
An Invisible Boundary between Geographic Ranges of Cryptic Species of Narrow-Headed Voles (Stenocranius, Lasiopodomys, Cricetidae) in Transbaikalia

 ,
,  ,
,
Abstract
:
1. Introduction
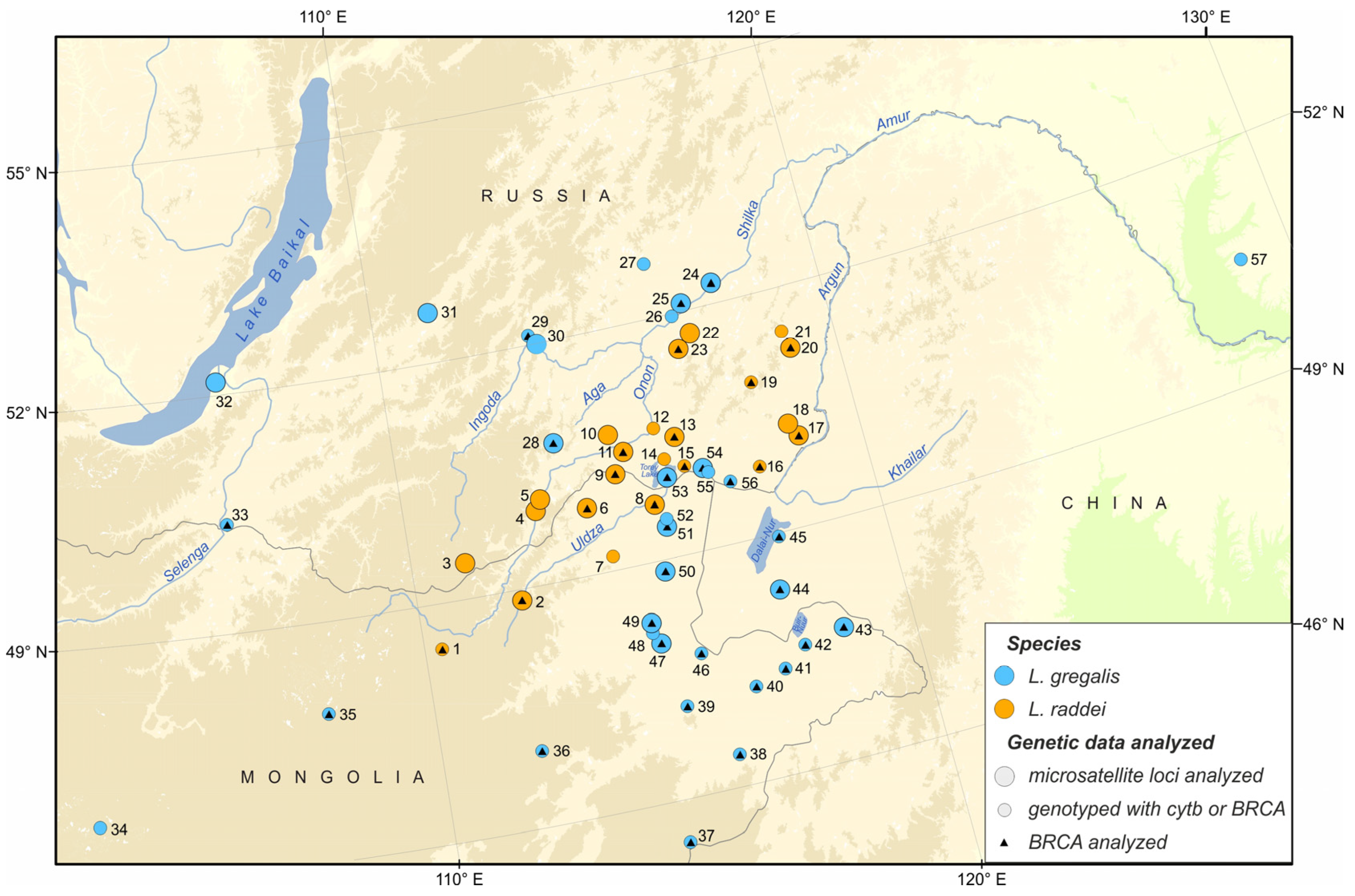
2. Materials and Methods
2.1. Sampling and DNA Isolation
2.2. Amplification of Mitochondrial Cytb and Nuclear BRCA1
2.3. Amplification of Microsatellite Loci
2.4. Cytb Phylogenetic Reconstruction and BRCA1 Genotyping
2.5. Microsatellite Loci Analyses
2.5.1. Quality Control and Grouping of Samples
2.5.2. Genetic Diversity Analyses
2.5.3. Population Structure Analysis
2.6. Species Distribution Modeling
2.6.1. Environmental Data
2.6.2. Occurrence Data and Target Group Backgrounds
2.6.3. Modeling
3. Results
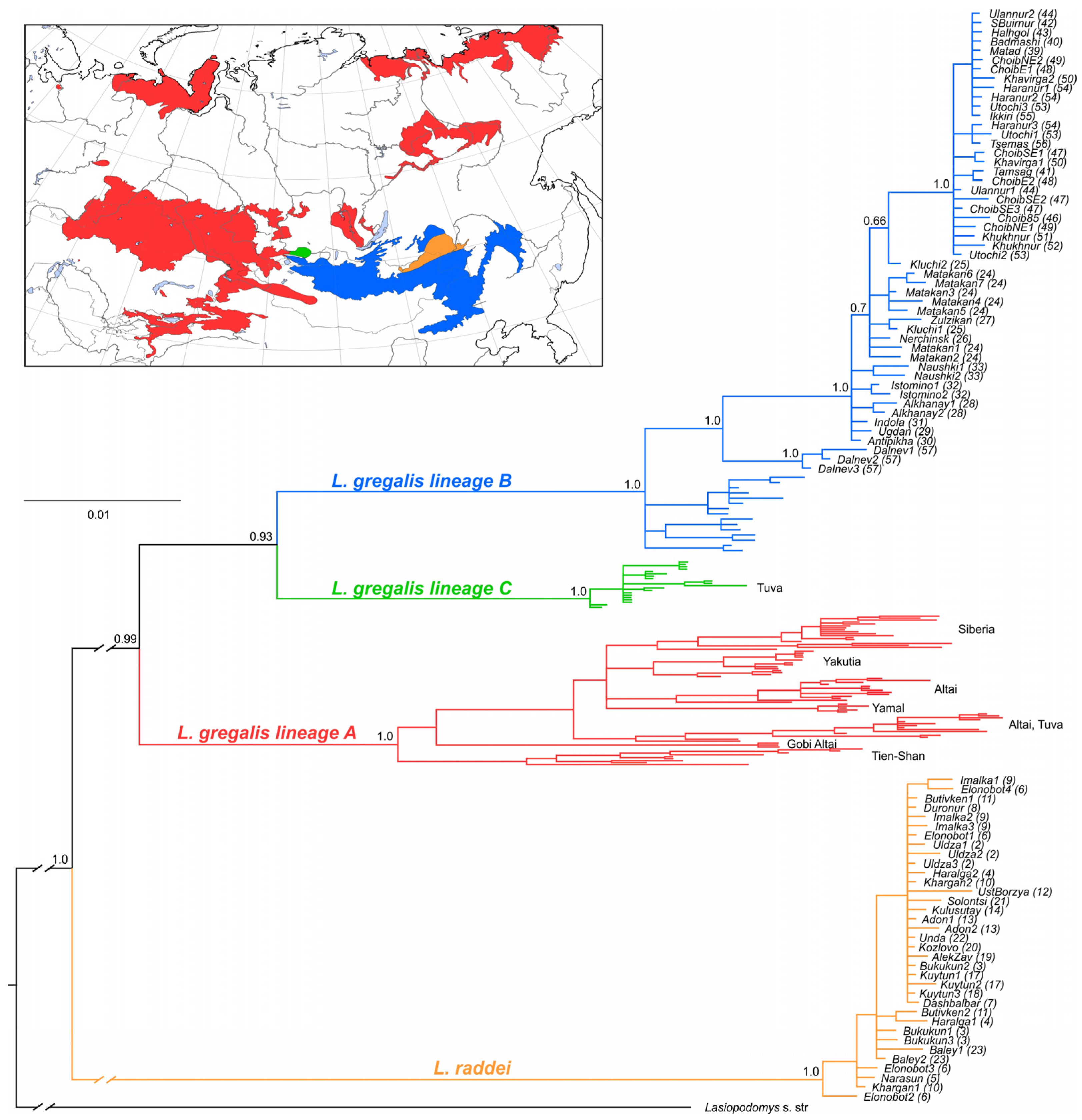
3.1. Species Identification by Means of Cytb and BRCA1
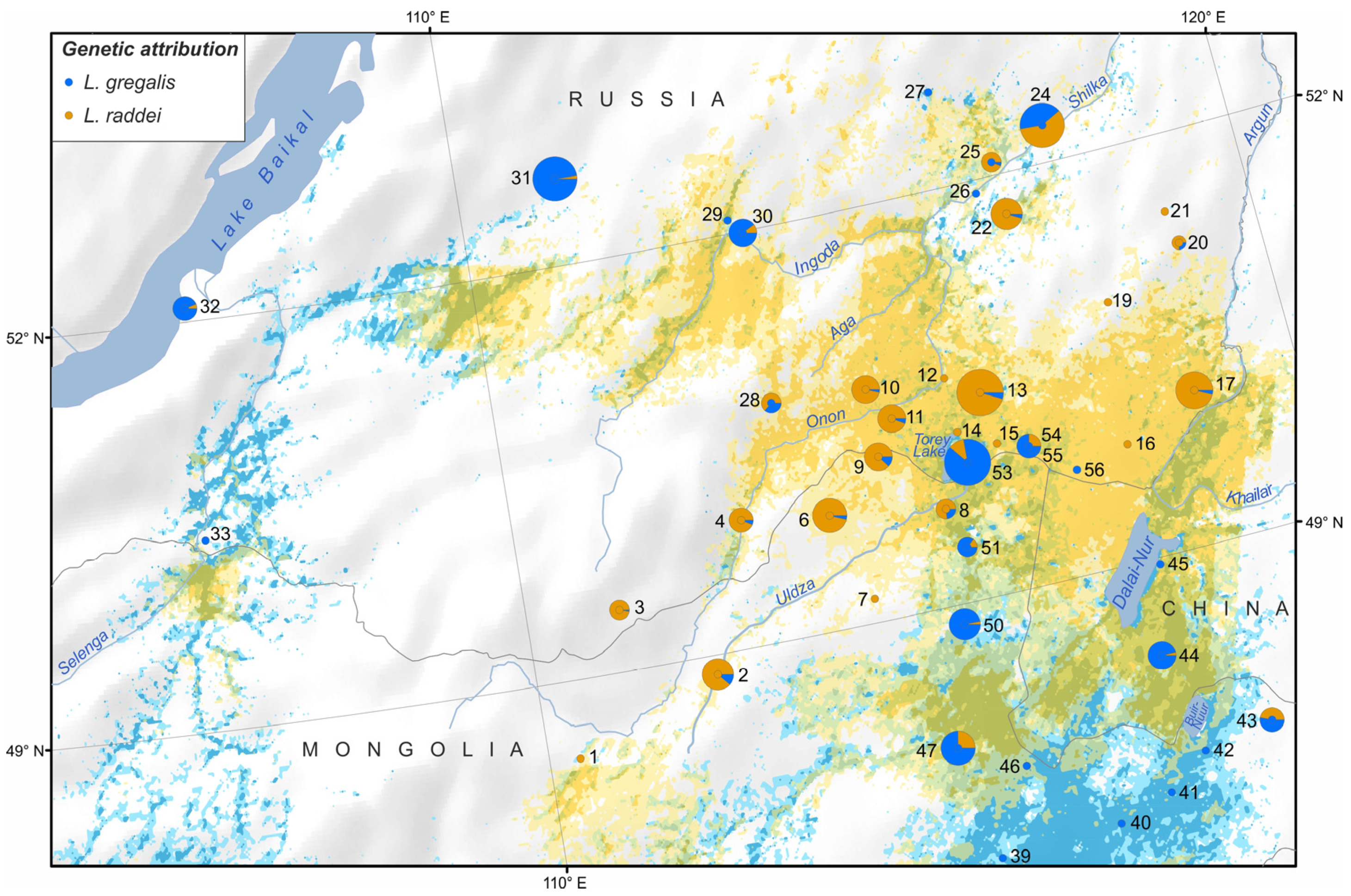
3.2. Population Structure Revealed by the Microsatellite Loci Analysis
3.2.1. Genetic Diversity Analysis
3.2.2. Population Diversity Analysis
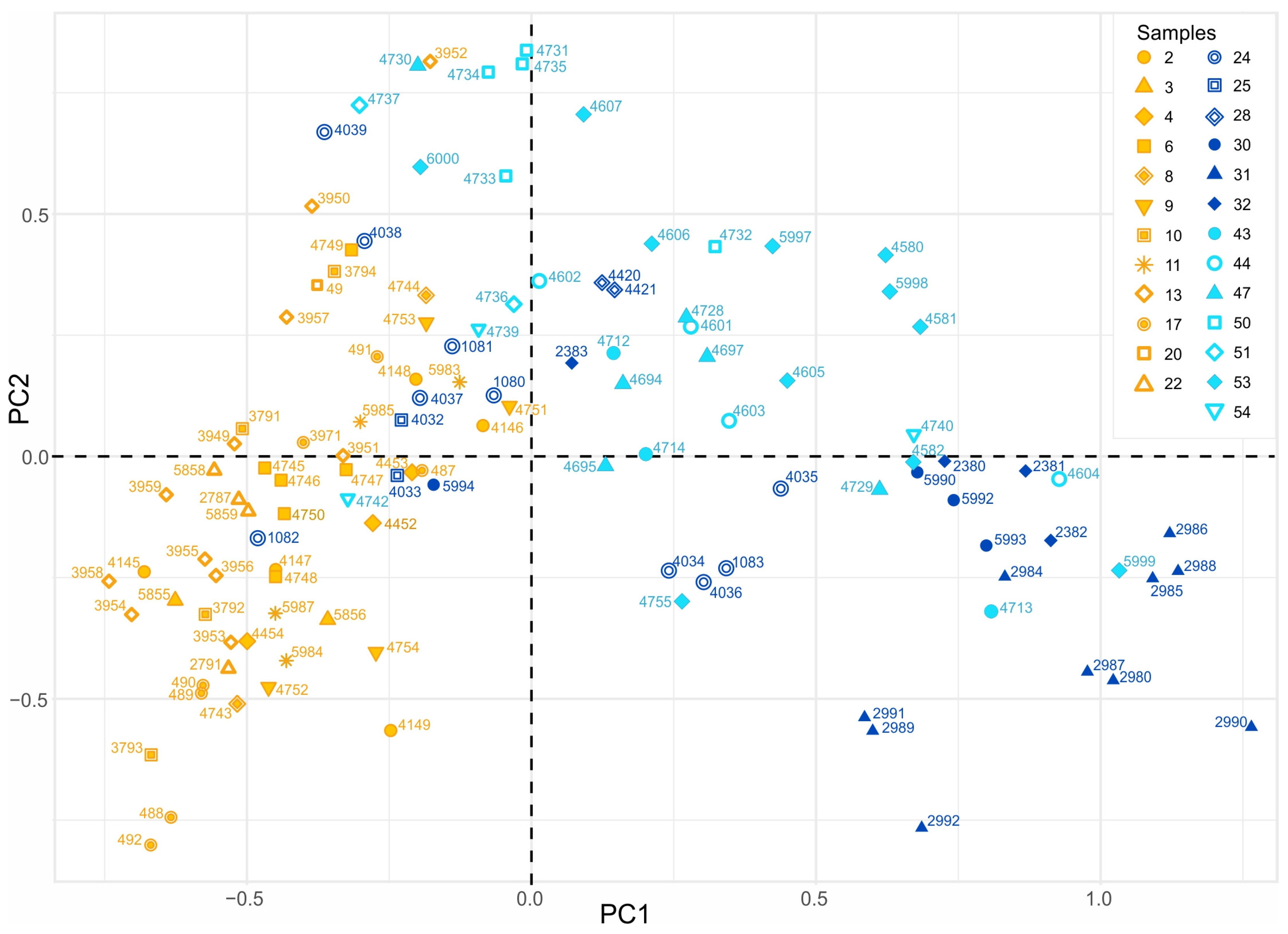
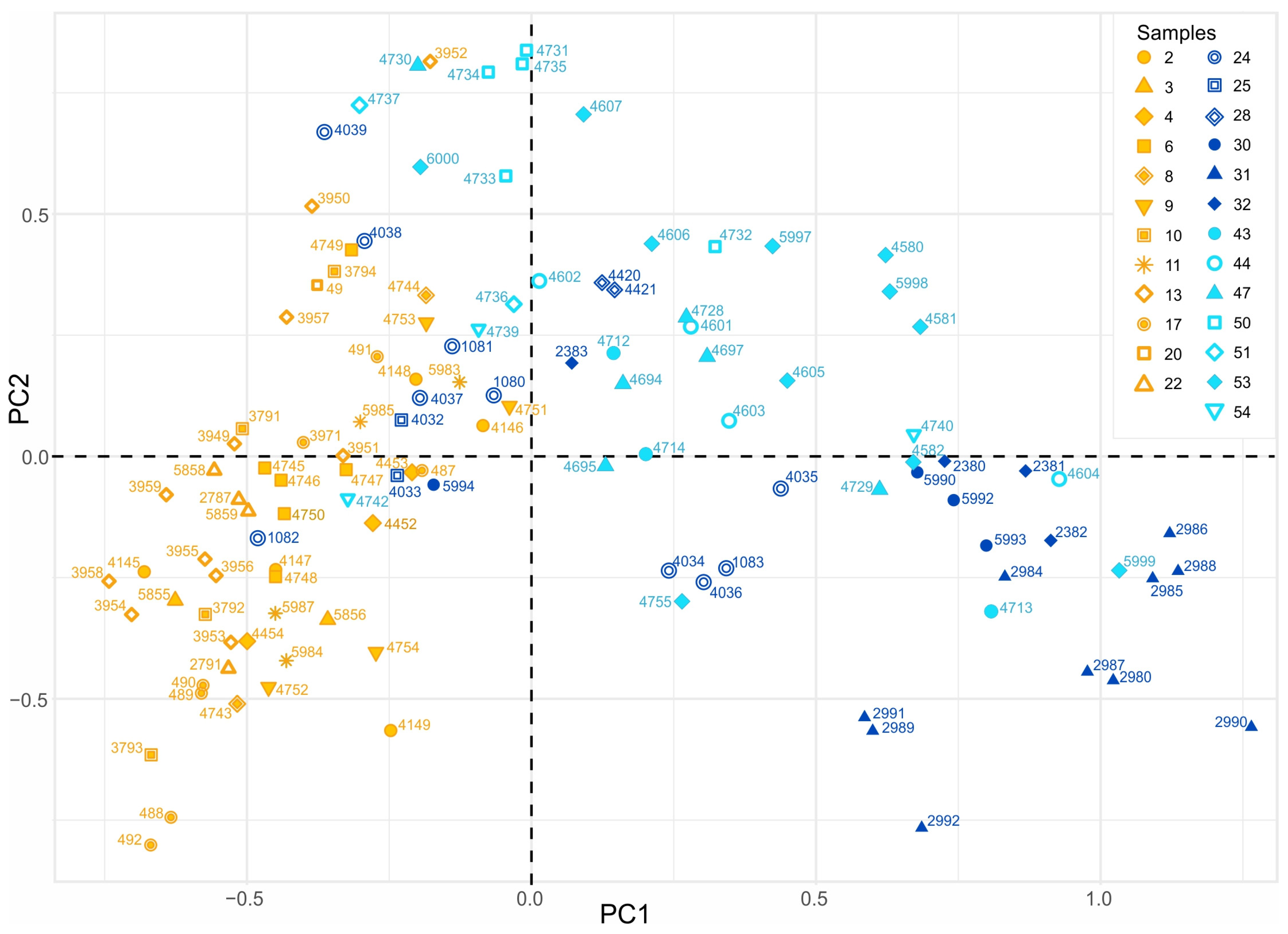
3.2.3. PCA
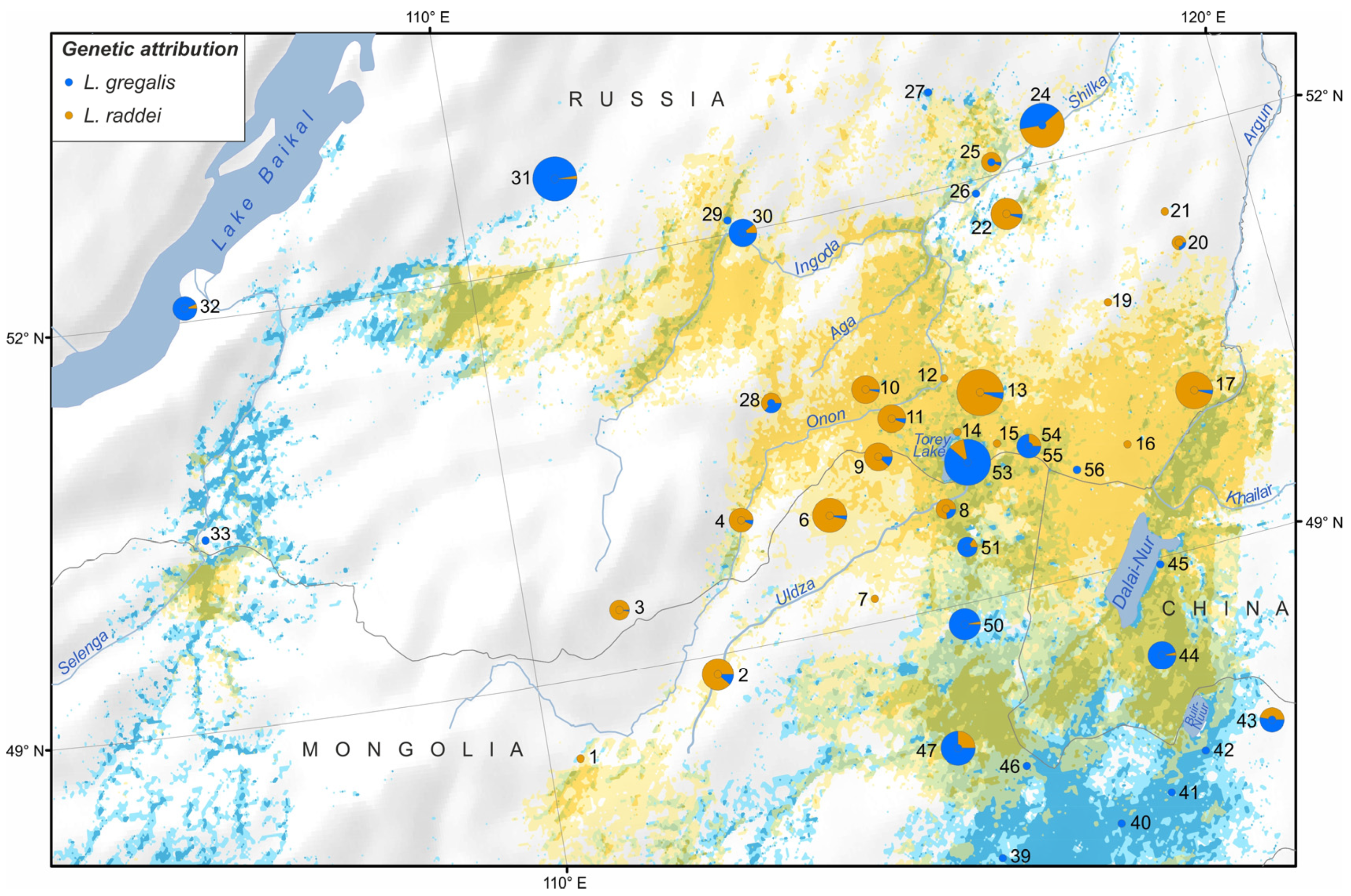
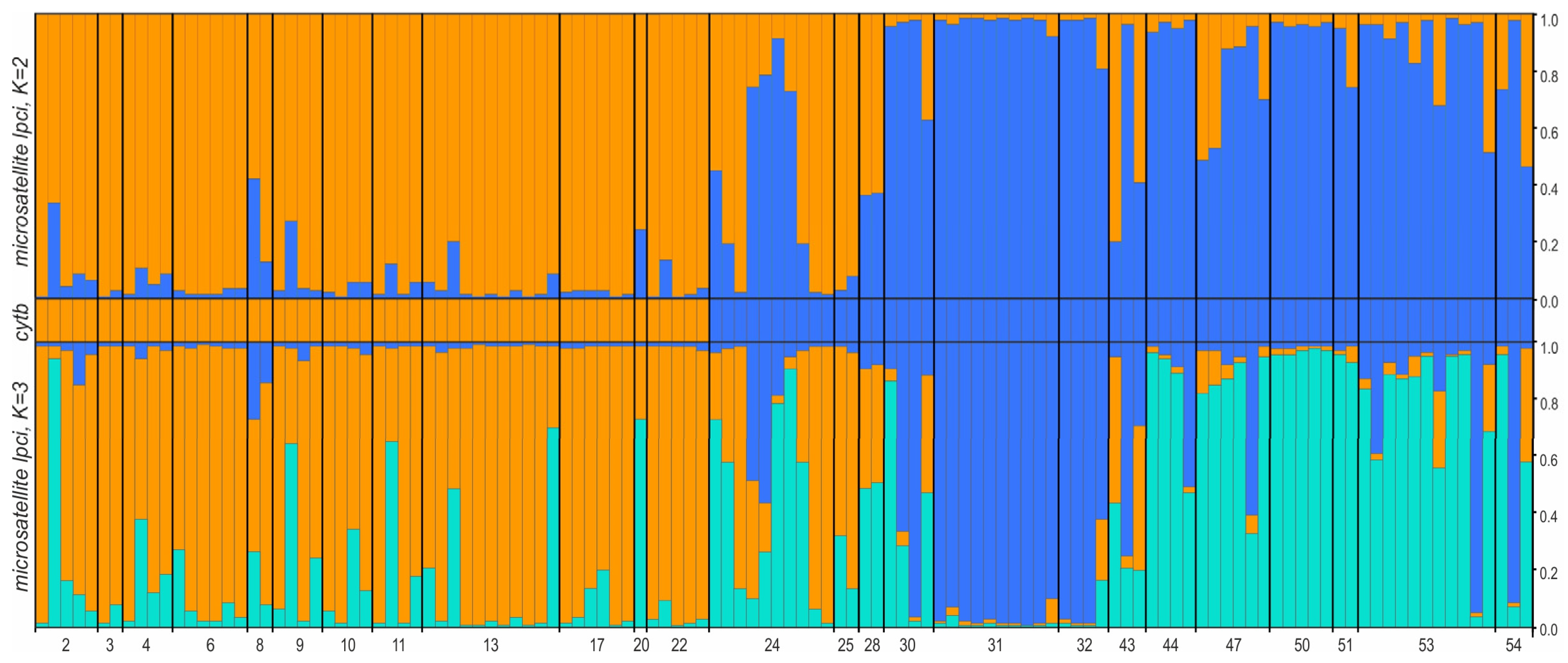
3.2.4. Population Structure
3.3. Species Distribution Prediction
4. Discussion
4.1. Clarification of Cryptic Species’ Ranges
4.2. Searching for Isolation Mechanisms
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hardin, G. The Competitive Exclusion Principle: An Idea That Took a Century to Be Born Has Implications in Ecology, Economics, and Genetics. Science 1960, 131, 1292–1297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gause, G.F. Experimental Studies on the Struggle for Existence. J. Exp. Biol. 1932, 9, 389–402. [Google Scholar] [CrossRef]
- Holt, R.D.; Grover, J.; Tilman, D. Simple Rules for Interspecific Dominance in Systems. Am. Nat. 1994, 144, 741–771. [Google Scholar] [CrossRef]
- Violle, C.; Nemergut, D.R.; Pu, Z.; Jiang, L. Phylogenetic Limiting Similarity and Competitive Exclusion: Phylogenetic Relatedness and Competition. Ecol. Lett. 2011, 14, 782–787. [Google Scholar] [CrossRef] [PubMed]
- Cothran, R.D.; Henderson, K.A.; Schmidenberg, D.; Relyea, R.A. Phenotypically Similar but Ecologically Distinct: Differences in Competitive Ability and Predation Risk among Amphipods. Oikos 2013, 122, 1429–1440. [Google Scholar] [CrossRef]
- Vodă, R.; Dapporto, L.; Dincă, V.; Vila, R. Why Do Cryptic Species Tend Not to Co-Occur? A Case Study on Two Cryptic Pairs of Butterflies. PLoS ONE 2015, 10, e0117802. [Google Scholar] [CrossRef] [Green Version]
- Barton, N.H.; Hewitt, G.M. Analysis of Hybrid Zones. Annu. Rev. Ecol. Syst. 1985, 16, 113–148. [Google Scholar] [CrossRef]
- Endler, J.A. Geographic Variation, Speciation, and Clines; Princeton University Press: Princeton, NJ, USA, 1977. [Google Scholar]
- Hewitt, G.M. Hybrid Zones-Natural Laboratories for Evolutionary Studies. Trends Ecol. Evol. 1988, 3, 158–167. [Google Scholar] [CrossRef]
- Gerasimov, I.P.; Velichko, A.A. Paleogeography of Europe During the Last 100,000 Years (Atlas-Monograph); Nauka: Moscow, Russia, 1982. [Google Scholar]
- Kowalski, K. Pleistocene Rodents of Europe. Folia Quat. 2001, 72, 389. [Google Scholar]
- Markova, A.; van Kolfschoten, T. Evolution of the European Ecosystems During Pleistocene-Holocene Transition (24–8 Kyr BP); KMK Scientific Press: Moscow, Russia, 2008. [Google Scholar]
- Shenbrot, G.; Krasnov, B. An Atlas of the Geographic Distribution of the Arvicoline Rodents of the World (Rodentia, Muridae: Arvicolinae); Pensoft: Sofia, Bulgaria, 2005. [Google Scholar]
- Petrova, T.V.; Zakharov, E.S.; Samiya, R.; Abramson, N.I. Phylogeography of the Narrow-Headed Vole Lasiopodomys (Stenocranius) gregalis (Cricetidae, Rodentia) Inferred from Mitochondrial Cytochrome b Sequences: An Echo of Pleistocene Prosperity. J. Zool. Syst. Evol. Res. 2015, 53, 97–108. [Google Scholar] [CrossRef]
- Lissovsky, A.A.; Obolenskaya, E.V.; Petrova, T.V. Morphological and Genetic Variation of Narrow-Headed Voles Lasiopo-domys gregalis from South-East Transbaikalia. Russ. J. Theriol. 2013, 12, 83–90. [Google Scholar] [CrossRef]
- Petrova, T.V.; Tesakov, A.S.; Kowalskaya, Y.M.; Abramson, N.I. Cryptic Speciation in the Narrow-Headed Vole Lasiopodomys (Stenocranius) gregalis (Rodentia: Cricetidae). Zool. Scr. 2016, 45, 618–629. [Google Scholar] [CrossRef]
- Abramson, N.I.; Rodchenkova, E.N.; Kostygov, A.Y. Genetic Variation and Phylogeography of the Bank Vole (Clethrionomys glareolus, Arvicolinae, Rodentia) in Russia with Special Reference to the Introgression of the MtDNA of a Closely Related Species, Red-Backed Vole (Cl. rutilus). Russ. J. Genet. 2009, 45, 533–545. [Google Scholar] [CrossRef]
- Bannikova, A.A.; Sighazeva, A.M.; Malikov, V.G.; Golenishchev, F.N.; Dzuev, R.I. Genetic Diversity of Chionomys Genus (Mammalia, Arvicolinae) and Comparative Phylogeography of Snow Voles. Russ. J. Genet. 2013, 49, 561–575. [Google Scholar] [CrossRef]
- Rudá, M.; Žiak, D.; Gauffre, B.; Zima, J.; Martínková, N. Comprehensive Cross-Amplification of Microsatellite Multiplex Sets across the Rodent Genus Microtus. Mol. Ecol. Resour. 2009, 9, 974–978. [Google Scholar] [CrossRef]
- Petrova, T.V.; Genelt-Yanovskiy, E.A.; Lissovsky, A.A.; Chash, U.-M.G.; Masharsky, A.E.; Abramson, N.I. Signatures of genetic isolation of the three lineages of the narrow-headed vole Lasiopodomys gregalis (Cricetidae, Rodentia) in a mosaic steppe landscape of South Siberia. Mamm. Biol. 2021, 101, 275–285. [Google Scholar] [CrossRef]
- Thompson, J.D.; Higgins, D.G.; Gibson, T.J. CLUSTAL W: Improving the Sensitivity of Progressive Multiple Sequence Alignment through Sequence Weighting, Position-Specific Gap Penalties and Weight Matrix Choice. Nucleic Acids Res. 1994, 22, 4673–4680. [Google Scholar] [CrossRef] [Green Version]
- Hall, T.A. BioEdit: A User-Friendly Biological Sequence Alignment Editor and Analysis Program for Windows 95/98/NT. Nucleic Acids Symp. Ser. 1999, 41, 95–98. [Google Scholar]
- Ronquist, F.; Teslenko, M.; van der Mark, P.; Ayres, D.L.; Darling, A.; Höhna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. MrBayes 3.2: Efficient Bayesian Phylogenetic Inference and Model Choice Across a Large Model Space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef] [Green Version]
- Rambaut, A.; Drummond, A.J.; Xie, D.; Baele, G.; Suchard, M.A. Posterior Summarization in Bayesian Phylogenetics Using Tracer 1.7. Syst. Biol. 2018, 67, 901–904. [Google Scholar] [CrossRef] [Green Version]
- Jombart, T. Adegenet: A R Package for the Multivariate Analysis of Genetic Markers. Bioinformatics 2008, 24, 1403–1405. [Google Scholar] [CrossRef] [Green Version]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2021. [Google Scholar]
- RStudio Team. RStudio: Integrated Development for R. RStudio; RStudio, PBC: Boston, MA, USA, 2020. [Google Scholar]
- Adamack, A.T.; Gruber, B. PopGenReport: Simplifying Basic Population Genetic Analyses in R. Methods Ecol. Evol. 2014, 5, 384–387. [Google Scholar] [CrossRef]
- Chakraborty, R.; Zhong, Y.; Jin, L.; Budowle, B. Nondetectability of Restriction Fragments and Independence of DNA Fragment Sizes within and between Loci in RFLP Typing of DNA. Am. J. Hum. Genet. 1994, 55, 391–401. [Google Scholar] [PubMed]
- Brookfield, J.F.Y. A Simple New Method for Estimating Null Allele Frequency from Heterozygote Deficiency. Mol. Ecol. 1996, 5, 453–455. [Google Scholar] [CrossRef] [PubMed]
- Weir, B.S.; Cockerham, C.C. Estimating F-Statistics for the Analysis of Population Structure. Evolution 1984, 38, 1358. [Google Scholar] [CrossRef]
- Jost, L. G ST and Its Relatives Do Not Measure Differentiation. Mol. Ecol. 2008, 17, 4015–4026. [Google Scholar] [CrossRef] [PubMed]
- Goudet, J. Hierfstat, a Package for r to Compute and Test Hierarchical F-Statistics. Mol. Ecol. Notes 2005, 5, 184–186. [Google Scholar] [CrossRef] [Green Version]
- Paradis, E. Pegas: An R Package for Population Genetics with an Integrated-Modular Approach. Bioinformatics 2010, 26, 419–420. [Google Scholar] [CrossRef] [Green Version]
- Pritchard, J.K.; Stephens, M.; Donnelly, P. Inference of Population Structure Using Multilocus Genotype Data. Genetics 2000, 155, 945–959. [Google Scholar] [CrossRef]
- Earl, D.A.; vonHoldt, B.M. STRUCTURE HARVESTER: A Website and Program for Visualizing STRUCTURE Output and Implementing the Evanno Method. Conserv. Genet. Resour. 2012, 4, 359–361. [Google Scholar] [CrossRef]
- Karger, D.N.; Conrad, O.; Böhner, J.; Kawohl, T.; Kreft, H.; Soria-Auza, R.W.; Zimmermann, N.E.; Linder, H.P.; Kessler, M. Climatologies at High Resolution for the Earth’s Land Surface Areas. Sci. Data 2017, 4, 170122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barber, R.A.; Ball, S.G.; Morris, R.K.A.; Gilbert, F. Target-group Backgrounds Prove Effective at Correcting Sampling Bias in Maxent Models. Divers. Distrib. 2022, 28, 128–141. [Google Scholar] [CrossRef]
- Muscarella, R.; Galante, P.J.; Soley-Guardia, M.; Boria, R.A.; Kass, J.M.; Uriarte, M.; Anderson, R.P. ENMeval: An R Package for Conducting Spatially Independent Evaluations and Estimating Optimal Model Complexity for Maxent Ecological Niche Models. Methods Ecol. Evol. 2014, 5, 1198–1205. [Google Scholar] [CrossRef] [Green Version]
- Kass, J.M.; Muscarella, R.; Galante, P.J.; Bohl, C.L.; Pinilla-Buitrago, G.E.; Boria, R.A.; Soley-Guardia, M.; Anderson, R.P. ENMeval 2.0: Redesigned for Customizable and Reproducible Modeling of Species’ Niches and Distributions. Methods Ecol. Evol. 2021, 12, 1602–1608. [Google Scholar] [CrossRef]
- Phillips, S.J.; Dudík, M.; Schapire, R.E. Maxent Software for Modeling Species Niches and Distributions (Version 3.4.1). 2019. Available online: http://biodiversityinformatics.amnh.org/open_source/maxent/ (accessed on 2 April 2019).
- Phillips, S.J. Package: Maxnet, Version 0.1.4. 2021. Available online: https://github.com/mrmaxent/maxnet (accessed on 6 June 2022).
- Brown, J.L.; Carnaval, A.C. A Tale of Two Niches: Methods, Concepts, and Evolution. Front. Biogeography 2019, 11, e44158. [Google Scholar] [CrossRef] [Green Version]
- Schoener, T.W. The Anolis Lizards of Bimini: Resource Partitioning in a Complex Fauna. Ecology 1968, 49, 704–726. [Google Scholar] [CrossRef]
- Warren, D.L.; Glor, R.E.; Turelli, M. ENMTools: A Toolbox for Comparative Studies of Environmental Niche Models. Ecography 2010, 33, 607–611. [Google Scholar] [CrossRef]
- Lissovsky, A.A.; Dudov, S.V.; Obolenskaya, E.V. Species-Distribution Modeling: Advantages and Limitations of Its Application. 1. General Approaches. Biol. Bull. Rev. 2021, 11, 254–264. [Google Scholar] [CrossRef]
- Kryštufek, B.; Shenbrot, G. Voles and Lemmings (Arvicolinae) of the Palaearctic Region; University of Maribor, University Press: Maribor, Slovenia, 2022; ISBN 978-961-286-611-2. [Google Scholar]
- Macarthur, R.H.; Wilson, E.O. The Theory of Island Biogeography; REV-Revised.; Princeton University Press: Princeton, NJ, USA, 1967; ISBN 978-0-691-08836-5. [Google Scholar]
- Thomas, O. The Duke of Bedford’s Zoological Exploration in Eastern Asia.—IX. List of Mammals from the Mongolian Plateau. Proc. Zool. Soc. Lond. 1908, 78, 104–110. [Google Scholar] [CrossRef]
- Poljakov, I.S. A systematic review of voles inhabiting Siberia. Proc. Acad. Sci. St. Petersburg 1881, 39, 87–94. [Google Scholar]
- Ma, Y. A New Subspecies of the Narrow-Skulled Vole from Inner Mongolia, China. Acta Zootaxonomica Sin. 1965, 2, 183–186. [Google Scholar]
- Waters, J.M.; Fraser, C.I.; Hewitt, G.M. Founder Takes All: Density-Dependent Processes Structure Biodiversity. Trends Ecol. Evol. 2013, 28, 78–85. [Google Scholar] [CrossRef] [PubMed]
- Petrova, T.; Skazina, M.; Kuksin, A.; Bondareva, O.; Abramson, N. Narrow-headed voles species complex (Cricetidae, Rodentia): Evidence for species differentiation inferred from transcriptome data. Diversity 2022, 14, 512. [Google Scholar] [CrossRef]
- Rutovskaya, M.V.; Nikoljski, A.A. The sound signals of the narrow-skulled vole (Microtus gregalis Pall.). Sensornye Sist. 2014, 28, 76–83. [Google Scholar]
- Rutovskaya, M.V. The sound signals of Brandt’s vole (Lasiopodomys brandti). Sensornye Sist. 2012, 26, 31–38. [Google Scholar]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Locus | Range (bp) | Na | Ho | He | Fna1 | Fna2 |
|---|---|---|---|---|---|---|
| Mar049 | 202–248 | 21 | 0.582 | 0.909 | 0.220 | 0.207 |
| Mar080 | 188–240 | 19 | 0.786 | 0.897 | 0.066 | 0.062 |
| Mar076 | 103–138 | 18 | 0.817 | 0.907 | 0.052 | 0.049 |
| MSMoe02 | 142–206 | 19 | 0.739 | 0.880 | 0.087 | 0.081 |
| MSMM2 | 167–202 | 22 | 0.802 | 0.927 | 0.073 | 0.070 |
| Sampling Site | Label | N | Ar | Np | Ho | He | FIS |
|---|---|---|---|---|---|---|---|
| Uldza | 2 | 5 | 3.845 | 4 | 0.710 | 0.747 | 0.156 |
| Elon-Obot | 6 | 6 | 4.144 | 1 | 0.917 | 0.803 | −0.046 |
| Adon-Chelon | 13 | 11 | 3.543 | 1 | 0.771 | 0.740 | 0.008 |
| Kuytun | 17 | 7 | 3.915 | 4 | 0.767 | 0.764 | 0.076 |
| Matakan | 24 | 10 | 4.211 | 7 | 0.715 | 0.822 | 0.188 |
| Indola | 31 | 10 | 2.888 | 3 | 0.548 | 0.574 | 0.071 |
| Choibalsan | 47 | 6 | 3.717 | 1 | 0.770 | 0.742 | 0.040 |
| Khavirga | 50 | 5 | 2.986 | 1 | 0.853 | 0.650 | −0.205 |
| Utochi | 53 | 11 | 3.937 | 3 | 0.704 | 0.775 | 0.162 |
| Sampling Site | Label | 2 | 6 | 13 | 17 | 24 | 31 | 47 | 50 | 53 |
|---|---|---|---|---|---|---|---|---|---|---|
| Uldza | 2 | |||||||||
| Elon-Obot | 6 | 0.021 * | ||||||||
| Adon-Chelon | 13 | 0.066 | 0.047 * | |||||||
| Kuytun | 17 | 0.039 * | 0.028 * | 0.055 | ||||||
| Matakan | 24 | 0.062 | 0.044 | 0.084 | 0.068 | |||||
| Indola | 31 | 0.249 | 0.250 | 0.293 | 0.267 | 0.195 | ||||
| Choibalsan | 47 | 0.116 * | 0.085 | 0.120 | 0.105 | 0.077 * | 0.174 | |||
| Khavirga | 50 | 0.188 | 0.143 | 0.193 | 0.195 | 0.172 | 0.312 | 0.152 | ||
| Utochi | 53 | 0.138 | 0.116 | 0.149 | 0.137 | 0.080 | 0.151 | 0.038 * | 0.094 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Petrova, T.V.; Dvoyashov, I.A.; Bazhenov, Y.A.; Obolenskaya, E.V.; Lissovsky, A.A. An Invisible Boundary between Geographic Ranges of Cryptic Species of Narrow-Headed Voles (Stenocranius, Lasiopodomys, Cricetidae) in Transbaikalia. Diversity 2023, 15, 439. https://doi.org/10.3390/d15030439
Petrova TV, Dvoyashov IA, Bazhenov YA, Obolenskaya EV, Lissovsky AA. An Invisible Boundary between Geographic Ranges of Cryptic Species of Narrow-Headed Voles (Stenocranius, Lasiopodomys, Cricetidae) in Transbaikalia. Diversity. 2023; 15(3):439. https://doi.org/10.3390/d15030439
Chicago/Turabian StylePetrova, Tatyana V., Ivan A. Dvoyashov, Yury A. Bazhenov, Ekaterina V. Obolenskaya, and Andrey A. Lissovsky. 2023. "An Invisible Boundary between Geographic Ranges of Cryptic Species of Narrow-Headed Voles (Stenocranius, Lasiopodomys, Cricetidae) in Transbaikalia" Diversity 15, no. 3: 439. https://doi.org/10.3390/d15030439
APA StylePetrova, T. V., Dvoyashov, I. A., Bazhenov, Y. A., Obolenskaya, E. V., & Lissovsky, A. A. (2023). An Invisible Boundary between Geographic Ranges of Cryptic Species of Narrow-Headed Voles (Stenocranius, Lasiopodomys, Cricetidae) in Transbaikalia. Diversity, 15(3), 439. https://doi.org/10.3390/d15030439