
Haplotype Disequilibrium in the TLR Genes of Czech Red Pied Cattle


Abstract
1. Introduction
2. Materials and Methods
2.1. Animals
2.2. DNA Samples
2.3. Next Generation Sequencing (NGS)
2.4. Sequencing Data Processing
2.5. Characterisation of SNPs
2.6. Genotyping
2.7. Haplotype Determination
2.8. Haplotype Graph Construction
3. Results
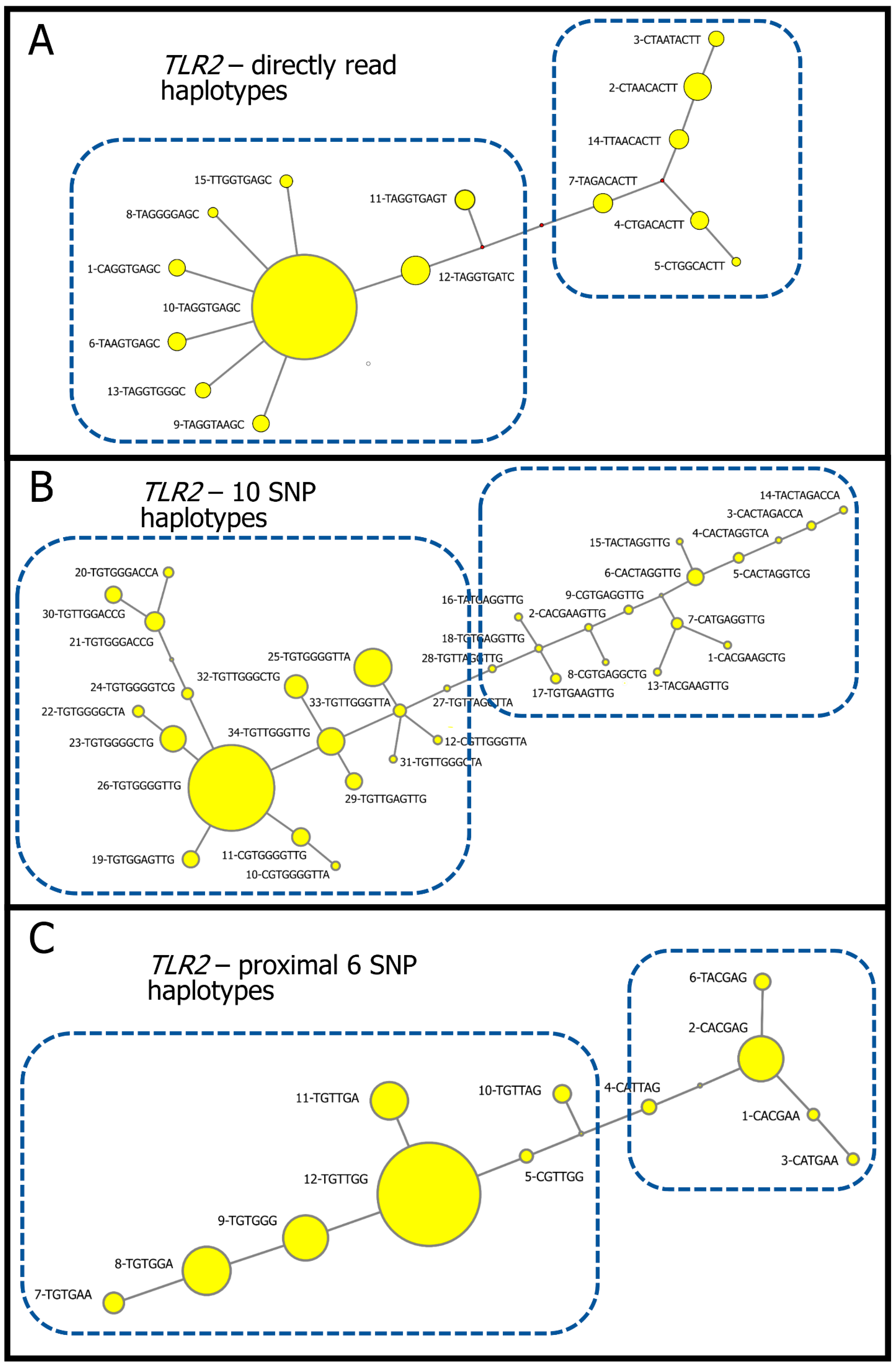
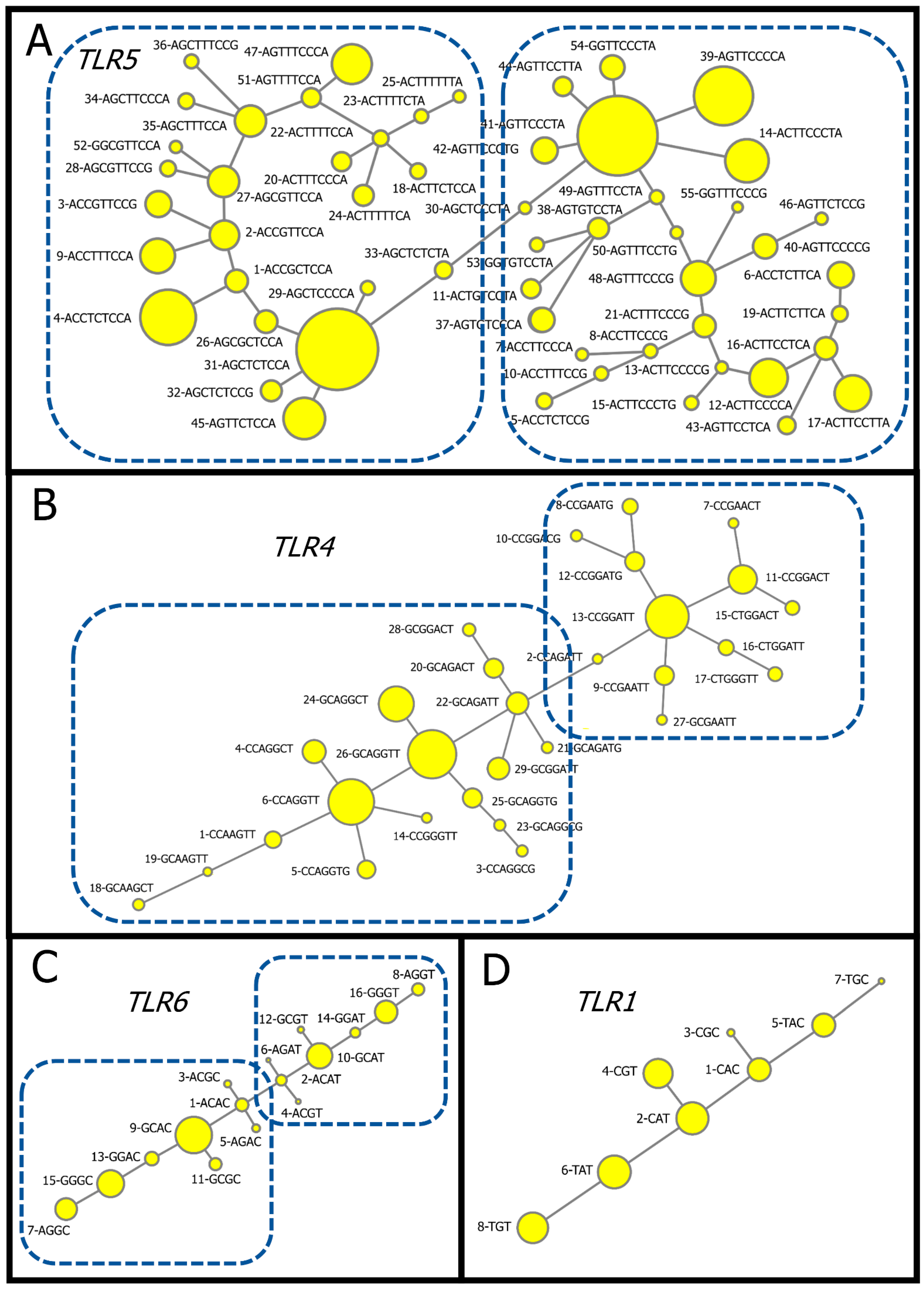
3.1. Directly Read Haplotypes
3.2. Reconstructed Haplotypes
4. Discussion
4.1. Context of the Previous Knowledge
4.2. Possible Reasons for the Observed Haplotype Clustering
4.3. A Dual Function Hypothesis
4.4. Approaches to the Discrimination of the Causative Mechanisms
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kawai, T.; Akira, S. The role of pattern-recognition receptors in innate immunity: Update on Toll-like receptors. Nat. Immunol. 2010, 11, 373–384. [Google Scholar] [CrossRef] [PubMed]
- Stein, D.; Roth, S.; Vogelsang, E.; Nüsslein-Volhard, C. The polarity of the dorsoventral axis in the Drosophila embryo is defined by an extracellular signal. Cell 1991, 65, 725–735. [Google Scholar] [CrossRef] [PubMed]
- Lemaitre, B.; Nicolas, E.; Michaut, L.; Reichhart, J.M.; Hoffman, J.A. The dorsoventral regulatory gene cassette spatzle/Toll/cactus controls the potent antifungal response in Drosophila adults. Cell 1996, 86, 973–983. [Google Scholar] [CrossRef] [PubMed]
- Wiens, M.; Korzhev, M.; Perovic-Ottstadt, S.; Luthringer, B.; Brandt, D.; Klein, S.; Mueller, W.E.G. Toll-like receptors are part of the innate immune defense system of sponges (Demospongiae: Porifera). Mol. Biol. Evol. 2007, 24, 792–804. [Google Scholar] [CrossRef]
- Zipfel, C.; Robatzek, S.; Navarro, L.; Oakeley, E.J.; Jones, J.D.G.; Felix, G.; Boller, T. Bacterial disease resistance in Arabidopsis through flagellin perception. Nature 2004, 428, 764–767. [Google Scholar] [CrossRef]
- Kannaki, T.R.; Shanmugam, M.; Verma, P.C. Toll-like receptors and their role in animal reproduction. Anim. Reprod. Sci. 2011, 125, 1–12. [Google Scholar] [CrossRef]
- Wahid, H.H.; Dorian, C.L.; Chin, P.Y.; Hutchinson, M.R.; Rice, K.C.; Olson, D.M.; Moldenhauer, L.M.; Robertson, S.A. Toll-like receptor 4 is an essential upstream regulator of on-time parturition and perinatal viability in mice. Endocrinology 2015, 156, 3828–3841. [Google Scholar] [CrossRef]
- Boichard, D.; Ducrocq, V.; Fritz, S. Sustainable dairy cattle selection in the genomic era. J. Anim. Breed. Genet. 2015, 132, 135–143. [Google Scholar] [CrossRef]
- Maurić Maljković, M.; Vlahek, I.; Piplica, A.; Ekert Kabalin, A.; Sušić, V.; Stevanović, V. Prospects of toll-like receptors in dairy cattle breeding. Anim. Genet. 2023, 2023, 1–10. [Google Scholar] [CrossRef]
- Jungi, T.W.; Farhat, K.; Burgener, I.A.; Werling, D. Toll-like receptors in domestic animals. Cell Tissue Res. 2011, 343, 107–120. [Google Scholar] [CrossRef]
- Jann, O.C.; Werling, D.; Chang, J.S.; Haig, D.; Glass, E.J. Molecular evolution of bovine Toll-like receptor 2 suggests substitutions of functional relevance. BMC Evol. Biol. 2008, 8, 288. [Google Scholar] [CrossRef]
- Seabury, C.M.; Seabury, P.M.; Decker, J.E.; Schnabel, R.D.; Taylor, J.F.; Womack, J.E. Diversity and evolution of 11 innate immune genes in Bos taurus taurus and Bos taurus indicus cattle. Proc. Natl. Acad. Sci. USA 2010, 107, 151–156. [Google Scholar] [CrossRef]
- Fisher, C.A.; Bhattarai, E.K.; Osterstock, J.B.; Dowd, S.E.; Seabury, P.M.; Vikram, M.; Whitlock, R.H.; Schukken, Y.H.; Schnabel, R.D.; Taylor, J.F.; et al. Evolution of the bovine TLR gene family and member associations with Mycobacterium avium subspecies paratuberculosis infection. PLoS ONE 2011, 6, 11. [Google Scholar] [CrossRef]
- Sharma, B.S.; Leyva, I.; Schenkel, F.; Karrow, N.A. Association of Toll-Like receptor 4 polymorphisms with somatic cell score and lactation persistency in Holstein bulls. Dairy Sci. 2006, 89, 3626–3635. [Google Scholar] [CrossRef]
- Zhang, L.P.; Gan, Q.F.; Ma, T.H.; Li, H.D.; Wang, X.P.; Li, J.Y.; Gao, X.; Chen, J.B.; Ren, H.Y.; Xu, S.Z. Toll-like receptor 2 gene polymorphism and its relationship with SCS in dairy cattle. Anim. Biotech. 2009, 20, 87–95. [Google Scholar] [CrossRef]
- Koets, A.; Santema, W.; Mertens, H.; Oostenrijk, D.; Keestra, M.; Overdijk, M.; Labouriau, R.; Franken, P.; Frijters, A.; Nielen, M.; et al. Susceptibility to paratuberculosis infection in cattle is associated with single nucleotide polymorphisms in Toll-like receptor 2 which modulate immune responses against Mycobacterium avium subspecies paratuberculosis. Prev. Vet. Med. 2010, 93, 305–315. [Google Scholar] [CrossRef]
- Bhaladhare, A.; Sharma, D.; Kumar, A.; Sonwane, A.; Chauhan, A.; Singh, R.; Kumar, P.; Yadav, R.; Baqir, M.; Bhushan, B.; et al. Single nucleotide polymorphisms in toll-like receptor genes and case-control association studies with bovine tuberculosis. Vet. World 2016, 9, 458–464. [Google Scholar] [CrossRef]
- Bilgen, N.; Kul, B.C.; Offord, V.; Werling, D.; Ertugrul, O. Determination of genetic variations of Toll-like receptor (TLR) 2, 4, and 6 with next-generation sequencing in native cattle breeds of Anatolia and Holstein Friesian. Diversity 2016, 8, 23. [Google Scholar] [CrossRef]
- Ruiz-Larrañaga, O.; Manzano, C.; Iriondo, M.; Garrido, J.M.; Molina, E.; Vazquez, P.; Juste, R.A.; Estonba, A. Genetic variation of toll-like receptor genes and infection by Mycobacterium avium ssp. paratuberculosis in Holstein-Friesian cattle. J. Dairy Sci. 2011, 94, 3635–3641. [Google Scholar] [CrossRef]
- Beecher, C.; Daly, M.; Childs, S.; Berry, D.P.; Magee, D.A.; McCarthy, T.V.; Giblin, L. Polymorphisms in bovine immune genes and their associations with somatic cell count and milk production in dairy cattle. BMC Genet. 2010, 11, 99. [Google Scholar] [CrossRef]
- Abdel-Shafy, H.; Bortfeldt, R.H.; Tetens, J.; Brockmann, G.A. Single nucleotide polymorphism and haplotype effects associated with somatic cell score in German Holstein cattle. Genet. Sel. Evol. 2014, 46, 35. [Google Scholar] [CrossRef] [PubMed]
- Novák, K.; Bjelka, M.; Samake, K.; Valčíková, T. Potential of TLR-gene diversity in Czech indigenous cattle for resistance breeding as revealed by hybrid sequencing. Arch. Anim. Breed. 2019, 62, 477–490. [Google Scholar] [CrossRef] [PubMed]
- White, S.N.; Taylor, K.H.; Abbey, C.A.; Gill, C.A.; Womack, J.E. Haplotype variation in bovine Toll-like receptor 4 and computational prediction of a positively selected ligand-binding domain. Proc. Natl. Acad. Sci. USA 2003, 100, 10364–10369. [Google Scholar] [CrossRef] [PubMed]
- Seabury, C.M.; Cargill, E.J.; Womack, J.E. Sequence variability and protein domain architectures for bovine Toll-like receptors 1, 5, and 10. Genomics 2007, 90, 502–515. [Google Scholar] [CrossRef] [PubMed]
- Sim, N.L.; Kumar, P.; Hu, J.; Henikoff, S.; Schneider, G.; Ng, P.C. SIFT web server: Predicting effects of amino acid substitutions on proteins. Nucl. Acids Res. 2012, 40, W452–W457. [Google Scholar] [CrossRef]
- Hoogendoorn, B.; Owen, M.J.; Oefner, P.J.; Williams, N.; Austin, J.; O’Donovan, M.C. Genotyping single nucleotide polymorphisms by primer extension and high performance liquid chromatography. Human Genet. 1999, 104, 89–93. [Google Scholar] [CrossRef]
- Bjelka, M.; Novák, K. Association of TLR gene variants in a Czech Red Pied cattle population with reproductive traits. Vet. Immunol. Immunopathol. 2020, 220, 109997. [Google Scholar] [CrossRef]
- Stephens, M.; Smith, N.J.; Donnelly, P. A new statistical method for haplotype reconstruction from population data. Am. J. Hum. Genet. 2001, 68, 978–989. [Google Scholar] [CrossRef]
- Bandelt, H.-J.; Forster, P.; Röhl, A. Median-joining networks for inferring intraspecific phylogenies. Mol. Biol. Evol. 1999, 16, 37–48. [Google Scholar] [CrossRef]
- Polzin, T.; Daneschmand, S.V. On Steiner trees and minimum spanning trees in hypergraphs. Oper. Res. Lett. 2003, 31, 12–20. [Google Scholar] [CrossRef]
- Kloch, A.; Wenzel, M.A.; Laetsch, D.R.; Michalski, O.; Bajer, A.; Behnke, J.M.; Welc-Falęciak, R.; Piertney, S.B. Signatures of balancing selection in toll-like receptor (TLRs) genes—Novel insights from a free-living rodent. Sci. Rep. 2018, 8, 8361. [Google Scholar] [CrossRef]
- Tschirren, B.; Andersson, M.; Scherman, K.; Westerdahl, H.; Mittl, P.R.E.; Råberg, L. Polymorphisms at the innate immune receptor TLR2 are associated with Borrelia infection in a wild rodent population. Proc. R. Soc. B Biol. Sci. 2013, 280, 20130364. [Google Scholar] [CrossRef]
- Morger, J.; Råberg, L.; Hille, S.M.; Helsen, S.; Štefka, J.; Al-Sabi, M.M.; Kapel, C.M.O.; Mappes, T.; Essbauer, S.; Ulrich, R.G.; et al. Distinct haplotype structure at the innate immune receptor Toll-like receptor 2 across bank vole populations and lineages in Europe. Biol. J. Linn. Soc. 2015, 116, 124–133. [Google Scholar] [CrossRef]
- Zhong, X.; Lundberg, M.; Råberg, L. Divergence in coding sequence and expression of different functional categories of immune genes between two wild rodent species. Genome Biol. Evol. 2021, 13, evab023. [Google Scholar] [CrossRef]
- Quéméré, E.; Hessenauer, P.; Galan, M.; Fernandez, M.; Merlet, J.; Chaval, Y.; Morellet, N.; Verheyden, H.; Gilot-Fromont, E.; Charbonnel, N. Pathogen-mediated selection favours the maintenance of innate immunity gene polymorphism in a widespread wild ungulate. J. Evol. Biol. 2021, 34, 1156–1166. [Google Scholar] [CrossRef]
- Yang, L.; Seki, E. Toll-like receptors in liver fibrosis: Cellular crosstalk and mechanisms. Front. Physiol. 2012, 3, 138. [Google Scholar] [CrossRef]
- Regan, T.; Nally, K.; Carmody, R.; Houston, A.; Shanahan, F.; MacSharry, J.; Brint, E. Identification of TLR10 as a key mediator of the inflammatory response to Listeria monocytogenes in intestinal epithelial cells and macrophages. J. Immunol. 2013, 191, 6084–6092. [Google Scholar] [CrossRef]
- Coffey, T.J.; Werling, D. Therapeutic targeting of the innate immune system in domestic animals. Cell Tissue Res. 2011, 343, 251–261. [Google Scholar] [CrossRef]
- Kurokawa, K.; Kim, M.-S.; Ichikawa, R.; Ryu, K.-H.; Dohmae, N.; Nakayama, H.; Lee, B.L. Environment-mediated accumulation of diacyl lipoproteins over their triacyl counterparts in Staphylococcus aureus. J. Bacteriol. 2012, 194, 3299–3306. [Google Scholar] [CrossRef]
- Lippi, C.A.; Ryan, S.J.; White, A.L.; Gaff, H.D.; Carlson, C.J. Trends and opportunities in tick-borne disease geography. J. Med. Entomol. 2021, 58, 2021–2029. [Google Scholar] [CrossRef]
- Song, Y.; Shou, L.M.; Ai, L.-Y.; Bei, Y.; Chen, M.-T. Mini-review: The non-immune functions of Toll-like receptors. Crit. Rev. Eukaryot. Gene Expr. 2019, 29, 37–45. [Google Scholar] [CrossRef] [PubMed]
- Kadam, L.; Gomez-Lopez, N.; Mial, T.N.; Kohan-Ghadr, H.R.; Drewlo, S. Rosiglitazone regulates TLR4 and rescues HO-1 and NRF2 expression in myometrial and decidual macrophages in inflammation-induced preterm birth. Reprod. Sci. 2017, 24, 1590–1599. [Google Scholar] [CrossRef] [PubMed]
- Francisco, S.; Billod, J.-M.; Merino, J.; Punzón, C.; Gallego, A.; Arranz, A.; Martin-Santamaria, S.; Fresno, M. Induction of TLR4/TLR2 interaction and heterodimer formation by low endotoxic atypical LPS. Front. Immunol. 2022, 12, 748303. [Google Scholar] [CrossRef] [PubMed]
- Mazzone, P.; Di Paolo, A.; Petrucci, L.; Torricelli, M.; Corneli, S.; Sebastiani, C.; Ciullo, M.; Sebastianelli, M.; Costarelli, S.; Scoccia, E.; et al. Evaluation of single nucleotide polymorphisms (SNPs) associated with genetic resistance to bovine paratuberculosis in Marchigiana beef cattle, an Italian native breed. Animals 2023, 13, 587. [Google Scholar] [CrossRef]
- Roe, D.; Vierra-Green, C.; Pyo, C.-W.; Eng, K.; Hall, R.; Kuang, R.; Spellman, S.; Ranade, S.; Geraghty, D.E.; Maiers, M. Revealing complete complex KIR haplotypes phased by long-read sequencing technology. Genes Immun. 2017, 18, 127–134. [Google Scholar] [CrossRef]
- Low, W.Y.; Tearle, R.; Liu, R.; Koren, S.; Rhie, A.; Bickhart, D.M.; Rosen, B.D.; Kronenberg, Z.N.; Kingan, S.B.; Tseng, E.; et al. Haplotype-resolved genomes provide insights into structural variation and gene content in Angus and Brahman cattle. Nat. Commun. 2021, 11, 2071. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Gene | SNP Identifier | Chromosomal Position | SNP a | SNaPshot Primer (Sequence 5′→3′) | Multiplex | SNP Type | Predicted Effect b | Variant Frequency |
|---|---|---|---|---|---|---|---|---|
| TLR1 | rs43702940 | 6_59688857 | 798C>T | CGCCAAACCAACTGGAGGATCGT | A1 | synonymous | low | 0.473 |
| TLR1 | rs210538093 | 6_59687893 | 1762G>A | CAGCCCAGGGACCACAATGGTGA | C1 | Val523Ile | moderate | 0.380 |
| TLR1 | rs109456287 | 6_59687558 | 2097T>C | (T4)CTCTGGACAAAGTTGGGAGACAAGAC | C2 | synonymous | low | 0.780 |
| TLR2 | rs68343162 | 17_3953930 | 115T>C | (T10)GAAATAACCAAGAGGGAAATGGAAT | A2 | intron | modifier | 0.827 |
| TLR2 | 17_3953036 | 1009A>G | ACACACCTCTGCAGGTCTCTGTTGC | B1 | Ala151Thr | tolerated | 0.260 | |
| TLR2 | rs68268249 | 17_3953001 | 1044T>C | (T9)GTGAAAAGCCTTGACCTGTCCAACAA | B3 | synonymous | low | 0.847 |
| TLR2 | rs55617172 | 17_3952998 | 1047G>T | (T12)GCAGGTCTCTGTTGCYGACATAGGTGAT | B4 | missense, 63Glu>Asp | moderate | 0.490 |
| TLR2 | rs68268250 | 17_3952985 | 1060G>A | (T5)CACACCTCTGCAGGTCTCTGTTGC | B2 | Gly68Ser | moderate | 0.700 |
| TLR2 | rs43706434 | 17_3952732 | 1313G>A | (T18)CTGTTACTATTTCCTACTTTTAGGGTC | B5 | Arg152Gln in LRR5 c | moderate | 0.787 |
| TLR2 | rs68268260 | 17_3951499 | 2546G>A | (T12)GGTCGACTGGCCCGATGACTACC | C3 | Arg563His in LRR20c | moderate | 0.877 |
| TLR2 | rs41830058 | 17_3951480 | 2565T>C | (T16)CTACCRCTGTGACTCTCCCTCCCA | C4 | synonymous | low | 0.688 |
| TLR2 | rs68343171 | 17_3951162 | 2883T>C | (T18)CCACTTGCCAGGAATGAAGTCTCGCTT | C5 | synonymous | low | 0.118 |
| TLR2 | rs68268268 | 17_3950839 | 3206G>A | (T17)GGTTAAATTTGAGAGCTGCAATAA | F1 | Arg782Lys | tolerated | 0.833 |
| TLR4 | rs29017188 | 8_108829143 | 245G>C | CTTCTTCTTCCTCTAACTTCCCCTC | D1 | 5′-UTR | changing expression | 0.401 |
| TLR4 | rs43578094 | 8_108829508 | 610C>T | (T5)GGGCCCAGCACAGGGAAACTGAGCA | D2 | intron | modifier | 0.931 |
| TLR4 | rs8193046 | 8_108833985 | 5087A>G | (T10)GCTAAGGTGCATGCAGGAAGACACC | D3 | intron | modifier | 0.575 |
| TLR4 | rs8193047 | 8_108834032 | 5134G>A | (T13)GATTTTGTAGAGATTCAGCTCCATGCA | D4 | synonymous | low | 0.849 |
| TLR4 | rs43578100 | 8_108836897 | 7999A>G | (T21)GGTTTCCTATTCAGCAGAAATATT | D5 | intron | modifier | 0.463 |
| TLR4 | rs8193060 | 8_108838320 | 9422C>T | ACTCGCTCCGGATCCTAGACTGCAG | E1 | synonymous | low | 0.356 |
| TLR4 | rs8193072 | 8_108839208 | 10310T>G | (T25)CCACCTGAGGAGGAGAATCCCCTGA | E6 | 3′-UTR | low | 0.839 |
| TLR5 | 16_27307966 | 305A>G | GCATGGTAAC TCGTGTACAC CATCAGA | F1 | upstream | modifier | 0.385 | |
| TLR5 | 16_27307783 | 488C>G | (T6)CCAGGGATGAAACCCRTGTCTCCTG | E2 | upstream | modifier | 0.383 | |
| TLR5 | rs55617223 | 16_27307726 | 545C>T | (T10)CCAGGGAAGTCTTGCTGGCCTACTG | E3 | upstream | modifier | 0.407 |
| TLR5 | ss73689429 | 16_27307652 | 619T>G | CCACAGCACCTTTGAGGCTGTGAC | G1 | upstream | modifier | 0.853 |
| TLR5 | ss73689443 | 16_27306535 | 1736C>T | (T2)GTACTTACAAYCATGCTTGCTATTTTT | G2 | upstream | modifier | 0.617 |
| TLR5 | rs55617187 | 16_27304557 | 3714T>C | (T15)GATTGAGCCAATGGATAAAAGCACT | E4 | synonymous | low | 0.481 |
| TLR5 | rs55617178 | 16_27304380 | 3891C>T | (T18)CACGAGGAACAGAGTCAAGGTGACAGT | E5 | synonymous | low | 0.907 |
| TLR5 | rs55617288 | 16_27303645 | 4626C>T | (T9)GGGGTCGCAAAGAGTAGGACATGACC | G3 | downstream | modifier | 0.690 |
| TLR5 | 14_27302910 | 5144A>G | (T16)CGTTTCCAGAGGGGCTGGTCAGTG | H4 | downstream | modifier | 0.10 | |
| TLR6 | rs43702941 | 6_59706074 | 855G>A | (T3)CCCAAATAGCTTTTTCTCTGTCCAAGTG | F2 | Asp214Asn | moderate | 0.714 |
| TLR6 | 6_59706064 | 865G>C | (T10)GTCAGTTGTAAGCACSCTAAACTATTC | F3 | Gly217Ala | moderate | 0.316 | |
| TLR6 | 6_59705939 | 990G>A | (T15)CCTTACTAAATTTTACCCTCAACCAC | F4 | Val259Met | moderate | 0.484 | |
| TLR6 | rs68268274 | 6_59705592 | 1337T>C | (T18)GATAAGTGTCTCCAATCTAGCTAAAGT | F5 | Asp374Glu | moderate | 0.333 |
| Gene | SNP Identifier | Chromosomal Position | SNP a | Multiplex | Effect Prediction | Variant Frequency |
|---|---|---|---|---|---|---|
| TLR2 | rs68343162 | 17_3953930 | 115T>C | intron | low | 0.163 |
| TLR2 | rs68268242 | 17_3953823 | 222A>T | intron | moderate | 0.168 |
| TLR2 | rs68343164 | 17_3953633 | 412G>A | intron | low | 0.159 |
| TLR2 | rs378996667 | 17_3953622 | 423G>A | intron | modifier | 0.176 |
| TLR2 | rs68268244 | 17_3953586 | 459T>C | intron | modifier | 0.150 |
| TLR2 | rs68343166 | 17_3953517 | 528G>A | intron | tolerated | 0.173 |
| TLR2 | rs68268246 | 17_3953507 | 538A>C | intron | low | 0.176 |
| TLR2 | rs68268247 | 17_3953421 | 624G>T | intron | moderate | 0.19 |
| TLR2 | rs68268248 | 17_3953388 | 657C>T | intron | moderate | 0.168 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Samaké, K.; Novák, K. Haplotype Disequilibrium in the TLR Genes of Czech Red Pied Cattle. Diversity 2023, 15, 811. https://doi.org/10.3390/d15070811
Samaké K, Novák K. Haplotype Disequilibrium in the TLR Genes of Czech Red Pied Cattle. Diversity. 2023; 15(7):811. https://doi.org/10.3390/d15070811
Chicago/Turabian StyleSamaké, Kalifa, and Karel Novák. 2023. "Haplotype Disequilibrium in the TLR Genes of Czech Red Pied Cattle" Diversity 15, no. 7: 811. https://doi.org/10.3390/d15070811
APA StyleSamaké, K., & Novák, K. (2023). Haplotype Disequilibrium in the TLR Genes of Czech Red Pied Cattle. Diversity, 15(7), 811. https://doi.org/10.3390/d15070811