ecological and evolutionary narratives [].
The functional profiles identified from the gut bacterial communities, despite the lack of significant variations among the species, underscore the pivotal roles these bacteria play in underpinning the biological and ecological roles of the flies. Functional redundancy, in essence, emphasizes the overlapping of indispensable roles across organisms or taxa. Such redundancy can be seen in the predominant functions related to metabolic processes and environmental information processing. This redundancy suggests these bacteria play a central role in processing nutrients and mediating interactions with the external environment [,]. Key functional categories like carbohydrate and amino acid metabolism, membrane transport, and signal transduction can directly influence the survival, adaptability, and reproductive efficacy of the flies []. This intricate web of host–microbe interactions provides a deeper understanding of how these flies have adapted to their necrophagous lifestyle and the potential evolutionary pressures that may have shaped their gut microbiota [].
4.3. Correlation between Phylogenetic Relationship and Gut Bacterial Community Distances
The discovery of a strong correlation between the phylogenetic distances of the fly species and the distances within their gut bacterial communities validates our initial hypothesis, inferring that closely related fly species are more likely to harbor similar gut bacterial communities than those that are more distantly related []. These findings contribute significantly to our understanding of host–microbiota co-evolutionary dynamics, hinting that evolutionary history might, indeed, partially shape the gut bacterial communities of these necrophagous flies []. This could be a reflection of the shared physiological and ecological traits among closely related species, which might influence their symbiotic bacterial community compositions [,]. The entanglement analysis offers a quantitative perspective on this interconnectedness. Our findings suggest varying degrees of entanglement across the genera, Lucilia and Chrysomya. The higher entanglement score observed in Chrysomya implies evolutionary and ecological pressures might more tightly link its gut bacterial community to its host lineage, compared to Lucilia. Such distinctions may arise from differences in dietary preferences, habitat selection, or reproductive strategies between the two genera. This differential entanglement raises intriguing questions about the broader evolutionary dynamics at play, hinting at potential adaptive strategies employed by these flies in their respective ecological niches.
Despite the influence of host evolution on gut microbiota, it is crucial to acknowledge other contributing factors such as environmental impacts and individual variations [,,]. The concept of entanglement underscores that, while the association is moderate, the host’s evolutionary trajectory has an undeniable influence on the gut bacterial community. Alongside these conventional understandings, Mendelian randomization could offer another perspective. This analytical method utilizes genetic variants as instrumental variables to determine causality between exposures and outcomes. Employing Mendelian randomization in this context might help to untangle potential confounding factors and provide more direct insights into the causal relationships within host–microbe interactions. Thus, considering Mendelian randomization in future studies could further enhance our comprehension of these intricate relationships [,].
5. Conclusions
This research delves into the intricate relationship between necrophagous flies and their gut microbiota, unraveling the nuanced dynamics of insect–microbiota symbiosis. It suggests that beyond just their phylogenetic relationships, the ecological roles these flies play also influence their gut bacterial communities. Such insights can enrich our comprehension of how these flies contribute to the decomposition of organic matter and offer a deeper perspective on symbiotic relationships in insects. Recognizing the limitations of our sample size, we intend to expand our study in the future, ensuring our findings resonate with broader applicability and offer more extensive insights into this fascinating interplay.
Author Contributions
Conceptualization, Y.D.; Methodology, W.-B.P. and J.-K.P.; Software, W.-B.P. and Y.D.; Validation, Y.D.; Formal analysis, W.-B.P. and J.-K.P.; Investigation, W.-B.P.; Resources, Y.D.; Data curation, W.-B.P.; Writing—Original draft preparation, W.-B.P. and Y.D.; Writing—Review and editing, Y.D.; Visualization, W.-B.P. and J.-K.P.; Supervision, Y.D.; Project administration, Y.D.; Funding acquisition, Y.D. All authors have read and agreed to the published version of the manuscript.
Funding
This research was supported by a National Research Foundation of Korea (NRF) grant funded by the Korean government (MSIT) (No. 2022R1A2C1004240).
Institutional Review Board Statement
Not applicable.
Data Availability Statement
The data generated in this study are available upon reasonable request from the corresponding author.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Losey, J.E.; Vaughan, M.J.B. The economic value of ecological services provided by insects. Bioscience 2006, 56, 311–323. [Google Scholar] [CrossRef]
- Hung, K.-L.J.; Kingston, J.M.; Albrecht, M.; Holway, D.A.; Kohn, J.R. The worldwide importance of honey bees as pollinators in natural habitats. Proc. R. Soc. B 2018, 285, 20172140. [Google Scholar] [CrossRef] [PubMed]
- Brundage, A.; Bros, S.; Honda, J.Y. Seasonal and habitat abundance and distribution of some forensically important blow flies (Diptera: Calliphoridae) in Central California. Forensic Sci. Int. 2011, 212, 115–120. [Google Scholar] [CrossRef] [PubMed]
- Hwang, C.; Turner, B. Spatial and temporal variability of necrophagous Diptera from urban to rural areas. Med. Vet. Entomol. 2005, 19, 379–391. [Google Scholar] [CrossRef] [PubMed]
- Prado e Castro, C.; Serrano, A.; Martins da Silva, P.; García, M. Carrion flies of forensic interest: A study of seasonal community composition and succession in Lisbon, Portugal. Med. Vet. Entomol. 2012, 26, 417–431. [Google Scholar] [CrossRef] [PubMed]
- Benbow, M.; Lewis, A.; Tomberlin, J.; Pechal, J. Seasonal necrophagous insect community assembly during vertebrate carrion decomposition. J. Med. Entomol. 2013, 50, 440–450. [Google Scholar] [CrossRef] [PubMed]
- Crooks, E.R.; Bulling, M.T.; Barnes, K.M. Microbial effects on the development of forensically important blow fly species. Forensic Sci. Int. 2016, 266, 185–190. [Google Scholar] [CrossRef] [PubMed]
- Durvasula, R.V.; Sundaram, R.K.; Kirsch, P.; Hurwitz, I.; Crawford, C.V.; Dotson, E.; Beard, C.B. Genetic transformation of a Corynebacterial symbiont from the Chagas disease vector Triatoma infestans. Exp. Parasitol. 2008, 119, 94–98. [Google Scholar] [CrossRef]
- Deel, H.L.; Montoya, S.; King, K.; Emmons, A.L.; Huhn, C.; Lynne, A.M.; Metcalf, J.L.; Bucheli, S.R. The microbiome of fly organs and fly-human microbial transfer during decomposition. Forensic Sci. Int. 2022, 340, 111425. [Google Scholar] [CrossRef]
- Wei, T.; Ishida, R.; Miyanaga, K.; Tanji, Y. Seasonal variations in bacterial communities and antibiotic-resistant strains associated with green bottle flies (Diptera: Calliphoridae). Appl. Microbiol. Biotechnol. 2014, 98, 4197–4208. [Google Scholar] [CrossRef]
- Wohlfahrt, D.; Woolf, M.S.; Singh, B. A survey of bacteria associated with various life stages of primary colonizers: Lucilia sericata and Phormia regina. Sci. Justice 2020, 60, 173–179. [Google Scholar] [CrossRef] [PubMed]
- Ma, Q.; Fonseca, A.; Liu, W.; Fields, A.T.; Pimsler, M.L.; Spindola, A.F.; Tarone, A.M.; Crippen, T.L.; Tomberlin, J.K.; Wood, T.K. Proteus mirabilis interkingdom swarming signals attract blow flies. ISME J. 2012, 6, 1356–1366. [Google Scholar] [CrossRef] [PubMed]
- McMullen, J.G.; Bueno, E.; Blow, F.; Douglas, A.E. Genome-inferred correspondence between phylogeny and metabolic traits in the wild Drosophila gut microbiome. Genome Biol. Evol. 2021, 13, evab127. [Google Scholar] [CrossRef] [PubMed]
- Nayduch, D. Special collection: Filth fly–microbe interactions. Ann. Entomol. Soc. Am. 2017, 110, 2–5. [Google Scholar] [CrossRef]
- Kaltenpoth, M. Actinobacteria as mutualists: General healthcare for insects? Trends Microbiol. 2009, 17, 529–535. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Longnecker, M.; Tarone, A.M.; Tomberlin, J.K. Responses of Lucilia sericata (Diptera: Calliphoridae) to compounds from microbial decomposition of larval resources. Anim. Behav. 2016, 115, 217–225. [Google Scholar] [CrossRef]
- Pichler, M.; Coskun, Ö.K.; Ortega-Arbulú, A.S.; Conci, N.; Wörheide, G.; Vargas, S.; Orsi, W.D. A 16S rRNA gene sequencing and analysis protocol for the Illumina MiniSeq platform. Microbiologyopen 2018, 7, e00611. [Google Scholar] [CrossRef]
- Allali, I.; Arnold, J.W.; Roach, J.; Cadenas, M.B.; Butz, N.; Hassan, H.M.; Koci, M.; Ballou, A.; Mendoza, M.; Ali, R. A comparison of sequencing platforms and bioinformatics pipelines for compositional analysis of the gut microbiome. BMC Microbiol. 2017, 17, 194. [Google Scholar] [CrossRef]
- Diniz-Filho, J.A.F.; Soares, T.N.; Lima, J.S.; Dobrovolski, R.; Landeiro, V.L.; Telles, M.P.d.C.; Rangel, T.F.; Bini, L.M. Mantel test in population genetics. Genet. Mol. Biol. 2013, 36, 475–485. [Google Scholar] [CrossRef]
- Sloan, W.T.; Lunn, M.; Woodcock, S.; Head, I.M.; Nee, S.; Curtis, T.P. Quantifying the roles of immigration and chance in shaping prokaryote community structure. Environ. Microbiol. 2006, 8, 732–740. [Google Scholar] [CrossRef]
- Matos, R.C.; Leulier, F. Lactobacilli-Host mutualism: “learning on the fly”. Microb. Cell Factories 2014, 13, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Kleerebezem, M.; Boekhorst, J.; Van Kranenburg, R.; Molenaar, D.; Kuipers, O.P.; Leer, R.; Tarchini, R.; Peters, S.A.; Sandbrink, H.M.; Fiers, M.W. Complete genome sequence of Lactobacillus plantarum WCFS1. Proc. Nati. Acad. Sci. USA 2003, 100, 1990–1995. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Han, G.; Kim, J.W.; Jeon, C.O.; Hyun, S. Taxon-specific effects of Lactobacillus on Drosophila host development. Microb. Ecol. 2020, 79, 241–251. [Google Scholar] [CrossRef] [PubMed]
- Iancu, L.; Angelescu, I.R.; Paun, V.I.; Henríquez-Castillo, C.; Lavin, P.; Purcarea, C. Microbiome pattern of Lucilia sericata (Meigen) (Diptera: Calliphoridae) and feeding substrate in the presence of the foodborne pathogen Salmonella enterica. Sci. Rep. 2021, 11, 15296. [Google Scholar] [CrossRef] [PubMed]
- Cirimotich, C.M.; Ramirez, J.L.; Dimopoulos, G. Native microbiota shape insect vector competence for human pathogens. Cell Host Microbe 2011, 10, 307–310. [Google Scholar] [CrossRef] [PubMed]
- Scott, J.J.; Oh, D.-C.; Yuceer, M.C.; Klepzig, K.D.; Clardy, J.; Currie, C.R. Bacterial protection of beetle-fungus mutualism. Science 2008, 322, 63. [Google Scholar] [CrossRef] [PubMed]
- Teixeira, L.; Ferreira, Á.; Ashburner, M. The bacterial symbiont Wolbachia induces resistance to RNA viral infections in Drosophila melanogaster. PLoS Biol. 2008, 6, e1000002. [Google Scholar] [CrossRef] [PubMed]
- Kaltenpoth, M.; Göttler, W.; Herzner, G.; Strohm, E. Symbiotic bacteria protect wasp larvae from fungal infestation. Curr. Biol. 2005, 15, 475–479. [Google Scholar] [CrossRef]
- Brownlie, J.C.; Johnson, K.N. Symbiont-mediated protection in insect hosts. Trends Microbiol. 2009, 17, 348–354. [Google Scholar] [CrossRef]
- Su, W.; Liu, J.; Bai, P.; Ma, B.; Liu, W. Pathogenic fungi-induced susceptibility is mitigated by mutual Lactobacillus plantarum in the Drosophila melanogaster model. BMC Microbiol. 2019, 19, 302. [Google Scholar] [CrossRef]
- Storelli, G.; Defaye, A.; Erkosar, B.; Hols, P.; Royet, J.; Leulier, F. Lactobacillus plantarum promotes Drosophila systemic growth by modulating hormonal signals through TOR-dependent nutrient sensing. Cell Metab. 2011, 14, 403–414. [Google Scholar] [CrossRef] [PubMed]
- Tomberlin, J.K.; Crippen, T.L.; Tarone, A.M.; Chaudhury, M.F.; Singh, B.; Cammack, J.A.; Meisel, R.P. A review of bacterial interactions with blow flies (Diptera: Calliphoridae) of medical, veterinary, and forensic importance. Ann. Entomol. Soc. Am. 2017, 110, 19–36. [Google Scholar] [CrossRef]
- De Smet, J.; Wynants, E.; Cos, P.; Van Campenhout, L. Microbial community dynamics during rearing of black soldier fly larvae (Hermetia illucens) and impact on exploitation potential. Appl. Environ. Microbiol. 2018, 84, e02717–e02722. [Google Scholar] [CrossRef] [PubMed]
- Thompson, C.R.; Brogan, R.S.; Scheifele, L.Z.; Rivers, D.B. Bacterial interactions with necrophagous flies. Ann. Entomol. Soc. Am. 2013, 106, 799–809. [Google Scholar] [CrossRef]
- Ridley, E.V.; Wong, A.C.; Westmiller, S.; Douglas, A.E. Impact of the resident microbiota on the nutritional phenotype of Drosophila melanogaster. PLoS ONE 2012, 7, e36765. [Google Scholar] [CrossRef] [PubMed]
- Douglas, A.E. The microbial dimension in insect nutritional ecology. Funct. Ecol. 2009, 23, 38–47. [Google Scholar] [CrossRef]
- Deguenon, J.M.; Travanty, N.; Zhu, J.; Carr, A.; Denning, S.; Reiskind, M.H.; Watson, D.W.; Michael Roe, R.; Ponnusamy, L. Exogenous and endogenous microbiomes of wild-caught Phormia regina (Diptera: Calliphoridae) flies from a suburban farm by 16S rRNA gene sequencing. Sci. Rep. 2019, 9, 20365. [Google Scholar] [CrossRef]
- Gibbons, S.M. Keystone taxa indispensable for microbiome recovery. Nat. Microbiol. 2020, 5, 1067–1068. [Google Scholar] [CrossRef]
- Singh, B.; Crippen, T.L.; Zheng, L.; Fields, A.T.; Yu, Z.; Ma, Q.; Wood, T.K.; Dowd, S.E.; Flores, M.; Tomberlin, J.K. A metagenomic assessment of the bacteria associated with Lucilia sericata and Lucilia cuprina (Diptera: Calliphoridae). Appl. Microbiol. Biotechnol. 2015, 99, 869–883. [Google Scholar] [CrossRef]
- Chandler, J.A.; Morgan Lang, J.; Bhatnagar, S.; Eisen, J.A.; Kopp, A. Bacterial communities of diverse Drosophila species: Ecological context of a host–microbe model system. PLoS Genet. 2011, 7, e1002272. [Google Scholar] [CrossRef]
- Fuhrman, J.A.; Cram, J.A.; Needham, D.M. Marine microbial community dynamics and their ecological interpretation. Nat. Rev. Microbiol. 2015, 13, 133–146. [Google Scholar] [CrossRef] [PubMed]
- Allison, S.D.; Martiny, J.B. Resistance, resilience, and redundancy in microbial communities. Proc. Natl. Acad. Sci. USA 2008, 105, 11512–11519. [Google Scholar] [CrossRef] [PubMed]
- Preston-Mafham, J.; Boddy, L.; Randerson, P.F. Analysis of microbial community functional diversity using sole-carbon-source utilisation profiles—A critique. FEMS Microbiol. Ecol. 2002, 42, 1–14. [Google Scholar] [PubMed]
- Pechal, J.L.; Crippen, T.L.; Tarone, A.M.; Lewis, A.J.; Tomberlin, J.K.; Benbow, M.E. Microbial community functional change during vertebrate carrion decomposition. PLoS ONE 2013, 8, e79035. [Google Scholar] [CrossRef] [PubMed]
- Malacrinò, A. Host species identity shapes the diversity and structure of insect microbiota. Mol. Ecol. 2022, 31, 723–735. [Google Scholar] [CrossRef] [PubMed]
- Brucker, R.M.; Bordenstein, S.R. The roles of host evolutionary relationships (genus: Nasonia) and development in structuring microbial communities. Evolution 2012, 66, 349–362. [Google Scholar] [CrossRef] [PubMed]
- Wong, A.C.-N.; Dobson, A.J.; Douglas, A.E. Gut microbiota dictates the metabolic response of Drosophila to diet. J. Exp. Biol. 2014, 217, 1894–1901. [Google Scholar]
- Lim, S.h.; Park, J.K.; Park, W.B.; Won, D.H.; Kim, M.S.; Hong, S.; Do, Y. Gut microbiome of three species of Odonata. Entomol. Res. 2023, 53, 167–172. [Google Scholar] [CrossRef]
- Lee, Y.H. Overview of Mendelian randomization analysis. J. Rheum. Dis. 2020, 27, 241–246. [Google Scholar] [CrossRef]
- Liu, X.; Tong, X.; Zou, Y.; Lin, X.; Zhao, H.; Tian, L.; Jie, Z.; Wang, Q.; Zhang, Z.; Lu, H.; et al. Mendelian randomization analyses support causal relationships between blood metabolites and the gut microbiome. Nat. Genet. 2022, 54, 52–61. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).